

Abstract
The morphology and composition of subnuclear organelles, such as Cajal bodies (CBs), nucleoli, and other nuclear bodies, is dynamic and can change in response to a variety of cell stimuli, including stress. We show that UV-C irradiation disrupts CBs and alters the distribution of a specific subset of CB components. The effect of UV-C on CBs differs from previously reported effects of transcription inhibitors. We demonstrate that the mechanism underlying the response of CBs to UV-C is mediated, at least in part, by PA28γ (proteasome activator subunit γ). The presence of PA28γ in coilin-containing complexes is increased by UV-C. Overexpression of PA28γ, in the absence of UV-C treatment, provokes a similar redistribution of the same subset of CB components that respond to UV-C. RNA interference–mediated knockdown of PA28γ attenuates the nuclear disruption caused by UV-C. These data demonstrate that CBs are specific nuclear targets of cellular stress-response pathways and identify PA28γ as a novel regulator of CB integrity.
Introduction
The mammalian cell nucleus is a complex structure containing distinct nuclear bodies (NBs), such as nucleoli, PML bodies, splicing speckles, and Cajal bodies (CBs; Lamond and Earnshaw, 1998; Dundr and Misteli, 2001; Handwerger and Gall, 2006). These bodies are typically enriched for specific proteins and nucleic acids, reflecting their function. Dynamic changes in NBs occur under both physiological and pathological conditions. For example, both the number and size of nucleoli vary between metabolically active and inactive cells, and PML bodies are altered in leukemic blasts and during virus infection (Zimber et al., 2004). The molecular events triggering such changes are not well characterized.
We examine the dynamic behavior of CBs and how their composition changes under stress conditions. CBs were discovered in 1903 by Santiago Ramón y Cajal (Gall, 2003) and are involved in the assembly and maturation of small nuclear ribonucleoproteins (snRNPs; Cioce and Lamond, 2005; Matera and Shpargel, 2006). Indeed, snRNPs are thought to accumulate in CBs upon their initial entry into the nucleus (Sleeman and Lamond, 1999), and a class of CB-specific modification guide RNAs (scaRNAs) are important for the sequence-specific modification of the snRNAs within CBs (Darzacq et al., 2002; Jady et al., 2003). CBs also contain survival of motor neuron (SMN), a protein linked to the neurodegenerative disease spinal muscular atrophy (Frugier et al., 2002). The SMN complex plays an important role in the cytoplasmic assembly of Sm core RNPs, (Eggert et al., 2006) and in their nuclear reimport and targeting to CBs (Narayanan et al., 2004; Ospina et al., 2005).
Other CB components include fibrillarin and NOPP140, proteins that localize in CBs before their subsequent accumulation in nucleoli. CBs likely have other functions besides snRNP maturation. For example, NPAT and PTFγ, which are proteins regulating histone and snRNA gene transcription, respectively, are found in CBs (Schul et al., 1998; Zhao et al., 2000). CBs can indeed associate with histone and snRNA gene loci (Frey and Matera, 1995), and they may also play a more general role in coordinating assembly of large multiprotein complexes in the nucleus (Gall, 2001). Interestingly, the presence in a subset of CBs of ZPR1 and FGF-2 suggests that CBs could be involved also in transducing proliferative signals to the nucleus (Claus et al., 2003; Gangwani et al., 2005).
Genetic evidence suggests that coilin, a nuclear phosphoprotein widely used as a marker for CBs, plays a role in the structural organization of CBs. Thus, in coilin knockout cells, CBs are disrupted and fail to accumulate snRNPs and SMN, whereas other CB components, such as fibrillarin, NOPP140, and scaRNAs, are redistributed in distinct subsets of remnant structures (Jady et al., 2003; Tucker et al., 2001). Posttranslational modifications of coilin can affect CB integrity. For example, changes in the phosphorylation state of coilin affect the number and integrity of CBs in mitotic and interphase cells (Carmo-Fonseca et al., 1993; Shpargel et al., 2003), and the extent of symmetrical dimethylation of arginine residues on coilin influences the targeting of SMN and, consequently, the accumulation of newly imported snRNPs in CBs (Boisvert et al., 2002; Hebert et al., 2002). However, coilin modification is not always linked with CB disassembly or turnover. For example, adenovirus infection causes fragmentation of CBs (Rebelo et al., 1996) without causing changes either in the levels or in the electrophoretic mobility of coilin.
UV irradiation represents a complex, multicomponent stress stimulus that subverts the metabolic activity of the cell nucleus. It affects different nuclear domains including nucleoli (Al-Baker et al., 2004; Kurki et al., 2004) and PML bodies (Kurki et al., 2003; Seker et al., 2003). UV light irradiation causes an immediate ligand-independent activation of receptor tyrosine kinases (i.e., EGF and PDGF receptors) caused by the inactivation of receptor-directed tyrosine phosphatases (Gross et al., 1999; Gulati et al., 2004). Subsequently, it triggers DNA damage caused by the formation of cyclobutane pyrimidine dimers, (6–4) photoproducts (Tornaletti and Pfeifer, 1996), and reactive oxygen species generation (Nishigori, 2006), and a complex transcriptional response involving modulation of genes associated with cell proliferation and repair of damaged DNA (Koch-Paiz et al., 2004). The next phase is characterized either by repair of DNA lesions, or by apoptosis of the cells that have not been able to start an appropriate response.
In this study, we show that CBs are also responsive to UV-C and characterize the molecular mechanism underlying this effect. We have identified PA28γ (proteasome activator subunit 28S γ) as a factor whose stable association with coilin-containing complexes is increased by UV-C treatment and show that PA28γ plays an important role in the mechanism underlying the disruption of CBs upon UV-C irradiation.
Results
UV-C treatment triggers disassembly of CBs and a redistribution of coilin
The effect of UV-C treatment on CB integrity was assessed by immunofluorescence labeling using antibodies specific for coilin (Fig. 1 A, top left). HaCaT (human immortalized keratinocytes) cells were immunolabeled at 6 h after brief exposure to UV-C (30 J/m2, 254 nm; see Materials and methods). This changed the number and appearance of CBs, with coilin redistributed to hundreds of microfoci clustered throughout the nucleoplasm (Fig. 1 A, bottom left). Splicing speckles were also affected by UV-C, although less dramatically than CBs; thus, UV-C caused splicing speckles to become more rounded, but with little or no change in the mean number per nucleus (Fig. 1 A, compare middle images).
Figure 1.

The subnuclear distribution of coilin and SC35 is affected by UV-C irradiation. (A) HaCaT cells, either mock (top) or UV-C–treated (bottom), were stained with anti-coilin (red) and -SC35 (green) antibodies and counterstained with DAPI to detect the nuclei. (B) Coilin is enriched in splicing speckles in UV-C–treated cells. UV-C–treated HaCaT cells stained with anti-coilin (red), -SC35, -Sm proteins (Y12), -TMG cap, and -U1A antibodies (green). (C) Time-course analysis. HaCaT cells stained with an anti-coilin antibody (red) at the indicated times after UV-C irradiation. Arrows denote intact CBs in mock-treated cells. Arrowheads denote the UV-C–induced, coilin-containing domains. Bar, 10 μM.
We noticed a similarity between the redistributed coilin labeling pattern and the rounded splicing speckles present after UV-C treatment. This was unexpected because coilin does not normally colocalize with splicing speckles (Lamond and Spector, 2003). To investigate this further, we double labeled UV-C–treated cells with anti-coilin antibodies (Fig. 1 B, left) and antibodies specific for known splicing speckle components, i.e., anti-SC35, anti-Sm (Y12), anti– trimethylguanosine (TMG)-cap, and anti-U1A (Fig. 1 B, middle). All four of these splicing speckle components showed a similar change into more rounded structures after UV-C treatment, and we observed a partial overlap between the reorganized coilin microfoci and the rounded splicing speckles (Fig. 1 B, compare left and middle). These experiments involved immunolabeling cells at 6 h after exposure to UV-C because this was empirically observed as causing the maximum effect on coilin redistribution (Fig. 1 C). Moreover, the UV-C effect is at least partially reversible, with recovery of the normal coilin distribution evident in ∼40% of the irradiated cells within 10–12 h. A subset of the irradiated cells, at 8–12 h after UV-C, formed coilin-containing perinucleolar caps (unpublished data). Further analysis confirmed that the majority of cells remained viable after UV-C exposure, with no correlation between coilin redistribution and UV-C–induced cell death (unpublished data). We conclude that UV-C treatment has a specific and reversible effect on subnuclear organization affecting CBs and splicing speckles.
