

Abstract
Barrier to autointegration factor (BAF) binds double-stranded DNA, selected histones, transcription regulators, lamins, and LAP2–emerin–MAN1 (LEM) domain proteins. During early Caenorhabditis elegans embryogenesis, BAF-1 is required to organize chromatin, capture segregated chromosomes within the nascent nuclear envelope, and assemble lamin and LEM domain proteins in reforming nuclei. In this study, we used C. elegans with a homozygous deletion of the baf-1 gene, which survives embryogenesis and larval stages, to report that BAF-1 regulates maturation and survival of the germline, cell migration, vulva formation, and the timing of seam cell fusion. In the seam cells, BAF-1 represses the expression of the EFF-1 fusogen protein, but fusion still occurs in C. elegans lacking both baf-1 and eff-1. This suggests the existence of an eff-1–independent mechanism for cell fusion. BAF-1 is also required to maintain the integrity of specific body wall muscles in adult animals, directly implicating BAF in the mechanism of human muscular dystrophies (laminopathies) caused by mutations in the BAF-binding proteins emerin and lamin A.
Introduction
Barrier to autointegration factor (BAF) was first discovered as a host component of retroviral preintegration complexes, which is required for integrase-mediated retroviral DNA insertion into target DNA in vitro (Chen and Engelman, 1998; Lee and Craigie, 1998; Lin and Engelman, 2003). BAF is highly conserved in metazoan evolution (52% identical between human and Caenorhabditis elegans; Margalit et al., 2005) and typically localizes at the nuclear periphery, nucleoplasm, and cytoplasm (Segura-Totten and Wilson, 2004). BAF binds directly to many different partners, including double-stranded DNA, histone H3, and certain linker histones, and to all characterized LAP2–emerin–MAN1 (LEM) domain proteins plus lamins and selected homeodomain transcription activators (Segura-Totten and Wilson, 2004; Montes de Oca et al., 2005). These interactions are regulated, at least in part, by the posttranslational modification of BAF (Bengtsson and Wilson, 2006; Gorjánácz et al., 2006; Nichols et al., 2006). In C. elegans, the nuclear envelope localization of BAF-1 protein (previously termed Ce-BAF) depends on its interaction with two inner nuclear membrane LEM domain proteins encoded by emr-1 (Ce-emerin) and lem-2 (Ce-lem2; formerly known as Ce-MAN1; Liu et al., 2003). In turn, BAF-1 is required to localize both LEM domain proteins and Ce-lamin in embryonic cells, suggesting mutual structural interdependence (Margalit et al., 2005). RNAi-mediated down-regulation in C. elegans of either baf-1 or lmn-1 or double down-regulation of both emr-1 and lem-2 caused chromosome segregation defects and failure to properly assemble daughter nuclei (Liu et al., 2003). In mammalian cells, the ectopic expression of mutant BAF that cannot bind DNA or LEM domain proteins dominantly blocked the recruitment of lamin A and the LEM domain proteins emerin and LAP2β but had no effect on B-type lamins or on LBR (lamin B receptor protein; Haraguchi et al., 2001).
Structural roles were also seen in Xenopus laevis egg extracts, in which nuclei can assemble in vitro; adding recombinant BAF either inhibited or enhanced nuclear assembly, depending on the amount of BAF added (Segura-Totten et al., 2002), suggesting important roles for BAF in organizing chromatin and the nucleus. Indeed, BAF is essential in both C. elegans (Margalit et al., 2005) and Drosophila melanogaster (Furukawa et al., 2003). BAF-null Drosophila die at the larval-pupal transition, when they run out of maternally deposited BAF, with phenotypes that include arrest at various stages of the cell cycle, chromatin clumping, abnormal lamin distribution, aberrant nuclear morphology, small brains, and missing imaginal discs (Furukawa et al., 2003). RNAi-mediated down-regulation of baf-1 in C. elegans revealed that the loss of both maternal and zygotic BAF-1 caused anaphase chromatin bridges, abnormal chromatin morphology, and chromosome missegregation as early as the two-cell stage, and embryos died at or before the ∼100-cell stage (Zheng et al., 2000; Margalit et al., 2005). The few embryos that escaped baf-1(RNAi) lethality grew into sterile adults with misplaced distal tip cells and gonads (Margalit et al., 2005).
To better understand the potential cellular and developmental roles of BAF, we studied a loss of function mutation in baf-1(gk324). In animals homozygous for the gk324 allele, the maternal supply of BAF-1 was sufficient to allow these animals to complete embryogenesis and larval stages, bypassing BAF-1's mitotic roles and allowing us to focus on later stages of C. elegans development. This genetic analysis reveals several novel tissue-specific roles for BAF-1 and sheds new light on the disease mechanisms of Emery-Dreifuss muscular dystrophy, which is caused by mutations in each of BAF's direct binding partners emerin and lamin A.
Results
BAF-1 is ubiquitously expressed and enriched at the nuclear envelope throughout development
In early C. elegans embryos, BAF-1 is enriched near the nuclear inner membrane (Margalit et al., 2005). To follow BAF-1 expression, localization, and dynamics in larval and adult stages, we prepared a construct in which the gfp ORF was fused to the 5′ end of the complete baf-1 ORF and driven by the baf-1 promoter (Fig. 1 A). Microparticle bombardment (Praitis et al., 2001) was used to create three independent stable transgenic lines expressing the GFP–BAF-1 fusion protein. GFP–BAF-1 localized primarily at the nuclear envelope, with weaker signals in both the nucleoplasm and cytoplasm (Fig. 1, B–E), as seen previously for endogenous BAF-1 (Margalit et al., 2005). GFP–BAF-1 was expressed ubiquitously throughout C. elegans development, as seen in gonads (Fig. 1 B, arrowhead), early embryos (Fig. 1 B, arrows), late embryos (not depicted), all larval stages (Fig. 1 C; shows L1), and adults (Fig. 1 D; adult vulva indicated by an arrow). The localization of GFP–BAF-1 at different stages of the cell cycle was similar to that of endogenous BAF-1 (Margalit et al., 2005; and unpublished data) and human BAF (Haraguchi et al., 2001), including its localization at the core region of chromosomes during late anaphase/telophase (unpublished data). The mobility of GFP–BAF-1 was measured by FRAP in worms lacking endogenous BAF-1 (Fig. 2; see the following section). GFP–BAF-1 recovered rapidly with a half-time of 2.24 ± 0.66 s (n = 4), which is somewhat less mobile than human BAF (0.26 s; Shimi et al., 2004). One possible explanation for this difference is that the mobility of human BAF was measured in the presence of endogenous BAF.
Figure 1.

GFP–BAF-1 protein expression. (A) Schematic view of the baf-1– gfp construct used for generating transgenic strains. It includes the baf-1 promoter driving the gfp gene fused to the baf-1 ORF followed by the baf-1 3′ untranslated region. (B–D) A stronger GFP–BAF-1 signal was detected at the nuclear periphery, whereas weaker signals were detected in the nucleoplasm and cytoplasm. GFP–BAF-1 was detected in the gonad (arrowhead in B) in young embryos (arrows in B) and in late embryos (not depicted), in L1 larvae (C), and in adult worms (arrow in D points toward the vulva). (E and F) DIC, left; GFP fluorescence, right. Although GFP–BAF-1 was localized normally at the nuclear periphery of control embryos (L4440; E), it was mislocalized in lmn-1(RNAi) embryos (arrowheads in F), where the GFP–BAF-1 was also found associated with anaphase chromatin bridges (arrow in F). The image in B was obtained with a confocal microscope, whereas images in C–F were viewed with an Axioplan II microscope (Carl Zeiss MicroImaging, Inc.). Bars, 10 μm.
Figure 2.

FRAP analysis of GFP–BAF-1 mobility. Cells from gk324/gk324 worms expressing GFP–BAF-1 were photobleached, and GFP–BAF-1 fluorescence recovery was measured. (A) Time-lapse images of a single cell in an adult living worm recorded at 0.12-s intervals starting 3.72 s before photobleaching and ending 32.16 s after photobleaching. Selected images are shown. GFP fluorescence was photobleached in the boxed area shown in the second panel. (B) The normalized fluorescence intensity in the bleached area shown in A is plotted as a function of time. Bar, 5 μm.
