

Abstract
There is a growing appreciation that the cyclic adenosine monophosphate (cAMP)–protein kinase A (PKA) signaling pathway is organized to form transduction units that function to deliver specific messages. Such organization results in the local activation of PKA subsets through the generation of confined intracellular gradients of cAMP, but the mechanisms responsible for limiting the diffusion of cAMP largely remain to be clarified. In this study, by performing real-time imaging of cAMP, we show that prostaglandin 1 stimulation generates multiple contiguous, intracellular domains with different cAMP concentration in human embryonic kidney 293 cells. By using pharmacological and genetic manipulation of phosphodiesterases (PDEs), we demonstrate that compartmentalized PDE4B and PDE4D are responsible for selectively modulating the concentration of cAMP in individual subcellular compartments. We propose a model whereby compartmentalized PDEs, rather than representing an enzymatic barrier to cAMP diffusion, act as a sink to drain the second messenger from discrete locations, resulting in multiple and simultaneous domains with different cAMP concentrations irrespective of their distance from the site of cAMP synthesis.
Introduction
cAMP is a ubiquitous second messenger and is responsible for a plethora of cellular effects and biological functions (Beavo and Brunton, 2002). The generation of cAMP occurs upon ligand binding to Gs protein–coupled receptors (GPCRs) and the consequent activation of a family of transmembrane adenylyl cyclases (ACs) localized at the plasma membrane (Cooper, 2005). An alternative source of cAMP is the soluble AC, an enzyme that has been shown to localize in different subcellular compartments, the activity of which is independent of GPCR stimulation and is regulated by bicarbonate and calcium ions (Zippin et al., 2003).
cAMP exerts its cellular functions via the activation of three different effectors: the cAMP-dependent PKA, cyclic nucleotide-gated ion channels (Biel et al., 1999), and exchange proteins directly activated by cAMP (Epac; Bos, 2003). The action of cAMP is terminated via its degradation by phosphodiesterases (PDEs), a large superfamily of enzymes grouped in 11 families with >40 isoenzyme variants (Soderling and Beavo, 2000; Francis et al., 2001; Houslay and Adams, 2003; Lugnier, 2006). Individual PDE enzymes exert specific functional roles as a consequence of the unique combination of regulatory mechanisms, intracellular localization, and enzyme kinetics (Houslay and Milligan, 1997; Beavo and Brunton, 2002; Baillie et al., 2005) and play an important role in shaping intracellular gradients of cAMP (Rich et al., 2001; Zaccolo and Pozzan, 2002; Rochais et al., 2004).
The notion of the spatial regulation of cAMP as a means of generating specific downstream responses is now well accepted (Tasken and Aandahl, 2004; Wong and Scott, 2004). Such a paradigm is grounded on increasing evidence that cAMP/PKA signaling is compartmentalized in discrete subcellular domains in which PKA is anchored to A kinase–anchoring proteins (AKAPs; Wong and Scott, 2004) in close proximity to its specific targets and is activated by restricted pools of cAMP (Buxton and Brunton, 1983; Jurevicius and Fischmeister, 1996; Zaccolo and Pozzan, 2002; Mongillo et al., 2004; Barnes et al., 2005; Dodge-Kafka et al., 2005). However, the concept of the restricted intracellular diffusion of cAMP contrasts with the evidence that the diffusion rate of this second messenger in the cytosol appears to be unrestrained (∼500–700 μm2s−1; Bacskai et al., 1993; Nikolaev et al., 2004). To explain the paradox of a signaling pathway organized in spatially segregated transduction units, the selective activation of which is committed to a freely diffusible second messenger, the hypothesis of either a physical or an enzymatic barrier restricting intracellular diffusion of cAMP has been formulated (Rich et al., 2000, 2001; Zaccolo et al., 2002; Cooper, 2003; Rochais et al., 2004; Willoughby et al., 2006; for review see Brunton, 2003).
Recently, new methods for monitoring intracellular cAMP concentration in single living cells have been developed. One approach using cyclic nucleotide-gated ion channel–based sensors relies on ion influx measurements as a readout of cAMP changes near the plasma membrane (Rich et al., 2001). Another approach uses fluorescence resonance energy transfer (FRET)–based indicators in which cAMP binding to PKA (Zaccolo et al., 2000) or Epac (DiPilato et al., 2004; Nikolaev et al., 2004; Ponsioen et al., 2004) proteins leads to a change in fluorescence emission that correlates with the intracellular concentration of cAMP. Such optical biosensors allow the detection of cAMP changes with submicrometer resolution in different intracellular compartments.