Next, we evaluated the effects of UV-C irradiation on a variety of cell lines, differing in tissue of origin, growth properties, and transformation status, to determine if the UV-C–induced effect on CB structure is either general, or specific for HaCaT cells (Table I). This shows a similar UV-C–induced redistribution of coilin in all the cell lines tested, including WI-38, HeLa, COS-7, HaCaT, MCF-7, SAOS-2, 293T, HCT116, and SW480, with little variation in the percentage of responsive cells between each cell type (Table I). A functional p53 gene is not required for the coilin redistribution in response to UV-C because whereas the SAOS-2 cells are p53 null, the HaCaT cells have a mutated, functionally impaired p53 gene, and the MCF-7 cells have a functional, wild-type p53 gene. In subsequent experiments, we concentrated on the HaCaT cells as the main experimental model to investigate the mechanisms of CB disassembly after UV-C irradiation because those cells were the most responsive to UV-C treatment and also because, among those tested, they represent the tissues most exposed to sunlight in vivo.
Table I.
Classification of the cell lines used in this study based on tissue of origin, transformation status, and CB status upon UV-C irradiation
| Cell line | Cell type | Tissue of origin | p53 status | Transformation status | UV-C–induced coilin redistribution |
|---|---|---|---|---|---|
| WI-38 | fibroblast | lung | wild type | not transformed | ++ |
| HCT-116 | epithelial-like | colon carcinoma | wild type | transformed | ++ |
| HeLa | epithelial | cervix adenocarcinoma | mut | transformed (HPV) | ++ |
| COS-7 | epithelial-like | kidney (simian) | mut | transformed (SV 40) | ++ |
| MCF-7 | epithelial-like | breast adenocarcinoma | wild type | transformed | ++ |
| 293 | epithelial | kidney | wild type (delocalized) | transformed (adenovirus) | ++ |
| HaCaT | epithelial | skin | mut | transformed (spontaneously) | +++ |
| SAOS-2 | epithelial-like | osteogenic sarcoma | null | transformed | ++ |
| SW 480 | epithelial-like | colon carcinoma | mut | transformed | ++ |
++ indicates that >50% of the observed cells show CB fragmentation upon UV-C treatment. +++ indicates that >80% of the observed cells show the CB fragmentation phenotype.
To better characterize the effect of UV-C irradiation on CBs, we next addressed whether UV-C irradiation affected other CB proteins, in both HaCaT and MCF-7 cells, namely SMN, snRNP components, fibrillarin, NOPP140, and NPAT (Fig. 2). After UV-C irradiation, both SMN and snRNP factors are no longer concentrated in CBs (Fig. 2, compare A and B). SMN and the U6 snRNP-associated factor SART3 show a widespread diffuse staining, whereas TMG-capped snRNAs and snRNP proteins show both nucleoplasmic staining and accumulation into rounded splicing speckles (Fig. 2, A and B; arrows and arrowheads indicate intact and rearranged CBs, respectively). Surprisingly, after UV-C irradiation, CB-like structures are still evident as bright, coilin-negative nucleoplasmic bodies when detected with antibodies against either NPAT, fibrillarin, or NOPP140 (Fig. 2 C, left, arrows). Thus, UV-C irradiation selectively affects a subset of CB components. The small CB-like structures, which remain after UV-C and lack coilin, SMN, and snRNPs, resemble in appearance and composition the residual CBs observed in coilin −/− mouse cells (Tucker et al., 2001; Jady et al., 2003).
Figure 2.

UV-C irradiation triggers a dramatic redistribution of the CB-associated pool of SMN, TMG cap-containing RNAs, and SART3. (A and B) HaCaT cells, either mock- (A) or UV-C–treated (B), were stained with anti-SMN, anti-TMG-cap and anti-SART3 (red), and anti-coilin (green) antibodies, respectively. Arrows denote intact CBs in mock-treated cells. Arrowheads denote the UV-C–induced coilin-containing domains. (C) UV-C irradiation does not trigger a redistribution of the CB-associated pool of NPAT, fibrillarin, or NOPP140. UV-C–treated HaCaT cells stained with anti-coilin (red), -NPAT, -fibrillarin, and -NOPP140 antibodies (green). Arrows denote NPAT, fibrillarin and NOPP140-containing remnant (coilin-negative) CBs in the irradiated cells. Bar, 10 μM.
It is known that transcription inhibitors disrupt CBs and cause speckles to enlarge and round up (for review see Lamond and Spector, 2003). This led us to examine whether the effect of UV-C exposure on transcription levels could explain the observed changes in CBs and in the distribution of coilin in the irradiated cells. A 5-fluorouridine (5-FU) incorporation assay (Fig. 3 A) confirmed, as expected, that there was a time-dependent general reduction in nuclear RNA synthesis after exposure to UV-C. The UV-C inhibition affected primarily nucleoplasmic transcription, with 5-FU incorporation within nucleoli still visible. Double-labeling with anti-coilin antibodies, again, showed a time-dependent disruption of CBs after UV-C treatment, with most coilin relocalized to nucleoplasmic microfoci. In some cells, a minor fraction of coilin also localizes in dots within nucleoli, distinct from perinucleolar caps (Fig. 3 A, insets). We compared the UV-C response directly with the effect of the transcription inhibitors 5,6-dichloro-1-β-D-ribobenzimidazole (DRB) and actinomycin D (Fig. 3 B and not depicted). Both inhibitors cause coilin to relocalize and accumulate in prominent perinucleolar caps (Fig. 3 B, bottom, arrowheads). However, we do not observe accumulation of coilin in nucleoplasmic microfoci or associated with splicing speckles after exposure to transcription inhibitors, which differs from the effect of UV-C exposure. Therefore, although some of the effects seen after UV-C exposure, such as the formation of large, rounded splicing speckles and possibly the appearance of coilin dots within nucleoli, may be attributed to the down-regulation of transcription, our data suggest that the UV-C disruption of CBs is not simply an indirect result of transcription inhibition.
Figure 3.

UVC irradiation triggers transcriptional inhibition. (A) HaCaT cells were UV-C–treated and stained with an anti-coilin antibody. Nuclei were labeled with 5-FU to monitor ongoing transcription at the indicated times following UV-C irradiation. (B) Inhibition of transcription does not trigger a UV-C–like fragmentation of CBs. HaCaT cells were either mock or DRB–treated for 3 h and stained with an anti-coilin antibody. Arrows denote intact CBs. Arrowheads denote the UV-C–induced, coilin-containing perinucleolar caps. Bars, 10 μM. (C) UV-C irradiation does not alter the levels, solubility, or subnuclear partitioning of coilin. Whole-cell lysates and subcellular fractions from either mock- (lanes 1–5) or UV-C–treated (lanes 6–10) HaCaT cells were analyzed by Western blot with an anti-coilin antibody. (D) UV-C irradiation triggers a reduction in the sDMA modification state of coilin. Nuclear extracts obtained from either mock or UV-C–treated HaCaT cells were immunoprecipitated with anti-coilin antibodies and an anti-GFP antibody (as a control), and the eluted material was analyzed by Western blot with anti-coilin and -sDMA rabbit polyclonal antibodies. (E) Whole-cell lysates obtained in denaturing conditions (9 M Urea) were probed with anti-coilin and -sDMA rabbit polyclonal antibodies.
Effect of UV-C irradiation on coilin
Because genetic depletion of murine coilin causes a similar change in CB appearance and composition to that detected here after UV-C treatment, we tested whether UV-C irradiation affected either the levels or modification state of coilin. Protein blotting analysis using anti-coilin antibodies to probe either whole-cell lysates, or soluble nuclear extracts prepared from both untreated and UV-C–treated cells, revealed little or no change in the relative levels of coilin, even under different extraction conditions (Fig. 3 C, compare lanes 1–5 and 6–10). Moreover, the absence of obvious changes in the electrophoretic mobility of coilin suggests that it does not undergo a major change in its phosphorylation status. In contrast, a clear shift in electrophoretic mobility of coilin was observed upon its hyperphosphorylation when cells enter mitosis (Carmo-Fonseca et al., 1993).
Symmetric dimethylarginine (sDMA) modification of coilin has been linked to the ability to recruit SMN to coilin-containing CBs. Either drug-induced hypomethylation of coilin (Boisvert et al., 2002; Hebert et al., 2002), or transient expression of mutant forms of the protein (Hebert et al. 2001), prevented SMN recruitment to CBs but, notably, did not cause CBs to fragment. Therefore, we investigated whether sDMA modification of coilin was affected by UV-C irradiation. Coilin was immunoprecipitated from nuclear extracts prepared from either control or UV-C irradiated cells, and the eluted proteins were blotted and probed with both anti-coilin and anti-sDMA antibodies (Fig. 3 D). This showed a minor reduction in sDMA levels. However, when whole-cell lysates obtained in denaturing conditions (9 M urea) from irradiated and mock-treated cells were separated by SDS-PAGE, transferred to nitrocellulose filters, and probed with anti-coilin and anti-sDMA antibodies, any differences in the extent of sDMA modification were less evident (Fig. 3 E). Given the absence of CB fragmentation in cells with hypomethylated coilin (Hebert et al. 2002), we infer that although minor changes in the sDMA status of coilin may take place in UV-C–treated cells, this does not likely play a major role in determining the fragmentation of CBs.