GFP–BAF-1 was mislocalized in embryos with RNAi–down-regulated Ce-lamin expression (Fig. 1 F, arrowheads) and associated with anaphase-bridged chromatin (Fig. 1 F, arrow), which is similar to BAF-1 behavior in embryos down-regulated for both emr-1 and lem-2 (Liu et al., 2003) and is consistent with the behavior of endogenous BAF-1 during mitosis (Margalit et al., 2005). GFP–BAF-1 localized normally in control animals fed with the empty L4440 vector (Fig. 1 E). Together with its ability to rescue most phenotypes caused by the lack of endogenous BAF-1 (see the following section), we conclude that the expression, localization, and dependence on lamins for its assembly of GFP–BAF-1 were comparable with those of endogenous BAF-1.
baf-1–null animals have diverse tissue-specific phenotypes
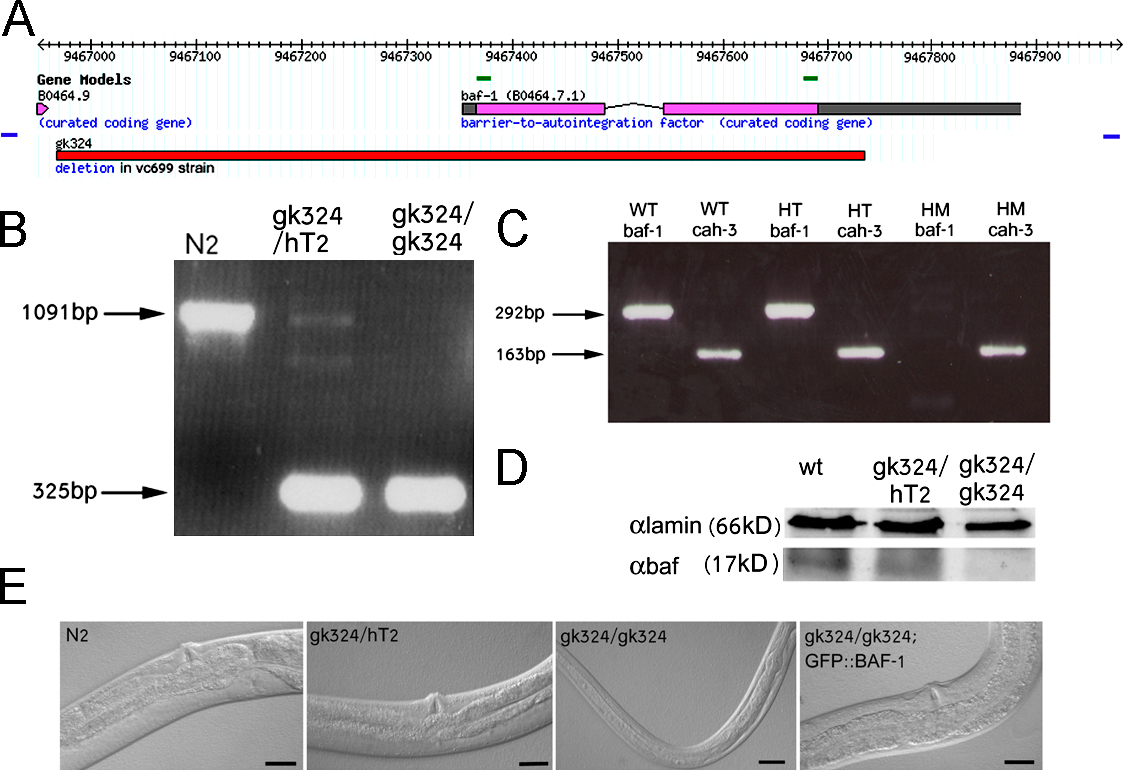
The VC699 strain contains the gk324 allele in which the baf-1 promoter and ORF are deleted (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200704049/DC1). This allele was outcrossed three times and balanced with the hT2 balancer, which carries an integrated pharyngeal GFP element. PCR analysis was used to confirm the 766-bp deletion in the gk324 allele (Fig. S1 B). baf-1 mRNA was undetectable by RT-PCR analysis of RNA from 4-d-old gk324/gk324 homozygous animals (Fig. S1 C). Controls showed that an unrelated transcript encoded by cah-3 was detected at similar levels in homozygous, heterozygous, and wild-type animals (Fig. S1 C). BAF-1 protein was not detected in Western blots of extracts from 4-d-old gk324/gk324 animals, whereas their Ce-lamin signal was comparable with wild type (Fig. S1 D). Heterozygous gk324/hT2 animals were indistinguishable from wild type (N2), with similar body size and normal brood size (unpublished data) despite their ∼40% reduced levels of BAF-1. In contrast, homozygous gk324/gk324 animals, which apparently used maternally supplied BAF-1 to complete embryogenesis and larval stages, were ∼50% thinner and ∼35% shorter and arrested at late L4/early adult stage with several tissue-specific phenotypes, as detailed in the following sections. The short/thin phenotype could be the result of normal numbers of smaller cells, fewer cells, and/or abnormal gonads.
BAF-1 is required to prevent the premature fusion of seam cells to the epidermis
Homozygous gk324/gk324 animals frequently exploded when touched, suggesting cuticle defects. Worms contain two syncytial rows of seam cells that interrupt the hypodermis and form alae on the cuticle surface during certain developmental stages (Fig. 3 A). During the L1–3 stages, the epithelial seam cells positioned along each side of the worm proliferate to create two cell types: epidermal daughter cells that fuse to the main body hyp7 hypodermis and ectoblastic daughter cells that remain unfused (Fig. 3 A; Sulston and Horvitz, 1977). During the L4 stage, these unfused ectoblastic seam cells, which extend from the tail to the head, fuse to each other laterally to form two syncytia on each side of the animal that eventually create cuticular structures (Fig. 3 A; Podbilewicz and White, 1994). To visualize the borders of the ectoblastic seam cells, we crossed the apical junction marker AJM-1–GFP into heterozygous gk324/hT2 worms (Shemer et al., 2000) and examined progeny that were homozygous or heterozygous for the baf-1 deletion. Until the late L3 stage, seam cells in both homozygous and heterozygous worms behaved like wild-type animals, forming one row of ∼12 cells on each side of the worm (unpublished data). In heterozygous gk324/hT2 animals, the ectoblastic seam cells remained unfused at the late L3/early L4 stage (Fig. 3 B, a; arrows), which is similar to wild-type animals (Podbilewicz and White, 1994). In contrast, the ectoblastic seam cells of homozygous gk324/gk324 animals fused prematurely with the epidermis, which was visualized by loss of the GFP signal at the apical borders of the seam cells (Fig. 3 B, b; arrows). Differential interference contrast (DIC) analysis revealed that at L3, after ectopic fusion, the number of seam cells remained at ∼12 (unpublished data). By late L4/early adult stage, all seam cells had fused to the epidermis instead of to each other in all tested worms (n = 20; Fig. 3 B, c; arrows). DIC analysis showed that after that stage, the seam cells disappeared. These cells did not stain for SYTO 11 (Invitrogen; Fridkin et al., 2004), indicating that their disappearance probably did not involve apoptosis (unpublished data). In addition, DIC analysis revealed no apoptotic bodies. This phenotype was specific for BAF-1 because in gk324/gk324 animals that expressed both GFP–BAF-1 and AJM-1–GFP transgenes, the seam cells remained unfused until the late L4 stage, as in heterozygous worms, and later fused correctly to each other to form lateral syncytia (Fig. 3 B, d; arrow). We conclude that BAF-1 is required to prevent the premature fusion of seam cells to the epidermis.
Figure 3.

Baf-1 deletion (gk324/gk324) homozygous animals have premature and ectopic fusion of the seam cells. (A) An illustration of seam cell fusion during larval development adapted from Shemer and Podbilewicz (2003). The epithelial seam cells divide during each larval molt, yielding epidermal daughter cells (which fuse to the hypodermis) and ectoblastic daughter cells, which stay unfused, forming single, lengthwise rows along the left and right sides (L1). These aligned cells fuse to themselves and form two syncytia that will form specific cuticle structures (L4/A). (B, a–d) Fluorescence micrographs of animals either homozygous or heterozygous for a baf-1 deletion expressing AJM-1–GFP. In gk324/hT2 animals, the ectoblastic seam cells stayed unfused at the late L3/early L4 stage (arrows in a). In gk324/gk324 animals, the seam cells ectopically fused to the epidermis (arrows in b), and, by the late L4/A stage, all of the seam cells fused to the epidermis (arrows in c). Transgenic expression of GFP–BAF-1 in gk324/gk324 animals rescued the cell fusion phenotype, allowing L4 stage seam cells to fuse to themselves and form two parallel syncytia (arrow in d); the GFP–BAF-1 nuclear signal is not in focus. Bars, 10 μm.
BAF-1 represses eff-1 in the seam cells
The premature fusion of the seam cells implies that BAF-1 represses the expression of key genes involved in cell fusion. EFF-1 is a cell surface protein that directly mediates most somatic cell fusion events in C. elegans (Shemer et al., 2004; Podbilewicz et al., 2006). Therefore, we tested whether the loss of BAF-1 expression causes the early expression of eff-1 in ectoblastic daughter seam cells. We generated a gk324/hT2 strain expressing GFP driven by the eff-1 promoter (Podbilewicz et al., 2006) and followed GFP expression in the seam cells of gk324/hT2 and gk324/gk324 offspring animals. At the L3 stage, the epidermal daughter cells of the seam cells in gk324/hT2 worms that fused to the hyp7 hypodermis expressed eff-1 (Fig. 4 A, arrow), whereas the ectoblastic daughter seam cells that remained unfused did not show a detectable GFP signal (Fig. 4 A, arrowhead), which is similar to wild-type animals (Shemer et al., 2004). In contrast, the eff-1–driven GFP expression was strong (>3.5-fold) in the presumed prematurely fused ectoblastic seam cells of gk324/gk324 L3-stage animals (Fig. 4 B, arrow).
Figure 4.

BAF-1 represses EFF-1 and can prevent premature fusion in an EFF-1–independent pathway in the seam cells. (A and B) Fluorescence micrographs of animals either homozygous or heterozygous for a baf-1 deletion expressing GFP driven by the eff-1 promoter. In gk324/hT2 L3 animals, the epidermal seam cells were fused and express low levels of EFF-1 (arrow in A), and the ectoblastic seam cells stayed unfused and did not express EFF-1 (arrowhead in A). On the other hand, in gk324/gk324, the ectoblastic seam cells did not form a longitude row, and the fused seam cells have strong GFP signal (arrow in B). (C–H) Fluorescence micrographs of gk324/hT2;eff-1(ok1021)/eff-1(ok1021) (heterozygous) or gk324/gk324;eff-1(ok1021)/eff-1(ok1021) (homozygous) animals expressing AJM-1–GFP at L2 (C and D), L3 (E and F), or L4 (G and H). At the L2 stage, the ectoblastic daughter seam cells in both baf-1 heterozygous and homozygous animals did not fuse because of the EFF-1 mutation (arrows in C and D), and, in heterozygous animals, the seam cells migrated (arrowhead in C). At the L3 and L4 stages, in baf-1 heterozygous worms, the unfused seam cells migrated ventrally and dorsally and formed several rows (arrows in E and G). In homozygous worms, the seam cells prematurely fuse and degenerated at the L3 stage (arrow in F). By L4, all seam cells fused or degenerated (arrow in H). (I) Suggested model of the function of BAF-1 in regulating the fusion of ectoblastic seam cells by repressing EFF-1 and AFF-1 expression and the consequence of mutations in baf-1. Bars, 10 μm.
GFP–BAF-1 binds to the eff-1 promoter
The ability of BAF-1 to bind DNA and chromatin (Margalit et al., 2007) and to repress eff-1 expression (see previous section) suggested that BAF-1 regulates eff-1 expression by binding to its promoter. To test this hypothesis, we used a GFP antibody and the chromatin immunoprecipitation (ChIP) assay on worms expressing GFP–BAF-1 (Yan et al., 2006; Ercan et al., 2007). We specifically analyzed the binding of GFP–BAF-1 to the eff-1 promoter (Fig. 5 A) compared with the coding and intronic regions of eff-1. A GFP antibody and the ChIP assay on wild-type worms and worms expressing AJM-1–GFP were used as controls for nonspecific binding to the same regions. The binding of BAF-1 to eff-1 promoter regions spanning from −2,165 to −2,057, −894 to −795, or −289 to −180 (Fig. 5, regions 4, 3, and 1) was 5.3–8.5-fold higher than its binding to eff-1 coding and intronic regions, 3.7–6.5-fold higher than nonspecific binding to the same region in wild-type animals, and 5.3–8.5-fold higher than its binding to the same region in worms expressing AJM-1–GFP. GFP–BAF-1 bound to two other regions in the promoter (−2,696 to −2,560 and −656 to −530; Fig. 5, regions 5 and 2) to the same extent as nonspecific antibody controls or to GFP control. Furthermore, GFP–BAF-1 did not show increased binding to two regions in the actin (act-1) promoter as compared with either coding and intronic regions of act-1 or with wild-type control (Fig. 5). We conclude that BAF-1 represses EFF-1 expression probably by binding directly to the eff-1 promoter.
Figure 5.

Testing BAF-1–binding sites in the eff-1, aff-1, and act-1 promoters. (A) Regions in act-1, eff-1, and aff-1 promoters were tested in wild-type, PS3729, YG1001, and YG1002 strains by ChIP assay for GFP–BAF-1 or AJM-1–GFP binding. The tested regions were as follows: (1) −580 to −435 and (2) −793 to −673 for act-1; (1) −279 to −180, (2) −656 to −530, (3) −894 to −795, (4) −2,165 to −2,057, and (5) −2,696 to −2,560 for eff-1; and (1) −329 to −209 and (2) −535 to −390 for aff-1. (B) Worms expressing AJM-1–GFP (gray bars), GFP–BAF-1 (checkered bars), or wild-type worms (black bars) were subjected to ChIP using GFP antibodies. The amount of precipitated DNA was quantified by quantitative PCR using specific primers and compared with that of the coding and intronic region. The graph represents the fold enrichment values for the different promoter regions in act-1, eff-1, and aff-1 genes normalized to the coding and intronic region in each gene. Data represent the mean and SD (error bars) of the fold enrichment from three to five experiments.
baf-1 deletion can cause the premature fusion of seam cells in an EFF-1–independent pathway
We next wanted to test whether the premature fusion of seam cells in gk324/gk324 animals depends only on EFF-1. Therefore, we introduced an eff-1–null deletion, eff-1(ok1021) (Podbilewicz et al., 2006), into gk324/hT2 animals expressing the apical junction marker AJM-1–GFP and tested AJM-1–GFP expression in gk324/hT2;eff-1(ok1021)/eff-1(ok1021) and gk324/gk324;eff-1(ok1021)/eff-1(ok1021) offspring animals. At the L2 stage, the seam cells remained unfused in both heterozygous and homozygous worms for the gk324 allele (Fig. 4, C and D; arrows). During the L3 stage, the seam cells of gk324/gk324;eff-1(ok1021)/eff-1(ok1021) double homozygous worms did not migrate but probably fused to the hypodermis (Fig. 4 F, arrow), and, during the L4 stage, the seam cells became very small and then disappeared (Fig. 4 H, arrow). These cells were SYTO 11 negative, suggesting that they did not undergo apoptosis (unpublished data). Likewise, DIC analysis could not find apoptotic bodies. In contrast, the seam cells of the L3- or L4-stage gk324/hT2;eff-1(ok1021)/eff-1(ok1021) baf-1 heterozygous worms did not fuse and continued to migrate, forming several rows of cells (Fig. 4, E and G; arrows; Mohler et al., 2002). We concluded that BAF-1 inhibits premature fusion of the seam cells probably by repressing both EFF-1 fusion–dependent and –independent pathways. AFF-1 fusogen was recently found to also be active in seam cell fusion independently of EFF-1 (Sapir et al., 2007). To test whether BAF-1 can bind aff-1 promoter regions, we used a GFP antibody and the ChIP assay on worms expressing GFP–BAF-1 (Fig. 5). The binding of BAF-1 to aff-1 promoter regions spanning from −535 to −390 or from −329 to −209 (regions 2 and 1, respectively; Fig. 5) was 4–4.6-fold higher than its binding to aff-1 intronic and coding regions. We concluded that BAF-1 inhibits premature fusion of the seam cells to the hypodermis probably by regulating both EFF-1 and AFF-1 pathways (Fig. 4 I).
BAF-1 is required for vulva formation
During the L3 stage, the vulva begins to form in the ventral epidermis when the anchor cell induces three (P5.p, P6.p, and P7.p) of the six vulval precursor cells (VPCs) to divide and to adopt vulval fates (Sharma-Kishore et al., 1999; Sommer, 2001). Strikingly, all gk324/gk324 animals were vulvaless (n = 50; Fig. 6 D; normal vulva in the control worm is indicated by an arrow in Fig. 6 C). To determine whether the lack of vulva formation is caused by a missing anchor cell, we introduced the anchor cell–specific marker CDH-3–GFP (Pettitt et al., 1996) into gk324/hT2 worms. During the L3 and L4 stages, CDH-3–GFP expression was detected in the anchor cell in both baf-1 heterozygous and homozygous animals (Fig. 6, A–D). We next followed the VPCs in the offspring of AJM-1–GFP;gk324/hT2 animals and wild-type worms expressing AJM-1–GFP. In early L3 stage, the six VPCs, P3.p to P8.p, were present in both wild-type and baf-1 homozygous gk324/gk324 worms (Fig. 6, E and F; arrows). During the L3 and L4 stages, the VPCs continued to divide in wild-type worms and formed a normal vulva structure (Fig. 6 G; arrow indicates vulva at the late L3 stage). However, in 73% of gk324/gk324 worms, the second VPC division did not occur, and, in 27% of the worms, only one division occurred, usually that of P6.p (Fig. 6 H, arrowhead). During the L4 stage, all VPCs degenerated but did not stain for SYTO 11 (unpublished data). The vulvaless phenotype of gk324/gk324 animals was specific to the loss of BAF-1 expression because vulva formation was fully rescued in strains expressing the GFP–BAF-1 transgene, as seen in L4 larvae (Fig. 6, I and J; arrows) and adults (Fig. 6 K, arrow). We conclude that BAF-1 is required for VPC divisions and vulva formation either at the level of anchor cell signaling, defects in responding cells, or both.
Figure 6.

BAF-1 is required for vulva formation. (A–D) L3 (A and B) and L4 (C and D) larvae imaged using DIC (left) or epifluorescence (right). CDH-3–GFP is expressed at the correct position in both heterozygous gk324/hT2 animals (arrows in A and C) and baf-1–null homozygous gk324/gk324 animals (arrows in B and D) at both larval stages. The anchor cell fused to the ventral uterine π cells in heterozygous animals (C) but not in homozygous animals (D). (E–H) AJM-1–GFP expression in vulva precursor cells (VPCs) of wild-type (E and G) and gk324/gk324 (F and H) animals. At the early L3 stage, all six VPCs were visualized in both wild-type (E) and gk324/gk324 worms (F). In wild-type worms at late L3, VPCs formed a normal vulva structure (G; 18-cell stage). In most gk324/gk324 worms, the VPCs either did not divide at all (not depicted) or had a partial division of P6.p (arrowhead in H). Arrows in E–H indicate VPCs. (I–K) Vulva formation in baf-1–null homozygous animals is rescued by the transgenic expression of GFP–BAF-1, as shown at early L4 (arrow in I), late L4 (arrow in J), and young adult (arrow in K). Bars, 10 μm.