UV-C induces changes in the protein composition of coilin-containing complexes
We tested whether UV-C irradiation causes a change in the protein composition of nuclear complexes containing coilin. Coilin complexes were affinity purified from cells expressing FLAG-tagged coilin (see Materials and methods). Silver staining of the eluted, affinity-purified material from both mock-treated and UV-C–treated cells revealed that a protein band of ∼32 kD was enriched specifically in the eluate from the irradiated cells (Fig. 4 A, arrow). Peptide mass fingerprinting was performed on the enriched band isolated from a large-scale preparation of UV-C–treated cells and identified as proteasome activator subunit γ (PA28γ). To validate the identification of PA28γ as a novel component of coilin complexes, and to test whether coilin and PA28γ associate in the absence of UV-C, immunoprecipitation experiments were performed with anti-PA28γ polyclonal antibodies and extracts from nonirradiated HaCaT and MCF-7 cells. The immunoprecipitated material was separated by SDS-PAGE, transferred to a nitrocellulose membrane, and probed with anti-coilin antibodies. Coilin was coimmunoprecipitated with PA28γ (Fig. 4 B, compare lanes 2 and 3), confirming that coilin and PA28γ are present in a common complex in vivo, even in the absence of UV-C treatment. Furthermore, the complex containing PA28γ and coilin was resistant to 1 M KCl, indicating a stable interaction (Fig. 4 B lanes 4 and 5,).
Figure 4.

The composition of coilin-containing complexes changes upon UV-C treatment. (A) Silver staining of the immunoaffinity eluates from either mock- or UV-C–treated 293 cells transiently transfected with a FLAG-coilin expression vector. Arrows indicate the protein band corresponding to FLAG-coilin and PA28γ, respectively (the latter is enriched in the complexes isolated from UV-C–treated cells). (B) Endogenous PA28γ and coilin form a high-salt–resistant complex in vivo. Nuclear extracts obtained from MCF-7 cells were immunoprecipitated with a mix of two anti-PA28γ polyclonal antibodies, as described in Materials and methods. Blotting analysis reveals the presence of both coilin and PA28γ in the immunoprecipitated material (lanes 2–3). The copurification of coilin and PA28γ is not prevented by high-salt treatment of the beads (lanes 4–5). (C) PA28γ is present in a subset of coilin-containing complexes. Nuclear extracts derived from 293 cells were fractionated by gel-filtration chromatography and subjected to Western blot analysis with anti-coilin and -PA28γ antibodies (upper and lower panels, respectively) showing that PA28γ cofractionates with a subset of coilin-containing complexes.
Gel filtration analysis of nuclear extracts reveals that, under steady-state conditions, coilin is present in two different regions of the column eluate (Fig. 4 C). The first region corresponds to very high molecular weight complexes, of ∼2 MD (Fig. 4 C, lanes 8–10). The second region corresponds to complexes ∼200–300 kD in size (Fig. 4 C, lanes 13–16). Consistent with previous studies (Tanahashi et al., 1997), we found PA28γ in molecular weight fractions corresponding to a size range of ∼250 kD (Fig. 4 C, lanes 12–17). Therefore, PA28γ cofractionates with the smaller-sized pool of coilin complexes.
Immunoprecipitation with anti-PA28γ antibodies in extracts prepared from mock-treated and UV-C–treated cells confirmed the result shown in Fig. 4 A, i.e., that the level of PA28γ isolated in a stable complex with coilin is significantly increased by UV-C treatment (Fig. 5 A, compare lanes 5 and 7; and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200604099/DC1). Thus, the amount of coilin complexed with PA28γ is maximal at 6 h after UV-C treatment and decreases at later time points (12 h; Fig. 5 A, lane 9), when the majority of the cells show complete or partial recovery of the normal coilin labeling pattern. Under these conditions we observe only a small increase in the total cellular level of PA28γ after UV-C, as judged by immunoblotting (Fig. 5 D), and infer that the enhanced PA28γ association with coilin is unlikely to arise only through changes in PA28γ protein levels (see Discussion).
Figure 5.

UV-C irradiation enhances the formation of the PA28γ-coilin complex. (A) Nuclear extracts obtained at the indicated times from irradiated HaCaT cells were immunoprecipitated with polyclonal anti-PA28γ antibodies. Western Blot analysis shows that the amount of coimmunoprecipitated coilin peaks at 6 h after UV-C irradiation. (B) PA28γ and coilin partially colocalize in UV-C–treated cells. HaCaT cells were either mock- or UV-C–treated and stained with anti-coilin (red) and -PA28γ (green) antibodies. Arrowheads indicate the UV-C–responsive domains containing both PA28γ and coilin. (C) PA28γ is partially colocalized with the splicing speckles of UV-C–treated cells. HaCaT cells were either mock- or UV-C–treated and stained with anti-PA28γ (red) and anti-SC35 (green) antibodies. (right) Merged image. Arrowheads indicate the UV-C–responsive domains containing both PA28γ and SC35. (D) UV-C irradiation triggers a slight increase in the levels of PA28γ. Western blot analysis with an anti-PA28γ antibody of whole-cell-lysates (lanes 1–2) obtained from mock- and UV-C–treated HaCaT cells reveals a minor accumulation of PA28γ in the irradiated cells (top). Ponceau staining of the transferred proteins was used as a loading control (bottom). Bars, 10 μM.
The observed association between coilin and PA28γ in control, nonirradiated cells likely does not occur within CBs, as we observe by immunofluorescence no accumulation of PA28γ in CBs (Fig. 5 B, top). However, upon UV-C treatment, coilin and PA28γ show enhanced colocalization in the irradiated cells, supporting the aforementioned biochemical evidence for a UV-C–induced increase in the association between coilin and PA28γ (Fig. 5 B, bottom, arrowheads). Consistent with our observation that coilin partially colocalizes with rounded splicing speckles in UV-C–treated cells (Fig. 1), a double-immunofluorescence labeling with anti-PA28γ and –SC35 antibodies revealed that PA28γ also shows a UV-C–induced partial colocalization with splicing speckles (Fig. 5 C, arrowheads). This is further supported by double-labeling experiments showing partial colocalization of PA28γ with SC35, U1A, and TMG cap RNAs in UV-C–treated cells (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200604099/DC1). Collectively, the data indicate that coilin and PA28γ are present in a common complex in vivo whose levels are increased by UV-C treatment.
Increased levels of PA28γ trigger an UV-C like redistribution of coilin
We hypothesized that the increased formation of complexes containing coilin and PA28γ upon UV-C treatment could contribute to the mechanism of UV-C–induced CB disruption and coilin redistribution. Therefore, we studied whether altering PA28γ expression levels could affect CB integrity and the distribution of coilin in the absence of UV-C treatment. Strikingly, transient overexpression of exogenous PA28γ in both HaCaT and MCF-7 cells triggered a similar redistribution of coilin to that observed in UV-C–irradiated cells (Fig. 6 A). Thus, the more brightly stained cells overexpressing PA28γ (Fig. 6 A, arrowheads) have lost the bright coilin foci (arrows) seen in untransfected cells. To test whether overexpression of PA28γ also induces a reduction in transcription, similar to that caused by UV-C treatment, we analyzed 5-FU incorporation in both transfected and untransfected cells (Fig. 6 B). This showed clearly that cells overexpressing PA28γ had similar levels of nuclear transcription to untransfected cells (Fig. 6 B, bottom). This thus demonstrates that the PA28γ-mediated disruption of CBs and relocalization of coilin is independent of changes in transcription levels.
Figure 6.

Increased levels of PA28γ triggers fragmentation of CBs, but does not block transcription. (A and B) MCF-7 cells, transfected with a FLAG-PA28γ expression vector and stained with anti-PA28γ (green) and anti-coilin (red) antibodies as indicated. (B) Cell nuclei were also stained with anti-PA28γ (green) antibodies and labeled with 5-FU (red) to monitor ongoing transcription (bottom). Arrows indicate intact CBs in the untransfected cells. Arrowheads indicate the transfected cells (revealed also by the higher intensity of staining for PA28γ compared with the untransfected cells). (C) A PDGF-stimulated increase in the levels of endogenous PA28γ triggers fragmentation of CBs. MCF-7 cells treated with PDGF and stained with anti-PA28γ (green) and anti-coilin (red) antibodies. In cells treated with PDGF for 30 h (bottom), increased levels of PA28γ correlate with the fragmentation of CBs (middle bottom). (D) A decrease in the levels of PA28γ promotes the restoration of CB integrity in FTC-133 cells. FTC-133 cells were either mock- or IFNγ-treated for 72 h, stained with anti-PA28γ (green) and -coilin (red) antibodies. In IFNγ-treated cells (bottom), the resulting reduction in the levels of PA28γ correlates with the reappearance of intact CBs (arrows).Bars, 10 μM.
To characterize further the effect of increased PA28γ expression levels, we used two experimental models.
First, because growth stimuli can increase PA28γ levels (Nikaido et al., 1989), we treated serum-starved MCF-7 cells for 72 h with either PDGF or EGF at the indicated doses for 30 h (Fig. 6 C and not depicted). The majority (>80%) of treated cells showed a brighter signal for PA28γ in the nucleus (Fig. 6 C, left), and almost all cells with an increased PA28γ signal showed a redistribution of coilin identical to that elicited by UV-C irradiation (Fig. 6 C, bottom).