BAF-1 is required for germline maintenance and maturation
Homozygous gk324/gk324 animals became sterile. The gonads of early L3 gk324/gk324 animals were similar in size to wild-type gonads, with similar numbers of germ cells compared with gk324/hT2 or wild-type animals (mean of 32 ± 5 germ cells in gk324/hT2 worms [n = 12] and 30 ± 9 germ cells in gk324/gk324 worms [n = 12]). However, during the L4 and adult stages, both gonad size and germ cell numbers were substantially reduced in gk324/gk324 animals (102 ± 30 germ cells in gk324/hT2 at L4 [n = 12], 58 ± 4 germ cells in gk324/gk324 worms at the L4 time stage [n = 12], 212 ± 26 germ cells in young adult gk324/hT2 [n = 12], and 36 ± 19 germ cells in gk324/gk324 worms at the young adult time stage [n = 12]; Fig. 7, C and D; DAPI stain) compared with gk324/hT2 animals (Fig. 7, A and B; DAPI stain). Fewer germ cells in adult gk324/gk324 gonads suggested germline cell proliferation defects, germ cell degeneration, or both. These findings were consistent with essential roles for BAF-1 in cell proliferation, as reported in Drosophila (Furukawa et al., 2003), and also with potentially essential roles in meiosis. Gametes were never seen in gk324/gk324 animals (unpublished data). The few surviving germ cells expressed the germline marker protein matefin/SUN-1 (Fig. 7 C; Fridkin et al., 2004), suggesting that BAF-1 was not essential for early germ cell differentiation. These phenotypes were specific for the loss of BAF-1 expression because gk324/gk324 animals expressing the GFP–BAF-1 transgene had normal sized gonads and apparently normal numbers of germ cells (Fig. 7 E) plus sperm cells that appeared to be wild type but were less confined to a specific area in the gonads. There were cells at the pachytene stage, but oocytes were not formed (unpublished data). The gonad size phenotype independently supported our previous conclusion based on animals that escaped the lethal consequences of RNAi-mediated baf-1 down-regulation that BAF-1 is required for gonad development (Margalit et al., 2005).
Figure 7.

Baf-1 deletion (gk324/gk324) homozygous animals have fewer germ cells, lack mature gametes, and show abnormal lamin organization. (A–E) gk324/hT2 (A and B), gk324/gk324 (C and D), or gk324/gk324 young adults expressing GFP–BAF-1 (E) were DAPI stained to visualize DNA and stained by indirect immunofluorescence using antibodies specific for either endogenous matefin/SUN-1 (A and C; Fridkin et al., 2004) or endogenous Ce-lamin (B, D, and E). HT, heterozygous; HM, homozygous. (F and G) Corresponding DIC images and transgenic Ce-lamin–GFP fluorescence in gk324/gk324 (F) and wild-type (G) adults at day 5 of development. Arrows in F indicate muscle nuclei with abnormal Ce-lamin localization. Insets (C–G, right) show enlarged nuclei. Bars, 10 μm.
BAF-1 is required for the second phase of DTC migration
The position of the gonads was abnormal in baf-1 homozygous animals (unpublished data) and closely resembled the abnormal gonad position in animals under conditions of incomplete RNAi of BAF-1 (Margalit et al., 2005). To specifically test distal tip cell (DTC) migration, we prepared a gk324/hT2 strain expressing lag-2–GFP, which expresses in DTC cells, and followed GFP expression at different stages in gk324/gk324 animals (Fig. 8). At the late L2 and early L3 stages, migration of the two DTCs away from the midbody occurred normally in both gk324/gk324 and gk324/hT2 animals (Fig. 8, A and B). At the late L3 stage, the ventral to dorsal migration of DTCs was normal in gk324/hT2 animals (Fig. 8 C) but failed to occur in gk324/gk324 animals (Fig. 8 D). The DTCs migrated back toward the midbody in gk324/hT2 animals but not in gk324/gk324 animals (unpublished data). The DTCs in gk324/gk324 animals had normal morphology (Fig. 9 B, arrow). DTCs migrated normally in gk324/gk324 animals expressing the GFP–BAF-1 transgene (unpublished data). These results confirmed our previous suggestion that BAF-1 is required for the second and third steps of DTC migration (Margalit et al., 2005) and further demonstrated that this phenotype is specific to BAF-1.
Figure 8.

DTC migration phenotypes in baf-1 deletion (gk324/gk324) homozygous animals. (A–D) Early L3- (A and B) and late L3 (C and D)-stage worms gk324/hT2 (A and C) or gk324/gk324 (B and D), which express GFP driven by lag-2. DIC and GFP fluorescence microscopy images were merged. The DTCs in gk324/hT2 animals migrated normally at phases 1 (A), 2 (C), and 3 (not depicted; Lehmann, 2001). In gk324/gk324 animals, DTCs migrated normally at phase 1 (B) but failed to migrate at phases 2 (D) and 3 (not depicted). Arrows illustrate the direction of DTC migration. Bars, 10 μm.
Figure 9.

Nuclear morphology in gk324/gk324 gonads. Visualization of gk324/hT2 and gk324/gk324 gonads at the L4 stage by thin section transmission electron microscopy. (A) Worms heterozygous for the baf-1 deletion had normal gonads and normal-looking nuclei (arrows in A). (B–D) Worms homozygous for the baf-1 deletion had one or two large-sized nuclei (arrowhead in B), but most gonadal nuclei were small and lobulated (arrows in C). Many of these small nuclei also had gaps in their nuclear envelope (arrowheads in C), additional nuclear membranes (arrow in D), and condensed chromatin (arrowheads in D). The DTC is indicated (arrow in B).
BAF-1 is required for postembryonic organization of the nuclear envelope and chromatin
BAF-1 is required to properly localize Ce-lamin at the nuclear envelope of early embryos (Margalit et al., 2005). To investigate this potential role for BAF-1 at later stages, we introduced Ce-lamin–GFP (Liu et al., 2000) into gk324/hT2 animals and localized Ce-lamin–GFP in gk324/gk324 somatic postembryonic nuclei. Until the L4 stage, Ce-lamin–GFP localization was similar to that of wild-type worms (unpublished data). However, afterward, the Ce-lamin–GFP signal coalesced into one to three strong patches at the nuclear periphery of body muscle cells (Fig. 7 F, inset), epidermal cells, and pharyngeal cells (not depicted). This redistribution was not seen in wild-type worms (Fig. 7 G, inset) and differed from the bright foci of the Ce-lamin–GFP signal that accompany normal aging in C. elegans (Haithcock et al., 2005). In the gk324/gk324 germ cells (Fig. 7 D, inset) but not in control gk324/hT2 germ cells (Fig. 7 B), the Ce-lamin signal was weak and localized primarily in the nuclear interior. This phenotype was fully rescued in gk324/gk324 animals expressing the GFP–BAF-1 transgene (Fig. 7 E, inset). We conclude that BAF-1 is required to organize Ce-lamin in both germline cells and adult somatic nuclei, including muscle nuclei.
The disorganization of Ce-lamin in the germ cell nuclei of gk324/gk324 animals suggested potential gross defects in nuclear architecture. Therefore, we used the thin section transmission electron microscopy method to visualize the nuclear membranes and chromatin of germ cells derived from gk324/gk324 and control animals (Fig. 9). Gonads of control gk324/hT2 animals had normal-appearing nuclei (Fig. 9 A, arrows) and were indistinguishable from wild-type (N2) gonads (not depicted). In contrast, the gonads of gk324/gk324 animals had only a few large nuclei (Fig. 9 B, arrowhead), whereas most nuclei were small and lobulated (Fig. 9 C, arrows). About 65% of these small nuclei (n = 26) had gaps in their nuclear envelope (Fig. 9 C, arrowheads), and 27% had extra layers of nuclear membranes (Fig. 9 D, arrow). In some cases, the chromatin was condensed in electron-dense patches (Fig. 9 D, arrowheads). We speculate that the more drastic phenotype of gapped nuclear envelopes seen in germline cells might be caused by proliferation-linked defects in nuclear assembly. Thus, for germline cells, we conclude that BAF-1 is required not only to organize Ce-lamin but also for nuclear envelope and chromatin organization. Loss of nuclear envelope integrity could account for germline failure, but our results did not distinguish whether this integrity was lost during germ cell mitotic proliferation, meiosis, interphase, or a combination of these events.