Second, because PA28γ levels are increased in thyroid neoplasms and correlate with the tumor stage (being low in differentiated and very high in anaplastic thyroid carcinoma; Okamura et al., 2003), we tested whether PA28γ levels were also elevated in a thyroid tumor–derived cell line, FTC133 (follicular thyroid carcinoma; Fig. 6 D). Immunofluorescence analysis revealed high levels of endogenous PA28γ in FTC133 nuclei in unstressed control cells (Fig. 6 D, top). Intriguingly, in most FTC133 cells CBs are fragmented and coilin is distributed in a widespread nucleoplasmic pattern similar to that observed in other cell lines only after UV-C irradiation. Furthermore, treatment of the FTC-133 cells with interferon γ (IFNγ), a stimulus known to down-regulate PA28γ levels (Tanahashi et al., 1997), for 72 h caused down-regulation of nuclear PA28γ and the reappearance of intact CBs in >70% of the treated cells (Fig. 6 D, bottom).
Overexpression of PA28γ recapitulates many, but not all, of the changes in subnuclear structure induced by UV-C treatment. Thus, TMG-capped snRNAs, SMN and SART3 (Fig. 7 A), were each relocalized out of CBs upon transient overexpression of PA28γ, whereas NPAT, fibrillarin, and NOPP140 remained localized in residual CBs (Fig. 7 B). However, it did not cause snRNPs to accumulate in enlarged splicing speckles. A likely explanation for this difference is that PA28γ overexpression, unlike UV-C, does not cause inhibition of transcription (Fig. 6 B).
Figure 7.
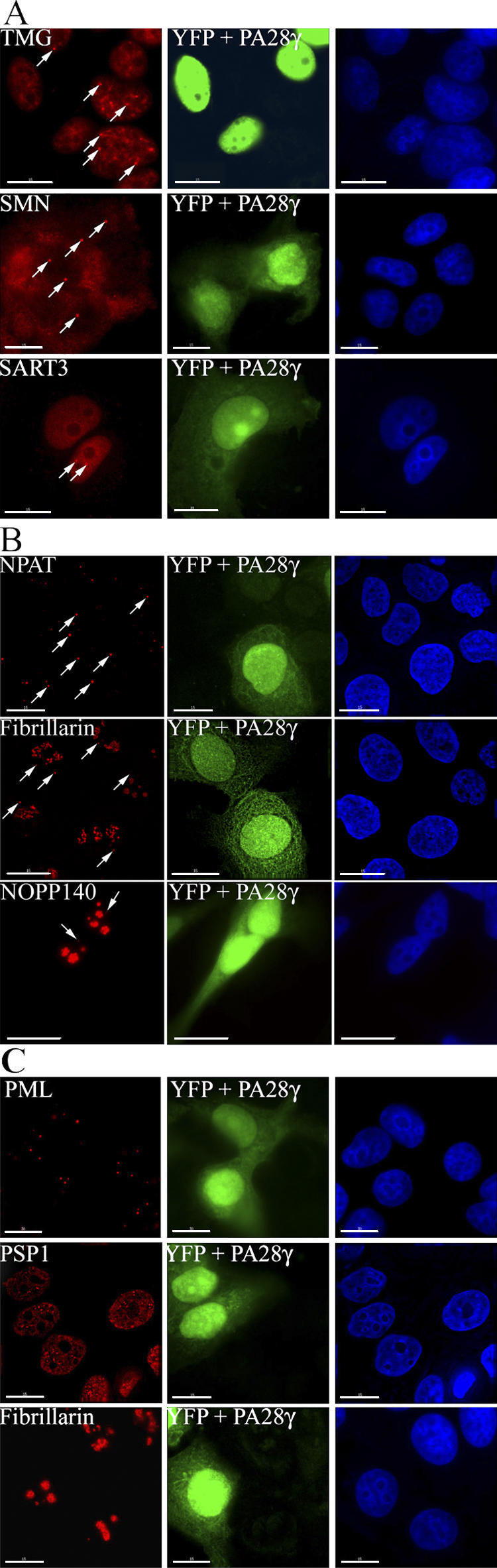
The overexpression of PA28γ alters the CB localization of TMG-capped RNAs, SMN, and SART3. (A) MCF-7 cells cotransfected with FLAG-PA28γ and pEYFP-C1 expression vectors (green) and stained with anti-TMG-cap, -SMN, and -SART3 antibodies (red). Arrows indicate intact CBs in the untransfected YFP-negative cells. (B) The overexpression of PA28γ does not alter the CB localization of NPAT, fibrillarin, and NOPP140. MCF-7 cells, cotransfected with FLAG-PA28γ and pEYFP-C1 expression vectors, were stained with anti-NPAT, anti-fibrillarin, and anti-NOPP140 antibodies (red). Arrows indicate intact CBs in both the untransfected (YFP-negative) and transfected (YFP-positive) cells. (C) Overexpression of PA28γ does not alter the integrity of PML bodies, paraspeckles, and nucleoli. MCF-7 cells cotransfected with FLAG-PA28γ and pEYFP-C1 expression vectors (green) were stained with anti-PML, anti-PSP1, and anti-fibrillarin antibodies (red). Little or no change was observed for any of these structures in the transfected YFP-positive cells. Bars,10 μM.
We observe that the effect of PA28γ is specific for CBs. Thus, transient overexpression of PA28γ had little or no effect on the number, integrity, or morphology of PML bodies, paraspeckles, or nucleoli in MCF-7 cells (Fig. 7 C). In summary, multiple independent lines of evidence strongly suggest that PA28γ can specifically affect CB integrity and coilin subnuclear localization.
Knock down of PA28γ attenuates the UV-C–induced redistribution of coilin
If PA28γ is the mediator of the UV-C effect on CBs, we postulated that RNA interference (RNAi)–mediated knock down of PA28γ should attenuate this effect. Therefore, we performed PA28γ knockdown experiments in both MCF-7 and HaCaT cells before and after UV-C treatment (Fig. 8 and not depicted). Initial experiments identified two double-stranded RNA oligonucleotides able to reduce the levels of PA28γ in transfected cells after 72 h. To monitor the transfection efficiency, a plasmid vector expressing YFP alone was cotransfected together with the RNAi duplexes. Protein blotting analysis showed that both RNAi duplexes specifically decreased PA28γ protein levels, but not the levels of either endogenous lamin B1 or transiently expressed YFP (Fig. 8 A). Immunofluorescence analysis with anti-coilin antibodies showed that the degree of UV-C–induced disassembly of CBs and coilin redistribution was reduced (>50% of the transfected cells do not show CB disassembly upon UV-C) in the cells transfected with either of the specific RNAi oligos (Fig. 8 B, arrows). In contrast, cells transfected with the control oligonucleotides are indistinguishable from the adjacent untransfected cells in terms of their UV-C response (Fig. 8 B, bottom). We note that neither the specific nor the control siRNAs altered the integrity of CBs in mock-treated cells (unpublished data). We conclude that PA28γ contributes to the mechanism that modulates the response and/or the sensitivity of CBs to UV-C irradiation.
Figure 8.

Knockdown of PA28γ attenuates the UV-C–induced redistribution of coilin. (A) MCF-7 cells were cotransfected with a pEYFP-C1 expression vector and with either PA28γ-specific RNAi oligonucleotides (oligos 1 and 2) or a control oligonucleotide (CT). Western blot analysis revealed a dramatic reduction in the PA28γ protein levels after 72 h in the cells transfected with the RNAi-specific oligonucleotides (top). Anti-LaminB1 and anti-GFP antibodies were used to assess equal loading of the samples and the efficiency of transfection, respectively (middle and bottom). (B) MCF-7 cells cotransfected with a pEYFP-C1 expression vector (green) and with either anti–PA28γ-specific RNAi oligonucleotides (top and middle) or a control oligonucleotide (bottom) were UV-C irradiated and stained 6 h later with an anti-coilin antibody (red). Arrows indicate intact CBs in the irradiated cells transfected with anti-PA28γ specific dsRNAs. Arrowheads denote the UV-C–induced, coilin-containing domains in the irradiated cells transfected with control dsRNAs. Bars, 10 μM.
Discussion
In this study, we have detected a novel effect of UV-C irradiation, showing that it disrupts CBs, causing a selective redistribution of a subset of CB components. Coilin, snRNPs, and other CB proteins involved in snRNP assembly and/or maturation are displaced from CBs after UV-C treatment, whereas other CB factors, including fibrillarin, NOPP140, and NPAT, remain in residual CB-like bodies. This effect is reversible, with maximum disruption evident within 6 h after treatment of cell lines in culture with UV-C and partial recovery within 12 h. We identify a novel coilin-associated factor, PA28γ, whose association with coilin complexes is increased by UV-C treatment, and we show that it is an important mediator of the molecular mechanism leading to UV-C–induced CB fragmentation. Furthermore, a transient increase in the level of PA28γ expression, in the absence of UV-C, is sufficient to trigger a similar CB disruption and relocalization of the same subset of CB components to that seen in response to UV-C.