Loss of BAF-1 causes rapid deterioration of body and tail muscles
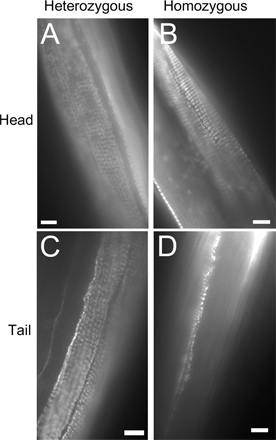
An unexpected finding was that baf-1–null animals had an uncoordinated (unc) phenotype. When grown at 20°C, the movement of gk324/gk324 animals was similar to that of gk324/hT2 or wild-type animals up to day 4 (Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200704049/DC1). From day 5 onwards, all homozygous gk324/gk324 animals (n = 146) developed an uncoordinated movement (Video 2 shows homozygous animals at day 5). In addition, a gradually increasing fraction of animals became paralyzed in the mid- and tail regions but not the head (Video 3 shows homozygous animals at day 12). The fraction of gk324/gk324 animals that could only move their head was 2.4% on day 7, increasing to 21.7% on day 11 and 88.5% on day 18 (n = 130, n = 104, and n = 76, respectively). Control gk324/hT2 animals did not develop uncoordinated movement, and the fraction of paralyzed animals was 0%, 0%, and 32.6% on days 7, 11, and 18, respectively (n = 144, n = 108, and n = 49, respectively). The paralysis phenotype of the gk324/hT2 controls differed from the gk324/gk324 animals because the control worms could still move their tail and were classified as class C aging animals, in which worms do not move even after prodding and can only move their head and/or tail or twitch in response to touch (Herndon et al., 2002). Both the uncoordinated and paralysis phenotypes of the gk324/gk324 animals were completely rescued by the GFP–BAF-1 transgene (unpublished data).
The Unc and paralyzed phenotypes could be caused either by nerve degeneration or muscle cell–intrinsic deterioration. To test the latter possibility, we used the thin section transmission electron microscopy method to examine the morphology of muscle cells in the head and tail regions of wild-type (N2), heterozygous gk324/hT2, and homozygous gk324/gk324 animals grown at 20°C on days 4, 8, and 12. In both control gk324/hT2 and homozygous gk324/gk324 animals, the head muscle tissues were similar to wild-type animals in each age group (Fig. 10, A–F). Muscle cells in the tail region of gk324/hT2 animals had normal morphology in all examined days, including day 12 (Fig. 10, G–I). In striking contrast, muscle cell morphology in gk324/gk324 animals was normal only at day 4 (Fig. 10 J) and deteriorated considerably by days 8 and 12 (Fig. 10, K and L). By day 12, the thin and thick filaments in the tail muscles became misorganized and contained dark aggregates (Fig. 10 L, arrows), which appeared already in day 8 (Fig. 10 K, arrow). Tail muscle cells in homozygous worms also had deteriorated nuclei (unpublished data). A previous study had shown that bodywall muscle cells are necessary for the normal distribution of myotactin, a protein that maintains the association between the muscle contractile apparatus and hypodermal fibrous organelles (Hresko et al., 1999). To further study the muscle deterioration, we immunostained gk324/hT2 and gk324/gk324 worms at day 12 with MH46, an antibody against myotactin. Myotactin distribution was normal at the tail and head of gk324/hT2 (Fig. S2, A and C; available at http://www.jcb.org/cgi/content/full/jcb.200704049/DC1) and at the head of gk324/gk324 worms (Fig. S2 B). However, myotactin distribution at the tail of gk324/gk324 worms was abnormal (Fig. S2 D). We conclude that BAF-1 is required to maintain the integrity of specific muscles in the body.
Figure 10.

Thin-section electron micrographs demonstrating the accelerated deterioration of muscles in the tail region of gk324/gk324 animals. Muscle morphology was normal in the head and tail regions of both gk324/hT2 and gk324/gk324 day 4 worms grown at 20°C (A, D, G, and J) as well as in gk324/hT2 worms at days 8 (B and H) and 12 (C and I). In the gk324/gk324 worms at days 8 and 12, the morphology of head muscles was normal (E and F). On day 8, the tail muscles of gk324/gk324 worms had deteriorated and started to have dense aggregates (arrow in K). At day 12, the tail muscle filaments were misorganized and had dense aggregates (arrows in L).
Discussion
BAF-1 is required for multiple developmental pathways
All phenotypes seen in homozygous baf-1–null (gk324/gk324) animals appeared after the larval L2 stage. These phenotypes affected specific cell types, including seam cells, vulva precursor cells, germ cells, DTCs, and selected muscle cells in the midbody and tail regions. These phenotypes were specific for BAF-1 because a transgene expressing GFP–BAF-1 fully rescued all phenotypes but one: transgenic BAF-1 did not restore the production of oocytes. We speculate that this failure might be caused by (1) insufficient expression of transgenic BAF-1 in germline cells, (2) incomplete activity of the GFP fusion protein, or (3) a second mutation in a gene located near baf-1, which was not removed by three to five outcrosses of the gk324/gk324 strain. Interestingly, C. elegans heterozygotes with a single copy of baf-1, which expressed ∼60% the normal level of BAF-1 protein, were apparently normal with brood sizes similar to wild-type animals (unpublished data). Furthermore, two extra copies of the GFP–BAF-1 transgene (seen in the YG1001-3 strains) also had no apparent effects, although protein levels were not measured. Thus, in contrast to nuclear assembly extracts derived from Xenopus eggs, C. elegans may tolerate a wider range of BAF-1 protein levels.
The mitotic and chromatin phenotypes seen in baf-1(RNAi) C. elegans embryos (Margalit et al., 2005) were seen to a limited extent at later stages of development, specifically in germ cells and in the VPCs, which might reflect a failure to assemble nuclei after mitosis. However, the successful embryonic development of homozygous gk324/gk324 embryos indicates that maternally contributed BAF-1 is sufficient for early development, when most somatic nuclear divisions occur. In this respect, our findings are consistent with findings in Drosophila, wherein maternal D-BAF was sufficient to complete all larval stages in flies homozygous for a baf deletion (Furukawa et al., 2003). At later stages of Drosophila development, the D-BAF deletion caused defects in chromatin organization, including clumped heterochromatin, which is similar to our findings for germ cell nuclei of baf-1–null C. elegans.
Perhaps there were defects in anaphase at postembryonic cells, but we could not see anaphase bridges in any cells examined either by DAPI staining, thin section EM, or DIC microscopy. The lack of anaphase chromatin bridges in somatic nuclei of post-L2 gk324/gk324 animals can be explained by the fact that most somatic cells are not dividing or by the activity of checkpoints that block entry into mitosis. Consistent with the first possibility, we saw severe nuclear morphology defects in proliferating germline cells after the L3 stage. Furthermore, the lack of mitotic figures in germline cells was consistent with the activity (in germline cells) of one or more checkpoints that are inactive in embryos (Encalada et al., 2005). We cannot rule out that many of the phenotypes of baf-1 mutants, including the lack of proliferation of germ cells and VPCs and the thin and uncoordinated phenotype, could be the results of defects in overall postembryonic cell divisions. On the other hand, it is likely that most phenotypes are probably caused by the aberrant regulation of BAF-1–regulated genes because these phenotypes were either absent or different in worms containing mutations in the cell cycle genes (Boxem et al., 1999; Fay and Han, 2000). In addition, the specific binding of BAF-1 to the eff-1 promoter confirms that BAF-1 is directly involved in gene regulation, as suggested previously in mammalian retinal cells (Wang et al., 2002). Given the differences in the timing of various developmental stages in C. elegans and Drosophila and the uncertain rate of loss of maternally provided BAF-1 protein, we are struck by the general similarities between the null phenotypes of these two organisms. In both organisms, BAF is required for efficient mitosis, chromatin organization, and nuclear envelope formation and also has partners involved in regulating tissue-specific roles during development. These results strongly support the hypothesis that many of BAF's roles are conserved in evolution. Our results suggest that BAF has additional partners involving the regulation of tissue-specific functions that remain to be discovered.
BAF-1 blocks the premature fusion of seam cells
In baf-1–null animals, the seam cells fused prematurely at the L3 stage. Most known genes involved in fusion and patterning of the epidermis are transcription factors (Podbilewicz, 2006). Ectopic and premature fusion of seam cells is also seen in worms deleted for ceh-16/engrailed (Cassata et al., 2005), which represses the transcription of a key gene, eff-1, encoding a cell surface protein that directly mediates most somatic cell fusion events in C. elegans (Shemer et al., 2004), including the ventral cell fusions required for vulva formation. The EFF-1 protein must be expressed in both cells for fusion to occur (Podbilewicz et al., 2006). Mutations in Ceh-16/engrailed derepress eff-1 and lead to abnormal fusion events during embryogenesis (Cassata et al., 2005). Ceh-16/engrailed normally blocks seam cell fusion to the syncytial hypodermis during embryogenesis. BAF is also involved in gene regulation. In mouse retinal cells, BAF inhibits gene expression by binding directly to Otx2, Crx, and other paired-like homeodomain transcription activators and blocks Crx-dependent gene expression in vivo (Wang et al., 2002). Our findings demonstrate that BAF-1 has more extensive roles in tissue-specific gene regulation because BAF-1 was required to prevent the premature fusion of seam cells. Our results suggest that BAF-1 normally represses eff-1 during embryogenesis and later stages of development by binding to the eff-1 promoter. It is also worth noting that BAF-1 is currently the only factor known to bind the eff-1 promoter directly.