PA28γ was originally discovered as a target of human autoantibodies in serum from patients suffering from systemic lupus erythematosus and as a proliferation-associated antigen (Ki antigen; Nikaido et al., 1989, 1990). It was named PA28γ because of its homology to the known proteasome activators PA28α and β, which share ∼40% amino acid identity. This similarity was also supported by the independent observation that PA28γ enhances the trypsin-like activity of the proteasome toward small peptides in vitro (Wilk et al., 2000). However, it remains unclear whether the only in vivo role of PA28γ is to activate the proteasome. For example, purified PA28γ activates the isolated proteasome to a lesser extent than PA28α and β, (Realini et al., 1997). Moreover, although expression of both PA28α and β is up-regulated by IFNγ, the protein levels of PA28γ, in contrast, drop dramatically after treatment with this chemokine (Tanahashi et al., 1997). This effect of IFNγ reducing the levels of PA28γ was also observed in this study using FTC133 cells. Another difference is provided by the phenotype of both PA28 α and β −/− mice, which have major immunological problems, including impaired antigen processing, cytotoxic T cell activation, and assembly of the proteasome subunits (Preckel et al., 1999). This phenotype is not evident in either of the two PA28γ knockout mice models generated, only one of which showed relatively modest immune defects, namely a slight reduction in the levels of CD8+ T cells and slower clearance of an experimental lung infection (Barton et al., 2004); in both cases, PA28γ −/− mice are viable (Murata et al., 1999; Barton et al., 2004).
We provide evidence demonstrating both a physical and a functional link between coilin and PA28γ. Evidence for a physical link came from biochemical affinity purification and proteomic analysis, which revealed PA28γ as the most prominent protein whose copurification with FLAG-tagged coilin was increased by UV-C irradiation. The endogenous forms of both proteins are also coimmunoprecipitated with antibodies specific for either coilin or PA28γ, and they form a stable, salt-resistant complex in vivo, even in the absence of UV-C irradiation. We examined whether PA28γ binds directly to coilin using several independent in vitro assays and purified recombinant proteins (unpublished data). In all cases, we see no evidence for direct binding. Therefore, although we cannot exclude that these negative results are caused by technical limitations in the binding assays, it is also possible that the association between coilin and PA28γ in vivo involves additional nuclear components and/or posttranslational modifications of one or both proteins.
Although PA28γ associates with coilin-containing complexes in the absence of UV-C treatment, we do not observe colocalization or accumulation of PA28γ in CBs. Instead, PA28γ in untreated cells is widely distributed throughout the nucleoplasm. Despite the obvious concentration of coilin in bright CB foci when viewed by immunofluorescence, it is known that the major fraction of coilin is also present in a diffuse nucleoplasmic pool (Ogg and Lamond, 2002). We infer from this that the complexes containing both coilin and PA28γ are present in this diffuse nucleoplasmic pool. Considering that our gel filtration chromatography experiments showed that only the smaller size class of coilin complexes cofractionates with PA28γ, it is possible that these smaller complexes may form at least part of this pool.
The level of complexes containing coilin and PA28γ clearly increases after UV-C treatment. Interestingly, the kinetics of this complex formation tightly parallel the kinetics of CB disruption, which is consistent with a functional connection. Further evidence for a functional role for PA28γ in CB integrity was provided by our discovery that RNAi-mediated knockdown of PA28γ either prevents or reduces the effect of UV-C treatment on CBs in irradiated cells. Although loss of PA28γ by RNAi prevents the UV-C response, we also show that an increase in the levels of PA28γ, either through transient overexpression, growth factor treatment, or in the transformed cell line FTC133, is sufficient to trigger a similar disruption of a subset of CB components to that induced by UV-C. However, it is unlikely that the effect of UV-C irradiation on CBs results from a large increase in intracellular PA28γ levels. Indeed, we observe only a small increase in PA28γ levels during the time course of the UV-C response. Instead UV-C may lead to an increase in affinity of PA28γ for the coilin complexes. For example a UV-C–induced phosphorylation mechanism could be involved. In this regard, it is interesting that a physical and functional interaction of PA28γ with MEKK3 has been reported (Hagemann et al., 2003). MEKK3 is a kinase involved in transducing both mitogenic (serum and FGF-2) and stress signals (UV-C and osmotic stress; Adams et al., 2002; Uhlik et al., 2003; Gilmore et al., 2004). Our preliminary results indicate that overexpression of MEKK3 increases the UV-C–induced interaction of PA28γ and coilin (unpublished data). Furthermore, we have also observed that hyperosmotic stress, induced by treating both HaCaT and MCF7 cells with 0.5 M sorbitol, resulted in a similar disruption of coilin from CBs to that caused by UV-C (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200604099/DC1). The fact that MEKK3 is known to be activated by both UV-C and hyperosmotic shock may not be a coincidence. Therefore, future studies will examine in more detail the role and mechanism of PA28γ in regulating CB integrity and the potential role of MEKK3 in signaling to CBs.
Interestingly, we observe that overexpression of PA28γ causes CB disruption without reducing nuclear transcription levels. It also acts specifically on CBs without changing the morphology or number of PML bodies, paraspeckles, or nucleoli. Based on our results, we infer that the novel effect of UV-C we have detected on CBs involves a specific PA28γ-dependent pathway. Although UV stress causes a complex set of cellular responses, affecting multiple targets and down-regulating nuclear transcription, our analysis of PA28γ should allow us to dissect a subset of these cellular events and the mechanisms involved.
Materials and methods
Cell culture, treatments, and transfections
WI 38, HeLa, COS-7, HaCaT, SAOS-2, and 293T cells were cultured as monolayers in DME (Invitrogen) supplemented with 10% fetal bovine serum, nonessential amino acids, penicillin-streptomycin, and l-glutamine (Invitrogen) in a humidified incubator at 37°C with 5% CO2. HCT116 and SW480 cells were cultured as monolayers in McCoy 5A medium (Invitrogen), supplemented like the aforementioned cells. MCF-7 cells were grown in RPMI medium (Invitrogen) supplemented as like the aforementioned cells. PDGF and EGF (Millipore) were added to the medium of serum-starved MCF-7 cells to a final concentration of 25 and 100 ng/ml, respectively. DRB was added to the cells at a final concentration of 30 μg/ml, for 3 h. Actinomycin D was added to the cells at a final concentration of 10 μg/ml for 2 h.
FTC-133 cells were treated with 500 U/ml of human recombinant IFN γ (PBL Laboratories). When indicated, cells were transfected with 250–500 ng /90-mm dish of the appropriate plasmid DNA using Effectene transfection reagent (QIAGEN) according to the manufacturer's instructions.
The PCDNA3-FLAG-PA28γ expression vector was a kind gift of M. Rechsteiner (University of Utah School of Medicine, Salt Lake City, UT). For PA28γ RNAi experiments, the following oligonucleotides targeting PA28γ have been used: OLIGO 1 (5-GAA GCC UUC CAA GGA ACC ATT-3) and OLIGO 2 (5-ACA UCC AUG ACC UAA CUC ATT-3; MWG Biotech). As a control (“off-target”), a dsRNA oligonucleotide targeting the Photinus pyralis luciferase gene was used (5-CGU ACG CGG AAU ACU UCGA-3). dsRNA oligonucleotides were transfected by using the RNAiFect transfection reagent (QIAGEN). dsRNAs were cotransfected with a highly purified (CsCl gradient) plasmid vector encoding YFP (p-EYFP-C1; CLONTECH Laboratories, Inc.), at a molar ratio of 1:40 (YFP: RNAi oligonucleotide) to facilitate the identification of transfected cells and to evaluate the effect of the transfection on the viability of the cells.
Reagents
Except when stated otherwise, all of the chemicals used were from Sigma-Aldrich.
Biochemical fractionation of isolated nuclei
Subcellular fractions were obtained from either mock- or UV-C–treated cells as follows: in brief, cell nuclei isolated in hypotonic/detergent containing buffer were subjected to DNase/RNase treatment and high salt extraction with (NH4)2SO4 and NaCl, respectively. The remaining material was solubilized with urea-containing buffer (10 mM Tris-Cl, pH 8.0, 9 M urea, IGEPAL AC-630, 100 mM KCl, 50 mM DTT, 50 mM NaF, 1 mM NaVO4, 1 mM MgCl2). Before SDS-PAGE analysis, the final salt concentration of the samples was normalized by the addition of 2 M (NH4)2SO4.
UV-C irradiation
In brief, semiconfluent cells were washed with PBS and incubated for 8–12 h in DME (or RPMI or McCoy 5A) supplemented with 0.5% FBS (serum starvation). Medium was collected and kept at 37°C and the PBS-washed cells irradiated in a UV Stratalinker 2400 oven with 254-nm bulbs (Stratagene) at 30 J/m2. After that, the old medium was quickly added back into the dishes and the cells were incubated for the indicated times.
Sorbitol treatment
Semiconfluent cells were washed with PBS and incubated for 8–12 h in DME (or RPMI or McCoy 5A) supplemented with 0.5% FBS (serum starvation). After that, medium was collected and cells were washed once with PBS and either mock-treated (old medium added back) or incubated with 0.5 M sorbitol (Sigma-Aldrich) dissolved in the previously collected medium for the indicated times.
Cell fixation and immunofluorescence microscopy
Adherent cells grown on glass coverslips were fixed for 10 min in freshly prepared PBS/3.7% paraformaldehyde at RT. Permeabilization was performed with PBS /1% IGEPAL AC-630 for 10 min at RT. After extensive washing, samples were blocked with PBS /2% BSA (Sigma-Aldrich) for at least 1 h at RT, and then incubated with the indicated antibodies at RT. When indicated, cell nuclei were stained with DAPI (0.3 μg/ml; Sigma- Aldrich) diluted in PBS for 3 min at RT. Coverslips were mounted in Vectashield medium (Vector Laboratories) and stored protected from light at 4°C.
Polyclonal antibodies used were as follows: anti-coilin 204/10 (1:300; Bohmann et al., 1995; Fig. 1, A and B [top and middle] and C; Fig. 2, A and B [top and middle] and C [middle and bottom]; Fig. 3, A and B; Fig. 5 B; Fig. 6, B–D; Fig. 8 B), anti-NPAT (1:500; a gift from J. Zhao, University of Rochester, Rochester, NY; Fig. 2 C; Fig. 7 B) anti–SART3 (1:200; a gift from D. Stanek and K. Neugebauer, Max Planck Institute, Dresden, Germany; Fig. 2, A and B; Fig. 7 A); anti-PSP1 (1:400; Fig. 7 C; Fox 2002); and anti-PA28γ (1:400; Affiniti Research Products; Fig. 5 C; Fig. 6 B; Fig. S2, top and bottom).
Monoclonal antibodies used were as follows: anti-coilin 5P10 (1:300; Fig. 1 B bottom; Fig. 2, A and B [bottom] and C [top]; Rebelo et al., 1996); anti-fibrillarin 72B9 (1:25; Fig. 2 C; Fig. 7, Band C; Reimer et al., 1987), anti-SC35 (1:400; Sigma-Aldrich; Fig. 1, A and B; Fig. 5 C; Fig. S2); anti PSME3 (PA28γ; 1:100; BD Biosciences; Fig. 5 B; Fig. 6, A, C, and D; Fig. S2, bottom); anti-SMN (1:100; BD Biosciences; Fig. 2, A and B; Fig. 7 A); anti-2,2,7 TMG (1:300; Oncogene; Fig. 2, A and B; Fig. 7 A; Fig. S2); anti-PML (1:100; Santa Cruz Biotechnologies, Inc.; Fig. 7 C).
Secondary antibodies used were FITC- or TRITC-conjugated goat anti–mouse or anti–rabbit IgGs (1:500; Jackson ImmunoResearch Laboratories).
5-FU incorporation assay
HaCaT or MCF-7 cells, either mock-treated or treated with UV-C or DRB for the indicated length of time, were incubated with 2 mM 5-FU (F5130) for 30 min at 37°C. Subsequently, cells were fixed, permeabilized, and incubated with primary anti-BrdU antibody (B2531; 1:500). Immunofluorescence microscopy was performed as indicated (see previous section). For transfected cells, 5-FU labeling was performed at 24 h after transfection.
Whole-cell lysate preparation and Western blotting
In brief, enzymatically detached cells were pelleted and resuspended in 2× reducing loading buffer (Invitrogen) and denatured at 80°C for 5 min. When indicated, cells were previously resuspended in urea-containing buffer (see Biochemical fractionation of isolated nuclei). For Western blot analysis, proteins resolved by SDS-PAGE were transferred to nitrocellulose filters (Schleicher and Schuell) and probed with specific primary antibodies and the corresponding horseradish peroxidase-conjugated secondary antibodies (GE Healthcare). An enhanced chemiluminescence reagent (ECL; GE Healthcare) was used to visualize protein bands, according to manufacturer's instructions. Red Ponceau staining of the transferred proteins was used to assess equal loading of the samples. Monoclonal antibodies used for Western blot analysis were as follows: anti-coilin 5P10 (1:100; Bohmann et al., 1995; Fig. 4 B; Fig. 5 A); anti-LaminB1 (1:200; Zymed Laboratories; Fig. 8); anti-GFP (1:500; Roche; Fig. 8), anti-PSME3 (PA28γ; 1:500; BD Biosciences; Fig. 4 B; Fig. 5, A and D). Polyclonal antibodies used for Western blot analysis were as follows: anti-coilin 204/10 (1:2,000 Fig. 3, C–E; Fig. 4 C; Fig. 8), anti-PA28γ (1:2,000; Affiniti Research Products; Fig. 4 C; Fig. 8); and anti-sDMA (SYM10; 1:500; Millipore; Fig. 3, D and E).
Nuclear extract preparation and immunoprecipitation
Enzymatically detached cells were pelleted and resuspended in hypotonic buffer (10 mM Tris-Cl, pH 7.1, 10% glycerol, 4 mM DTT, 50 mM NaF, 1 mM NaVO4, 1 mM MgCl2, and Roche protease inhibitors). IGEPAL AC-630 was added dropwise while mixing to a final concentration of 0.5%. Cells were gently resuspended for 3 min on ice and crude nuclei were pelleted by centrifugation (3,000 rpm for 5 min at 4°C) and resuspended in two bead volumes of ice-cold digestion buffer (2 mM Tris-Cl, pH 8.5, 20% glycerol, 10 mM DTT, 50 mM NaF, 1 mM NaVO4, 1 mM MgCl2, 5 mM CaCl2, Roche protease inhibitors, and 75 U/ml micrococcal nuclease; GE Healthcare). Samples were allowed to digest at 25°C for 15 min with continuous mixing, and then ice-cold extraction buffer (2 mM Tris-Cl, pH 8.5, 50 mM NaF, 1 mM NaVO4, 1 mM MgCl2, 20 mM EDTA, pH 8.0, 0.84 M KCl, and Roche protease inhibitors) was added vol/vol. Samples were then incubated on ice with frequent mixing for 20 min. Finally, the nuclear lysate was clarified by ultracentrifugation at 50,000 rpm for 30 min at 4°C in a fixed-angle rotor (TLA 100.3; Beckman Coulter). Supernatant was collected and used immediately or flash frozen in liquid nitrogen.
For size fractionation of nuclear extracts, cleared nuclear extract (0.5 ml) was loaded into a Superose 6 (GE Healthcare) gel filtration column equilibrated with 2 mM Tris-Cl, pH 8.5, 50 mM NaF, 1 mM NaVO4, 1 mM MgCl2, 20 mM EDTA, pH 8.0, 0.5 M KCl, and Roche protease inhibitors by using a FPLC system (GE Healthcare). 1-ml fractions were collected, and the size of the eluted complexes calculated according to the elution profile of known molecular weight markers (Fig. 4C).
For immunoprecipitation studies, in brief, unconjugated antibodies were added to the cleared nuclear extracts on ice for 45 min. After that, protein A– or G–agarose beads (GE Healthcare) preblocked for 30 min at RT with 2% BSA in hypotonic buffer, were added to the solution, and the mixture was incubated for 3 h at 4°C with slow agitation. The beads were then collected by low-speed centrifugation (1,000 rpm for 3 min at 4°C) and washed three times with high-salt washing buffer (2 mM Tris-Cl, pH 8.0, 10% glycerol, 50 mM NaF, 1 mM NaVO4, 1 mM MgCl2, 0.3 M KCl, 0.05% IGEPAL AC-630, protease inhibitor cocktail [Roche]) and twice with low-salt washing buffer (2 mM Tris-Cl, pH 8.0, 10% glycerol, 50 mM NaF, 1 mM NaVO4, 1 mM MgCl2, 0.1 M KCl, 0.2% IGEPAL AC-630, and Roche protease inhibitor cocktail). Immunoprecipitated material was eluted by incubating the beads with reducing loading buffer at 70°C for 5 min. Primary antibodies used for immunoprecipitation studies were as follows: a mixture (1:1) of mouse monoclonal anti-coilin antibodies (1:50 [BD Biosciences] and 1:50 [Sigma-Aldrich], respectively: Fig. 3 D); anti-FLAG M2 agarose conjugated (Sigma-Aldrich; Fig. 4 A), rabbit polyclonal anti-PA28γ antibodies (a mix of MBL [1:100] and Affiniti Research [1:150], respectively; Fig. 4 B and Fig. 5 A). For control immunoprecipitations, equivalent amounts of a rabbit polyclonal anti-GST antibody (GE Healthcare; Fig. 4 B) or a mouse monoclonal anti-GFP antibody (Roche; Fig. 3 D and Fig. 5 A) were used, respectively.
Image acquisition and manipulation
All images were acquired with a Deltavision Restoration Microscope (Applied Precision) and a camera (Micromax KAF1400; Kodak). Imaging was performed at RT using either a 40× Plan-Neofluar or a 63× Plan-Apochromat (Carl Zeiss MicroImaging, Inc.) objective lens. Images were acquired as TIFF files using SoftWorX (Applied Precision) and Photoshop (Adobe) used for composing the panels shown in the respective figures.