Although eff-1 expression was altered in gk324/gk324 worms, premature seam cell fusion still occurred in worms homozygous for mutations in both eff-1 and baf-1. The seam cells in these worms are smaller and disappear later. Thus, the eff-1 mutant background may cause additional deleterious effects. We speculate that there are other proteins mediating cell fusion, which are repressed by BAF-1. One such protein is AFF-1, which has fusogenic activities that do not involve EFF-1 activity, especially in anchor cell fusion and fusion between the lateral seam cells (Sapir et al., 2007). Our ChIP analysis suggests that BAF-1 also binds the promoter of aff-1 and, therefore, prevents seam cell fusion by regulating both the aff-1 and eff-1 promoters (Fig. 4 I).
baf-1–null adults develop dystrophic muscles
One of the most intriguing phenotypes of animals homozygous for a baf-1 deletion was the accelerated deterioration of specific muscles in aging animals. This suggests a role for BAF-1 in adult muscle integrity. This finding is consistent with the idea that BAF is required to efficiently localize emerin at the nuclear envelope of mammalian cells (Bengtsson and Wilson, 2006) and Ce-lamin in C. elegans cells (this study). The observed deterioration of muscle cells in C. elegans differed from normal muscle aging (Herndon et al., 2002) in several ways. First, the baf-1–null muscular dystrophy–like phenotype appeared only in midbody and tail muscles (which are innervated by the ventral or dorsal nerve cords), whereas head muscles (which synapse to the nerve ring) remained functional. Consequently, the head muscles often behave differently. During normal aging, C. elegans suffers from loss of muscle mass and the function of muscles in the body wall and pharynx (Herndon et al., 2002). We cannot rule out the possibility that the midbody and tail muscle phenotypes result from problems with the ventral or dorsal nerve cords. However, we disfavor this model because the morphology of the affected muscles was visibly defective on day 8, when the gk324/gk324 animals could still move, and, therefore, nerve cells were still functional. Furthermore, similar phenotypes of the disorganization of muscle cell filaments and gradual paralysis were previously reported in worms with mutations in muscle-related genes such as unc-54 (myosin heavy chain; Tajsharghi et al., 2005), unc-27 (Troponin I; Burkeen et al., 2004), or unc-52 (perlecan; Mackenzie et al., 1978), which also influence DTC migration (Merz et al., 2003). These genes might be regulated directly or indirectly by BAF-1. In addition, muscle attachment to hypodermal cells was also aberrant in the tail of the gk324/gk324 animals. Therefore, we hypothesize that loss of baf-1 function directly disrupts the function of selected muscle cells in C. elegans.
Possible involvement of BAF in laminopathies
In humans, mutations in A-type lamins cause several forms of muscular dystrophy, including autosomal dominant Emery-Dreifuss muscular dystrophy, Limb-Girdle muscular dystrophy, and dilated cardiomyopathy with conduction system defects (Decostre et al., 2005). Mutations in emerin, an inner nuclear membrane LEM domain protein that directly binds to lamin A, cause X-linked recessive Emery-Dreifuss muscular dystrophy, which is clinically indistinguishable from autosomal dominant Emery-Dreifuss muscular dystrophy (Decostre et al., 2005). Both proteins (lamin A and emerin) directly interact with BAF (Holaska et al., 2003), and these interactions are conserved in C. elegans: Ce-emerin and Ce-lamin (B type) each bind BAF-1 directly (Lee et al., 2001; Liu et al., 2003; unpublished data). The selective muscular dystrophy–like phenotype of baf-1–null animals strongly suggests a novel role for BAF in muscle cell integrity, potentially at the level of muscle-specific gene regulation. Future work will aim to identify putative BAF-regulated genes in muscles to shed new light on the mechanisms of Emery-Dreifuss muscular dystrophy and other laminopathies.
Materials and methods
C. elegans strains
C. elegans strains were handled as described previously (Brenner, 1974). Strains N2 (wild type), JK2049, PS3352, PS3729, eff-1(ok1021), and FC121 were obtained from the C. elegans genome center. Strain VC699 containing the gk324 deletion allele of baf-1 (baf-1(gk324) III/hT2[qIs48] (I;III)) was prepared by the C. elegans Reverse Genetics Core Facility at the University of British Columbia. It was outcrossed three times before balancing with hT2. Strain VC699 was crossed with the following strains: PS3352, JK2049, jcls, PS3729, PD4810, YG1001, YG1002, FC121, and eff-1(ok1021). The hT2 balancer was reintroduced to the strains by crossing to gk324/hT2 males. The gk324 genotype was determined by single-worm PCR analysis, and GFP expression was assessed by fluorescence microscopy. The three independent GFP-BAF–expressing strains (YG1001, YG1002, and YG1003) were generated by bombarding the construct pYG1001 (Fig. 1 A).
Microscopy and live cell imaging
Transmission electron microscopy analysis of C. elegans was performed as described previously (Haithcock et al., 2005). DIC and immunofluorescence images were taken either with a CCD camera (Axiocam; Carl Zeiss MicroImaging, Inc.) mounted on a microscope (Axioplan II; Carl Zeiss MicroImaging, Inc.) equipped for fluorescence and DIC or with a confocal scanhead (MRC-1024; Bio-Rad Laboratories) coupled to an inverted microscope (Axiovert 135M; Carl Zeiss MicroImaging, Inc.) equipped with a 63× NA 1.3 oil immersion objective (Carl Zeiss MicroImaging, Inc). For FRAP analysis, gk324/gk324 worms expressing GFP–BAF-1 were imaged using a confocal microscope (FV-1000; Olympus) equipped with an inverted microscope (IX81; Olympus) and a 60× NA 1.4 oil immersion objective (Olympus). GFP–BAF-1 fluorescence was photobleached by a 405-nm laser in a defined region of each cell and was imaged with a 488-nm laser line for excitation and a 505–525-nm filter for emission before, during, and after the photobleach. For FRAP analysis, fluorescence intensity in the bleached area, the backgroup area, and the total cell area were measured as a function of time after bleaching and were normalized essentially as described previously (Rabut and Ellenberg, 2005).
Antibodies, indirect immunofluorescence staining, and immunoblots
Adult C. elegans were fixed and stained by indirect immunofluorescence as described previously (Fridkin et al., 2004). MH46 (Francis and Waterston, 1991) was used at a 1:10 dilution. 135 4-d-old young adults were collected in 30 μl M9 buffer, mixed with 15 μl of 2× SDS sample buffer, boiled for 10 min, and subjected to protein blot analysis as described previously (Margalit et al., 2005).
ChIP
For extract preparations, N2, PS3729, YG1001, or YG1002 asynchronous population worms grown in six 9-cm plates were collected. Worms were washed twice with M9 and fixed with 2% formaldehyde for 30 min at room temperature, washed once with 100 mM Tris, pH 7.5, twice with M9 buffer, and once with homogenization buffer (50 mM Hepes/KOH, pH 7.5, 1 mM EDTA, 140 mM KCl, 0.5% NP-40, 10% glycerol, and 5 mM DTT with protease inhibitors), and frozen in liquid nitrogen. Worms were sonicated on ice 10 times for 30 s each with a sonicator (Sonic; Heat Systems Ultrasonic, Inc.) and centrifuged at 6,500 rpm for 20 min at 4°C. The supernatant was sonicated again to shear the DNA on ice five times for 30 s each and was centrifuged at 14,000 rpm for 20 min. The supernatant was collected and tested for the presence of GFP–BAF-1 or AJM-1–GFP by immunoblotting and frozen in liquid nitrogen. Lysates were incubated with 5 μg anti-GFP antibody (Roche) for 2 h, and cellular debris was removed by centrifugation at 6,500 rpm for 15 min at 4°C. Lysates were then centrifuged at 14,000 rpm for 10 min, and 50 μl of protein G–Sepharose (Roche) was added to the supernatant. Immunocomplexes were washed twice with each buffer: ChIP buffer (50 mM Hepes/KOH, pH 7.5, 1 mM EDTA, 0.5%, NP-40, and 5 mM DTT with protease inhibitors) with 100 mM KCl and ChIP buffer with 1 M KCl and Tris-EDTA. Complexes were eluted with elution buffer (1% SDS and 10 mM Tris-HCl, pH 8), and 16 μl of 5 M NaCl was added to the elution and heated at 65°C overnight. DNA was then isolated using a standard procedure (phenol-chloroform extraction) and was resuspended in 20 μl Tris-EDTA. The amount of eluted DNA was quantified using locus-specific primers. Quantitative PCR was used to monitor ChIP results. 20 μl of quantitative PCRs contained 1:2 SYBR green Mix (ABgene), 250 nM of each primer, and an appropriate amount of DNA. The quantitative PCR results were analyzed essentially as described previously (Wang et al., 2004).
PCR and RT-PCR analyses
Single-worm PCR analysis using primers 5′-AACCGAAATTCTCAGCCCTT-3′ and 5′-GATCGCGGCCGCCTTAGAAACACTCTTCAGGATCG-3′ to distinguish between wild-type, gk324/hT2, and gk324/gk324 worms (Fig. S1, A and B) was performed essentially as described previously (Williams et al., 1992). For RT-PCR, 100 wild-type (N2), gk324/hT2, or gk324/gk324 worms were collected from each strain in 700 μl of extraction mixture (0.1 M NaOAc, 50% phenol, 2 M guanidinium thiocyanate, 12 mM sodium citrate, pH 7.0, 0.25% Sarkosyl, and 50 mM β-mercaptoethanol), immediately frozen in liquid nitrogen, and incubated at −80°C for at least 20 min. Samples were thawed at 60°C, vortexed, and incubated for 1 min on ice. Total RNA was isolated using a standard procedure (phenol extraction) and digested with RNase-free DNase I (Promega), and cDNA was synthesized from ∼400 ng RNA using a 15-nt oligodT primer using Moloney murine leukemia virus reverse transcriptase (Promega) according to the manufacturer's instructions. From each 20 μl cDNA, 2 μl was analyzed by PCR using the following primers: baf-1 forward (5′-GATCGAATTCATGTCGACTTCTGTTAAGCATCG-3′), baf-1 reverse (5′-GATCGCGGCCGCCATGAACTGATCTGCCCACTCG-3′), cah-3 forward (5′-CACTTCCATTGGGGAGAGAA-3′), and cah-3 reverse (5′-ACAACGCCTTTCCCTCTTTT-3′).
Worm movement assays
N2, VC699, and gk324/gk324-expressing GFP–BAF-1 worms were collected, washed with M9 buffer, treated for 5 min with hypochlorite solution (1.1% hypochlorite and 0.62 M NaOH), and washed with M9, and the embryos were collected and grown on nematode growth medium plates at 16°C for 3 d until they reached the L4/young adult stage. The VC699 and gk324/gk324 worms that expressed GFP–BAF-1 were sorted into two classes representing the gk324/hT2 and gk324/gk324 genotypes based on the presence of GFP fluorescence in the pharynx. For each experiment, 120 worms from each line were transferred to new nematode growth medium plates at 20°C (∼40 worms per plate), and movement was classified essentially as described previously (Henderson et al., 1997). Worms were also processed for transmission electron microscopy at days 4, 8, and 12 from synchronized embryos.
Online supplemental material
Fig. S1 describes the gk324 allele and provides evidence that baf-1 is not expressed in L4 worms homozygous for the gk324 allele. Fig. S2 shows that muscle attachment to hypodermal cells is aberrant in the tail region of gk324/gk324 animals by staining gk324/gk324 animals with MH46 antibody. The supplemental text gives further details on primers that were used in this study. Videos 1–3 show movement of heterozygous and homozygous worms for the gk324 allele at days 5 and 12. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200704049/DC1.
Supplementary Material
Acknowledgments
We thank Yuval Nevo for assistance with RT-PCR, Orit German-Gruenbaum for helping with the movement assays, Amir Sapir, Gidi Shemer, and Meital Suissa for assistance with the seam cells and VPC fusion, Verena Jantsch for assistance with analysis of the baf-1 deletion rescue strain, Naomi Melamed-Book for assistance with FRAP experiments, Limor Broday for antibodies and helpful discussions, and Shai Melcer for critical comments. We are grateful to the C. elegans Gene Knockout Consortium and C. elegans Reverse Genetics Core Facility at the University of British Columbia for providing the baf-1 deletion allele (gk324). We also thank the C. elegans Genetic Center (which is funded by the National Institutes of Health [NIH] National Center for Research Resources) for strains.
This work was supported by the USA-Israel Binational Science Foundation, Israel Science Foundation (ISF), Israel Ministry of Health, European Union's FP6 Life Science, Genomics, and Biotechnology for Health (grant LSHM-CT-2005-018690 to Y. Gruenbaum), support from the ISF (grant to B. Podbilewicz), and support from NIH (grant RO1 GM48646 to K.L. Wilson).
Abbreviations list: BAF, barrier to autointegration factor; ChIP, chromatin immunoprecipitation; DIC, differential interference contrast; DTC, distal tip cell; LEM, LAP2–emerin–MAN1; VPC, vulval precursor cell.
References
- Bengtsson, L., and K.L. Wilson. 2006. BAF phosphorylation on Ser-4 regulates emerin binding to lamin A in vitro and emerin localization in vivo. Mol. Biol. Cell. 17:1154–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxem, M., D.G. Srinivasan, and S. van den Heuvel. 1999. The Caenorhabditis elegans gene ncc-1 encodes a cdc2-related kinase required for M phase in meiotic and mitotic cell divisions, but not for S phase. Development. 126:2227–2239. [DOI] [PubMed] [Google Scholar]
- Brenner, S. 1974. The genetics of Caenorhabditis elegans. Genetics. 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkeen, A.K., S.L. Maday, K.K. Rybicka, J.A. Sulcove, J. Ward, M.M. Huang, R. Barstead, C. Franzini-Armstrong, and T.S. Allen. 2004. Disruption of Caenorhabditis elegans muscle structure and function caused by mutation of troponin I. Biophys. J. 86:991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassata, G., G. Shemer, P. Morandi, R. Donhauser, B. Podbilewicz, and R. Baumeister. 2005. ceh-16/engrailed patterns the embryonic epidermis of Caenorhabditis elegans. Development. 132:739–749. [DOI] [PubMed] [Google Scholar]
- Chen, H., and A. Engelman. 1998. The barrier-to-autointegration protein is a host factor for HIV type 1 integration. Proc. Natl. Acad. Sci. USA. 95:15270–15274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decostre, V., R. Ben Yaou, and G. Bonne. 2005. Laminopathies affecting skeletal and cardiac muscles: clinical and pathophysiological aspects. Acta Myol. 24:104–109. [PubMed] [Google Scholar]
- Encalada, S.E., J. Willis, R. Lyczak, and B. Bowerman. 2005. A spindle checkpoint functions during mitosis in the early Caenorhabditis elegans embryo. Mol. Biol. Cell. 16:1056–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan, S., P.G. Giresi, C.M. Whittle, X. Zhang, R.D. Green, and J.D. Lieb. 2007. X chromosome repression by localization of the C. elegans dosage compensation machinery to sites of transcription initiation. Nat. Genet. 39:403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay, D.S., and M. Han. 2000. Mutations in cye-1, a Caenorhabditis elegans cyclin E homolog, reveal coordination between cell-cycle control and vulval development. Development. 127:4049–4060. [DOI] [PubMed] [Google Scholar]
- Francis, R., and R.H. Waterston. 1991. Muscle cell attachment in Caenorhabditis elegans. J. Cell Biol. 114:465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridkin, A., E. Mills, A. Margalit, E. Neufeld, K.K. Lee, N. Feinstein, M. Cohen, K.L. Wilson, and Y. Gruenbaum. 2004. Matefin, a C. elegans germ-line specific SUN-domain nuclear membrane protein, is essential for early embryonic and germ cell development. Proc. Natl. Acad. Sci. USA. 101:6987–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa, K., S. Sugiyama, S. Osouda, H. Goto, M. Inagaki, T. Horigome, S. Omata, M. McConnell, P.A. Fisher, and Y. Nishida. 2003. Barrier-to-autointegration factor plays crucial roles in cell cycle progression and nuclear organization in Drosophila. J. Cell Sci. 116:3811–3823. [DOI] [PubMed] [Google Scholar]
- Gorjánácz, M., E.P.F. Klerkx, V. Galy, R. Santarella, C. López-Iglesias, P. Askjaer, and I.W. Mattaj. 2006. C. elegans BAF-1 and its kinase VRK-1 participate directly in postmitotic nuclear envelope assembly. EMBO J. 26:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haithcock, E., Y. Dayani, E. Neufeld, A.J. Zahand, N. Feinstein, A. Mattout, Y. Gruenbaum, and J. Liu. 2005. Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 102:16690–16695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi, T., T. Koujin, M. Segura-Totten, K.K. Lee, Y. Matsuoka, Y. Yoneda, K.L. Wilson, and Y. Hiraoka. 2001. BAF is required for emerin assembly into the reforming nuclear envelope. J. Cell Sci. 114:4575–4585. [DOI] [PubMed] [Google Scholar]
- Henderson, S.T., D. Gao, S. Christensen, and J. Kimble. 1997. Functional domains of LAG-2, a putative signaling ligand for LIN-12 and GLP-1 receptors in Caenorhabditis elegans. Mol. Biol. Cell. 8:1751–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndon, L.A., P.J. Schmeissner, J.M. Dudaronek, P.A. Brown, K.M. Listner, Y. Sakano, M.C. Paupard, D.H. Hall, and M. Driscoll. 2002. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 419:808–814. [DOI] [PubMed] [Google Scholar]