Isolation of coilin-containing complexes
Semi-confluent 293 cells transiently transfected with a PCDNA3-FLAG-coilin expression vector, were either mock- or UV-C–treated. The derived cleared nuclear extracts (typically 50 mg of protein) were loaded into a 5-ml heparin–Sepharose CL 4B column (GE Healthcare) by using a FPLC system. The retained protein complexes were eluted with a gradient of increasing ionic strength (10 mM Tris-Cl, pH 7.1, 10% glycerol, 4 mM DTT, 50 mM NaF, 1 mM NaVO4, 1mM MgCl2, 0.15% IGEPAL AC-630, 50–600 mM KCl, and Roche protease inhibitors cocktail) and the coilin-containing fractions (eluted in a peak at ∼200–250 mM KCl) were subsequently incubated with M2 anti-FLAG antibody conjugated agarose beads (Sigma-Aldrich) for 3 h at 4°C with gentle agitation. Immunoprecipitated material was processed as previously described (see Nuclear extract preparation and immunoprecipitation) and collected by a three-step elution with two bead volumes of 100 mM glycine-HCl, pH 3.0., 1 M Tris-Cl, pH 8.5, and 100% glycerol (Sigma- Aldrich) were added to the eluted fractions to a final concentration of 100 mM and 10%, respectively, before further use or flash freezing in liquid nitrogen.
Mass spectrometry and identification of PA28γ
Purified protein complexes were separated on SDS-PAGE gels and stained with Colloidal Coomassie (Invitrogen). The chosen protein bands were excised and subjected to trypsin digestion, and the derived peptides were analyzed by matrix-assisted laser desorption/ionization time-of-flight (Perspective Biosystems) at the Peptide Mass Fingerprinting facility at the University of Dundee (http://www.dundee.ac.uk/biocentre/services_proteomics.htm). Protein identification was made with the ProteinProspector software MS-FIT (http://prospector.ucsf.edu/prospector/4.0.7/html/msfit.htm) using the NCBInr (nonredundant) and Swissprot databases.
Online supplemental material
Fig. S1 shows that UV-C enhances PA28γ in coilin complexes. Fig. S2 shows that PA28γ redistributes to splicing speckles in irradiated cells. Fig. S3 shows that hyperosmolar shock triggers fragmentation of CBs. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200604099/DC1.
Supplementary Material
Acknowledgments
We wish to thank J. Zhao, D. Stanek, and K.M. Neugebauer and M. Rechsteiner for the generous gift of anti-NPAT and anti-SART3 antibodies and a PCDNA3-FLAG-PA28γ expression vector, respectively. We thank E. Frittoli and G. Scita for their helpful discussions and suggestions. We are grateful to L. Trinkle-Mulcahy, D. Lleres, M. Denegri, and other members of the Lamond laboratory for their advice and support, and we thank Y.W Lam for sharing observations and preliminary results.
A.I. Lamond is a Wellcome Trust Principal Research Fellow. M. Cioce was partially supported by International Agency for Research on Cancer. S. Boulon is supported by a European Molecular Biology Organization long-term fellowship.
Mario Cioce's present address is Instituto di Ricerca di Biologia Molecolare P. Angeletti, Merck Research Laboratories Rome, 00040 Pomezia Rome, Italy.
Abbreviations used in this paper: CB, Cajal body; FU, fluoruridine; IFNγ, interferon γ; DRB, 5,6-dichloro-1-β-D-ribobenzimidazole; NB, nuclear body; RNAi, RNA interference; sDMA, symmetric dimethylarginine; SMN, survival of motor neuron; snRNP, small nuclear RNP; TMG, trimethylguanosine.
References
- Adams, D.G., N.A. Sachs, and R.R. Vaillancourt. 2002. Phosphorylation of the stress-activated protein kinase, MEKK3, at serine 166. Arch. Biochem. Biophys. 407:103–116. [DOI] [PubMed] [Google Scholar]
- Al-Baker, E.A., J. Boyle, R. Harry, and I.R. Kill. 2004. A p53-independent pathway regulates nucleolar segregation and antigen translocation in response to DNA damage induced by UV irradiation. Exp. Cell Res. 292:179–186. [DOI] [PubMed] [Google Scholar]
- Barton, L.F., H.A. Runnels, T.D. Schell, Y. Cho, R. Gibbons, S.S. Tevethia, G.S. Deepe Jr., and J.J. Monaco. 2004. Immune defects in 28-kDa proteasome activator gamma-deficient mice. J. Immunol. 172:3948–3954. [DOI] [PubMed] [Google Scholar]
- Bohmann, K., J.A. Ferreira, and A.I. Lamond. 1995. Mutational analysis of p80 coilin indicates a functional interaction between coiled bodies and the nucleolus. J. Cell Biol. 131:817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert, F.M., J. Cote, M.C. Boulanger, P. Cleroux, F. Bachand, C. Autexier, and S. Richard. 2002. Symmetrical dimethylarginine methylation is required for the localization of SMN in Cajal bodies and pre-mRNA splicing. J. Cell Biol. 159:957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo-Fonseca, M., J. Ferreira, and A.I. Lamond. 1993. Assembly of snRNP-containing coiled bodies is regulated in interphase and mitosis–evidence that the coiled body is a kinetic nuclear structure. J. Cell Biol. 120:841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioce, M., and A.I. Lamond. 2005. Cajal bodies: a long history of discovery. Annu. Rev. Cell Dev. Biol. 21:105–131. [DOI] [PubMed] [Google Scholar]
- Claus, P., F. Doring, S. Gringel, F. Muller-Ostermeyer, J. Fuhlrott, T. Kraft, and C. Grothe. 2003. Differential intranuclear localization of fibroblast growth factor-2 isoforms and specific interaction with the survival of motoneuron protein. J. Biol. Chem. 278:479–485. [DOI] [PubMed] [Google Scholar]
- Darzacq, X., B.E. Jady, C. Verheggen, A.M. Kiss, E. Bertrand, and T. Kiss. 2002. Cajal body-specific small nuclear RNAs: a novel class of 2'-O-methylation and pseudouridylation guide RNAs. EMBO J. 21:2746–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr, M., and T. Misteli. 2001. Functional architecture in the cell nucleus. Biochem. J. 356:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert, C., A. Chari, B. Laggerbauer, and U. Fischer. 2006. Spinal muscular atrophy: the RNP connection. Trends Mol. Med. 12:113–121. [DOI] [PubMed] [Google Scholar]
- Fox, A.H., Y.W. Lam, A.K. Leung, C.E. Lyon, J. Andersen, M. Mann, and A.I. Lamond. 2002. Paraspeckles: a novel nuclear domain. Curr. Biol. 12:13–25. [DOI] [PubMed] [Google Scholar]
- Frey, M.R., and A.G. Matera. 1995. Coiled bodies contain U7 small nuclear RNA and associate with specific DNA sequences in interphase human cells. Proc. Natl. Acad. Sci. USA. 92:5915–5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frugier, T., S. Nicole, C. Cifuentes-Diaz, and J. Melki. 2002. The molecular bases of spinal muscular atrophy. Curr. Opin. Genet. Dev. 12:294–298. [DOI] [PubMed] [Google Scholar]
- Gall, J.G. 2001. A role for Cajal bodies in assembly of the nuclear transcription machinery. FEBS Lett. 498:164–167. [DOI] [PubMed] [Google Scholar]
- Gall, J.G. 2003. The centennial of the Cajal body. Nat. Rev. Mol. Cell Biol. 4:975–980. [DOI] [PubMed] [Google Scholar]
- Gangwani, L., R.A. Flavell, and R.J. Davis. 2005. ZPR1 is essential for survival and is required for localization of the survival motor neurons (SMN) protein to Cajal bodies. Mol. Cell. Biol. 25:2744–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore, P.M., N. McCabe, J.E. Quinn, R.D. Kennedy, J.J. Gorski, H.N. Andrews, S. McWilliams, M. Carty, P.B. Mullan, W.P. Duprex, et al. 2004. BRCA1 interacts with and is required for paclitaxel-induced activation of mitogen-activated protein kinase kinase kinase 3. Cancer Res. 64:4148–4154. [DOI] [PubMed] [Google Scholar]
- Gross, S., A. Knebel, T. Tenev, A. Neininger, M. Gaestel, P. Herrlich, and F.D. Bohmer. 1999. Inactivation of protein-tyrosine phosphatases as mechanism of UV-induced signal transduction. J. Biol. Chem. 274:26378–26386. [DOI] [PubMed] [Google Scholar]