- Holaska, J.M., K.K. Lee, A.K. Kowalski, and K.L. Wilson. 2003. Transcriptional repressor germ cell-less (GCL) and barrier-to-autointegration factor (BAF) compete for binding to emerin in vitro. J. Biol. Chem. 278:6969–6975. [DOI] [PubMed] [Google Scholar]
- Hresko, M.C., L.A. Schriefer, P. Shrimankar, and R.H. Waterston. 1999. Myotactin, a novel hypodermal protein involved in muscle-cell adhesion in Caenorhabditis elegans. J. Cell Biol. 146:659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K.K., T. Haraguchi, R.S. Lee, T. Koujin, Y. Hiraoka, and K.L. Wilson. 2001. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 114:4567–4573. [DOI] [PubMed] [Google Scholar]
- Lee, M.S., and R. Craigie. 1998. A previously unidentified host protein protects retroviral DNA from autointegration. Proc. Natl. Acad. Sci. USA. 95:1528–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann, R. 2001. Cell migration in invertebrates: clues from border and distal tip cells. Curr. Opin. Genet. Dev. 11:457–463. [DOI] [PubMed] [Google Scholar]
- Lin, C.W., and A. Engelman. 2003. The barrier-to-autointegration factor is a component of functional human immunodeficiency virus type 1 preintegration complexes. J. Virol. 77:5030–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J., T. Rolef-Ben Shahar, D. Riemer, P. Spann, M. Treinin, K. Weber, A. Fire, and Y. Gruenbaum. 2000. Essential roles for Caenorhabditis elegans lamin gene in nuclear organization, cell cycle progression, and spatial organization of nuclear pore complexes. Mol. Biol. Cell. 11:3937–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J., K.K. Lee, M. Segura-Totten, E. Neufeld, K.L. Wilson, and Y. Gruenbaum. 2003. MAN1 and emerin have overlapping function(s) essential for chromosome segregation and cell division in C. elegans. Proc. Natl. Acad. Sci. USA. 100:4598–4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie, J.M.J., R.L. Garcea, J.M. Zengel, and H.F. Epstein. 1978. Muscle development in Caenorhabditis elegans: mutants exhibiting retarded sarcomere construction. Cell. 15:751–762. [DOI] [PubMed] [Google Scholar]
- Margalit, A., M. Segura-Totten, Y. Gruenbaum, and K.L. Wilson. 2005. Barrier-to-autointegration factor is required to segregate and enclose chromosomes within the nuclear envelope and assemble the nuclear lamina. Proc. Natl. Acad. Sci. USA. 102:3290–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margalit, A., A. Brachner, J. Gotzmann, R. Foisner, and Y. Gruenbaum. 2007. Barrier-to-autointegration factor - a BAFfling little protein. Trends Cell Biol. 17:202–208. [DOI] [PubMed] [Google Scholar]
- Merz, D.C., G. Alves, T. Kawano, H. Zheng, and J.G. Culotti. 2003. UNC-52/perlecan affects gonadal leader cell migrations in C. elegans hermaphrodites through alterations in growth factor signaling. Dev. Biol. 256:173–186. [DOI] [PubMed] [Google Scholar]
- Mohler, W.A., G. Shemer, J.J. del Campo, C. Valansi, E. Opoku-Serebuoh, V. Scranton, N. Assaf, J.G. White, and B. Podbilewicz. 2002. The type I membrane protein EFF-1 is essential for developmental cell fusion. Dev. Cell. 2:355–362. [DOI] [PubMed] [Google Scholar]
- Montes de Oca, R., K.K. Lee, and K.L. Wilson. 2005. Binding of barrier to autointegration factor (BAF) to histone H3 and selected linker histones including H1.1. J. Biol. Chem. 280:42252–42262. [DOI] [PubMed] [Google Scholar]
- Nichols, R.J., M.S. Wiebe, and P. Traktman. 2006. The vaccinia-related kinases phosphorylate the N' terminus of BAF, regulating its interaction with DNA and its retention in the nucleus. Mol. Biol. Cell. 17:2451–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt, J., W.B. Wood, and R.H. Plasterk. 1996. cdh-3, a gene encoding a member of the cadherin superfamily, functions in epithelial cell morphogenesis in Caenorhabditis elegans. Development. 122:4149–4157. [DOI] [PubMed] [Google Scholar]
- Podbilewicz, B. 2006. Cell fusion. WormBook, ed The C. elegans Research Community, WormBook. / 10.1895/wormbook.1.52.1 http://www.wormbook.org. [DOI]
- Podbilewicz, B., and J.G. White. 1994. Cell fusions in the developing epithelia of C. elegans. Dev. Biol. 161:408–424. [DOI] [PubMed] [Google Scholar]
- Podbilewicz, B., E. Leikina, A. Sapir, C. Valansi, M. Suissa, G. Shemer, and L.V. Chernomordik. 2006. The C. elegans developmental fusogen EFF-1 mediates homotypic fusion in heterologous cells and in vivo. Dev. Cell. 11:471–481. [DOI] [PubMed] [Google Scholar]
- Praitis, V., E. Casey, D. Collar, and J. Austin. 2001. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics. 157:1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabut, G., and J. Ellenberg. 2005. Photobleaching techniques to study mobility and molecular dynamics of proteins in live cells: FRAP, iFRAP and FLIP. In Live Cell Imaging: a Laboratory Manual. R.D. Goldman and D. Spector, editors. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 101–127. [DOI] [PubMed]
- Sapir, A., J. Choi, E. Leikina, O. Avi-Noam, C. Valansi, L.V. Chernomordik, A.P. Newman, and B. Podbilewicz. 2007. AFF-1, a novel FOS-1-regulated fusogen mediates fusion of the anchor-cell in C. elegans. Dev. Cell. 12:683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura-Totten, M., and K.L. Wilson. 2004. BAF: roles in chromatin, nuclear structure and retrovirus integration. Trends Cell Biol. 14:261–266. [DOI] [PubMed] [Google Scholar]
- Segura-Totten, M., A.K. Kowalski, R. Craigie, and K.L. Wilson. 2002. Barrier-to-autointegration factor: major roles in chromatin decondensation and nuclear assembly. J. Cell Biol. 158:475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma-Kishore, R., J.G. White, E. Southgate, and B. Podbilewicz. 1999. Formation of the vulva in Caenorhabditis elegans: a paradigm for organogenesis. Development. 126:691–699. [DOI] [PubMed] [Google Scholar]
- Shemer, G., and B. Podbilewicz. 2003. The story of cell fusion: big lessons from little worms. Bioessays. 25:672–682. [DOI] [PubMed] [Google Scholar]
- Shemer, G., R. Kishore, and B. Podbilewicz. 2000. Ring formation drives invagination of the vulva in Caenorhabditis elegans: Ras, cell fusion, and cell migration determine structural fates. Dev. Biol. 221:233–248. [DOI] [PubMed] [Google Scholar]
- Shemer, G., M. Suissa, I. Kolotuev, K.C. Nguyen, D.H. Hall, and B. Podbilewicz. 2004. EFF-1 is sufficient to initiate and execute tissue-specific cell fusion in C. elegans. Curr. Biol. 14:1587–1591. [DOI] [PubMed] [Google Scholar]
- Shimi, T., T. Koujin, M. Segura-Totten, K.L. Wilson, T. Haraguchi, and Y. Hiraoka. 2004. Dynamic interaction between BAF and emerin revealed by FRAP, FLIP, and FRET analyses in living HeLa cells. J. Struct. Biol. 147:31–41. [DOI] [PubMed] [Google Scholar]
- Sommer, R.J. 2001. As good as they get: cells in nematode vulva development and evolution. Curr. Opin. Cell Biol. 13:715–720. [DOI] [PubMed] [Google Scholar]
- Sulston, J.E., and H.R. Horvitz. 1977. Postembryonic cell lineages of the nematode Caenorhabditis elegans. Dev. Biol. 56:110–156. [DOI] [PubMed] [Google Scholar]
- Tajsharghi, H., M. Pilon, and A. Oldfors. 2005. A Caenorhabditis elegans model of the myosin heavy chain IIa E706K mutation. Ann. Neurol. 58:442–448. [DOI] [PubMed] [Google Scholar]
- Wang, J.C., M.K. Derynck, D.F. Nonaka, D.B. Khodabakhsh, C. Haqq, and K.R. Yamamoto. 2004. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc. Natl. Acad. Sci. USA. 101:15603–15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., S. Xu, C. Rivolta, L.Y. Li, G.H. Peng, P.K. Swain, C.H. Sung, A. Swaroop, E.L. Berson, T.P. Dryja, and S. Chen. 2002. Barrier to autointegration factor interacts with the cone-rod homeobox and represses its transactivation function. J. Biol. Chem. 277:43288–43300. [DOI] [PubMed] [Google Scholar]
- Williams, B.D., B. Schrank, C. Huynh, R. Shownkeen, and R.H. Waterston. 1992. A genetic mapping system in Caenorhabditis elegans based on polymorphic sequence-tagged sites. Genetics. 131:609–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, J., L. Xu, G. Crawford, Z. Wang, and S.M. Burgess. 2006. The forkhead transcription factor FoxI1 remains bound to condensed mitotic chromosomes and stably remodels chromatin structure. Mol. Cell. Biol. 26:155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, R., R. Ghirlando, M.S. Lee, K. Mizuuchi, M. Krause, and R. Craigie. 2000. Barrier-to-autointegration factor (BAF) bridges DNA in a discrete, higher-order nucleoprotein complex. Proc. Natl. Acad. Sci. USA. 97:8997–9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}