- Gulati, P., B. Markova, M. Gottlicher, F.D. Bohmer, and P.A. Herrlich. 2004. UVA inactivates protein tyrosine phosphatases by calpain-mediated degradation. EMBO Rep. 5:812–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann, C., R. Patel, and J.L. Blank. 2003. MEKK3 interacts with the PA28 gamma regulatory subunit of the proteasome. Biochem. J. 373:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handwerger, K.E., and J.G. Gall. 2006. Subnuclear organelles: new insights into form and function. Trends Cell Biol. 16:19–26. [DOI] [PubMed] [Google Scholar]
- Hebert, M.D., P.W. Szymczyk, K.B. Shpargel, and A.G. Matera. 2001. Coilin forms the bridge between Cajal bodies and SMN, the spinal muscular atrophy protein. Genes Dev. 15:2720–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert, M.D., K.B. Shpargel, J.K. Ospina, K.E. Tucker, and A.G. Matera. 2002. Coilin methylation regulates nuclear body formation. Dev. Cell. 3:329–337. [DOI] [PubMed] [Google Scholar]
- Jady, B.E., X. Darzacq, K.E. Tucker, A.G. Matera, E. Bertrand, and T. Kiss. 2003. Modification of Sm small nuclear RNAs occurs in the nucleoplasmic Cajal body following import from the cytoplasm. EMBO J. 22:1878–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch-Paiz, C.A., S.A. Amundson, M.L. Bittner, P.S. Meltzer, and A.J. Fornace Jr. 2004. Functional genomics of UV radiation responses in human cells. Mutat. Res. 549:65–78. [DOI] [PubMed] [Google Scholar]
- Kurki, S., L. Latonen, and M. Laiho. 2003. Cellular stress and DNA damage invoke temporally distinct Mdm2, p53 and PML complexes and damage-specific nuclear relocalization. J. Cell Sci. 116:3917–3925. [DOI] [PubMed] [Google Scholar]
- Kurki, S., K. Peltonen, and M. Laiho. 2004. Nucleophosmin, HDM2 and p53: players in UV damage incited nucleolar stress response. Cell Cycle. 3:976–979. [PubMed] [Google Scholar]
- Lamond, A.I., and W.C. Earnshaw. 1998. Structure and function in the nucleus. Science. 280:547–553. [DOI] [PubMed] [Google Scholar]
- Lamond, A.I., and D.L. Spector. 2003. Nuclear speckles: a model for nuclear organelles. Nat. Rev. Mol. Cell Biol. 4:605–612. [DOI] [PubMed] [Google Scholar]
- Matera, A.G., and K.B. Shpargel. 2006. Pumping RNA: nuclear bodybuilding along the RNP pipeline. Curr. Opin. Cell Biol. 18:317–324. [DOI] [PubMed] [Google Scholar]
- Murata, S., H. Kawahara, S. Tohma, K. Yamamoto, M. Kasahara, Y. Nabeshima, K. Tanaka, and T. Chiba. 1999. Growth retardation in mice lacking the proteasome activator PA28gamma. J. Biol. Chem. 274:38211–38215. [DOI] [PubMed] [Google Scholar]
- Narayanan, U., T. Achsel, R. Luhrmann, and A.G. Matera. 2004. Coupled in vitro import of U snRNPs and SMN, the spinal muscular atrophy protein. Mol. Cell. 16:223–234. [DOI] [PubMed] [Google Scholar]
- Nikaido, T., K. Shimada, Y. Nishida, R.S. Lee, A.B. Pardee, and Y. Nishizuka. 1989. Loss in transformed cells of cell cycle regulation of expression of a nuclear protein recognized by SLE patient antisera. Exp. Cell Res. 182:284–289. [DOI] [PubMed] [Google Scholar]
- Nikaido, T., K. Shimada, M. Shibata, M. Hata, M. Sakamoto, Y. Takasaki, C. Sato, T. Takahashi, and Y. Nishida. 1990. Cloning and nucleotide sequence of cDNA for Ki antigen, a highly conserved nuclear protein detected with sera from patients with systemic lupus erythematosus. Clin. Exp. Immunol. 79:209–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishigori, C. 2006. Cellular aspects of photocarcinogenesis. Photochem. Photobiol. Sci. 5:208–214. [DOI] [PubMed] [Google Scholar]
- Ogg, S.C. and A.I. Lamond. 2002. Cajal bodies and coilin—moving towards function. J. Cell Biol. 159:17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura, T., S. Taniguchi, T. Ohkura, A. Yoshida, H. Shimizu, M. Sakai, H. Maeta, H. Fukui, Y. Ueta, I. Hisatome, and C. Shigemasa. 2003. Abnormally high expression of proteasome activator-gamma in thyroid neoplasm. J. Clin. Endocrinol. Metab. 88:1374–1383. [DOI] [PubMed] [Google Scholar]
- Ospina, J.K., G.B. Gonsalvez, J. Bednenko, E. Darzynkiewicz, L. Gerace, and A.G. Matera. 2005. Cross-talk between snurportin1 subdomains. Mol. Biol. Cell. 16:4660–4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preckel, T., W.P. Fung-Leung, Z. Cai, A. Vitiello, L. Salter-Cid, O. Winqvist, T.G. Wolfe, M. Von Herrath, A. Angulo, P. Ghazal, et al. 1999. Impaired immunoproteasome assembly and immune responses in PA28−/− mice. Science. 286:2162–2165. [DOI] [PubMed] [Google Scholar]
- Realini, C., C.C. Jensen, Z. Zhang, S.C. Johnston, J.R. Knowlton, C.P. Hill, and M. Rechsteiner. 1997. Characterization of recombinant REGalpha, REGbeta, and REGgamma proteasome activators. J. Biol. Chem. 272:25483–25492. [DOI] [PubMed] [Google Scholar]
- Rebelo, L., F. Almeida, C. Ramos, K. Bohmann, A.I. Lamond, and M. Carmo-Fonseca. 1996. The dynamics of coiled bodies in the nucleus of adenovirus-infected cells. Mol. Biol. Cell. 7:1137–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer, G., I. Raska, E.M. Tan, and U. Scheer. 1987. Human autoantibodies: probes for nucleolus structure and function. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 54:131–143. [DOI] [PubMed] [Google Scholar]
- Schul, W., R. van Driel, and L. de Jong. 1998. Coiled bodies and U2 snRNA genes adjacent to coiled bodies are enriched in factors required for snRNA transcription. Mol. Biol. Cell. 9:1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seker, H., C. Rubbi, S.P. Linke, E.D. Bowman, S. Garfield, L. Hansen, K.L. Borden, J. Milner, and C.C. Harris. 2003. UV-C-induced DNA damage leads to p53-dependent nuclear trafficking of PML. Oncogene. 22:1620–1628. [DOI] [PubMed] [Google Scholar]
- Shpargel, K.B., J.K. Ospina, K.E. Tucker, A.G. Matera, and M.D. Hebert. 2003. Control of Cajal body number is mediated by the coilin C-terminus. J. Cell Sci. 116:303–312. [DOI] [PubMed] [Google Scholar]
- Sleeman, J.E., and A.I. Lamond. 1999. Newly assembled snRNPs associate with coiled bodies before speckles, suggesting a nuclear snRNP maturation pathway. Curr. Biol. 9:1065–1074. [DOI] [PubMed] [Google Scholar]
- Tanahashi, N., K. Yokota, J.Y. Ahn, C.H. Chung, T. Fujiwara, E. Takahashi, G.N. DeMartino, C.A. Slaughter, T. Toyonaga, K. Yamamura, et al. 1997. Molecular properties of the proteasome activator PA28 family proteins and gamma-interferon regulation. Genes Cells. 2:195–211. [DOI] [PubMed] [Google Scholar]
- Tornaletti, S., and G.P. Pfeifer. 1996. UV damage and repair mechanisms in mammalian cells. Bioessays. 18:221–228. [DOI] [PubMed] [Google Scholar]
- Tucker, K.E., M.T. Berciano, E.Y. Jacobs, D.F. LePage, K.B. Shpargel, J.J. Rossire, E.K. Chan, M. Lafarga, R.A. Conlon, and A.G. Matera. 2001. Residual Cajal bodies in coilin knockout mice fail to recruit Sm snRNPs and SMN, the spinal muscular atrophy gene product. J. Cell Biol. 154:293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlik, M.T., A.N. Abell, N.L. Johnson, W. Sun, B.D. Cuevas, K.E. Lobel-Rice, E.A. Horne, M.L. Dell'Acqua, and G.L. Johnson. 2003. Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat. Cell Biol. 5:1104–1110. [DOI] [PubMed] [Google Scholar]
- Wilk, S., W.E. Chen, and R.P. Magnusson. 2000. Properties of the nuclear proteasome activator PA28gamma (REGgamma). Arch. Biochem. Biophys. 383:265–271. [DOI] [PubMed] [Google Scholar]
- Zhao, J., B.K. Kennedy, B.D. Lawrence, D.A. Barbie, A.G. Matera, J.A. Fletcher, and E. Harlow. 2000. NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes Dev. 14:2283–2297. [PMC free article] [PubMed] [Google Scholar]
- Zimber, A., Q.D. Nguyen, and C. Gespach. 2004. Nuclear bodies and compartments: functional roles and cellular signalling in health and disease. Cell. Signal. 16:1085–1104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}