These new technologies led to the identification in human embryonic kidney (HEK) 293 cells of a subplasma membrane compartment showing a larger rise in cAMP concentration in response to prostaglandin 1 (PGE1) receptor stimulation as compared with the bulk cytosol (Rich et al., 2001; DiPilato et al., 2004), but the molecular and structural components responsible for such compartmentalization largely remain to be defined. Here, we set out to study local cAMP dynamics by using FRET-based biosensors that are selectively targeted to distinct subcellular compartments in HEK293 cells. We found that compartmentalized PDEs, rather than acting as barriers to cAMP diffusion from the plasma membrane to the bulk cytosol, act as a sink that drains cAMP concentration in defined domains by locally degrading the second messenger. As a result, intracellular cAMP concentration is not bound to change along a uniform gradient from the plasma membrane to the deep cytosol, but multiple contiguous domains with different concentrations of cAMP may coexist within the volume of the cell.
Results
Generation of a FRET-based sensor for cAMP targeted to the plasma membrane
We recently generated a FRET-based sensor for real-time imaging of cAMP. The sensor, called PKA-GFP, includes the regulatory (R) and catalytic (C) subunits of the PKA tagged with the CFP and YFP variants of GFP, respectively (Fig. 1 A). When overexpressed in HEK293 cells, PKA-GFP shows a uniform distribution in the cytosol (Fig. 1 A).
Figure 1.

The PKA-based sensor for cAMP. (A) Schematic representation of the R and C subunits of PKA fused to CFP and YFP, respectively (top), and confocal images showing their localization in HEK293 cells coexpressing the two subunits of the sensor (bottom). (B) Schematic representation of the membrane-targeted version of the PKA-based sensor (top) and distribution of the two subunits in HEK293 cells (bottom) as detected at the confocal microscope. Bars, 10 μm. (C) Apparent activation constants determination for PKA-GFP and mpPKA-GFP.
To compare cAMP dynamics in the bulk cytosol and in the subplasma membrane compartment, we generated a variant of the PKA-GFP sensor that is targeted to the plasma membrane. To this end, we fused to the N terminus of the R subunit a short polypeptide (mp) corresponding to the N-terminal targeting signal from the Lyn kinase (Resh, 1999). This sequence is posttranslationally myristoylated and palmitoylated and targets to the plasma membrane (Fig. 1 B). HEK293 cells cotransfected with mpR-CFP and C-YFP (mpPKA-GFP) show the predicted localization of both the R and C subunits at the plasma membrane (Fig. 1 B). To verify that modification of the R-CFP sequence with the mp peptide does not affect the sensitivity for cAMP, we determined activation constant (Ka) values both for PKA-GFP and mpPKA-GFP. As shown in Fig. 1 C, Ka values were identical for the two sensors (Ka = 0.77 μM). Modification at the N terminus of the R subunit does not affect its dynamic interactions with the C subunit, as shown by its complete release into the cytosol upon cAMP increase and binding to mpR-CFP. C-YFP is effectively resequestered to the plasma membrane upon cAMP removal (supplemental material and Video 1; available at http://www.jcb.org/cgi/content/full/jcb.200605050/DC1).
PGE1 stimulation generates a higher cAMP response under the plasma membrane as compared with the bulk cytosol
To verify whether GPCR stimulation generates different cAMP signals in the subplasma membrane compartment as compared with the bulk cytosol, we stimulated HEK293 cells transfected with either PKA-GFP or mpPKA-GFP with 10 μM PGE1 and measured cAMP-induced FRET changes in the two compartments. As shown in Fig. 2 (A–C), the cAMP response in the subplasma membrane compartment was almost twice as large as the response in the bulk cytosol (ΔR/R0 = 18.6 ± 2.1 [mean ± SEM; n = 30] vs. 9.6 ± 1.2% [n = 34]; P = 1.4 × 10−4). Furthermore, the time to reach half-maximal response to PGE1 was almost twice as fast in the subplasma membrane compartment as compared with the bulk cytosol (t 1/2 = 58.9 ± 7.3 [n = 30] and 110.6 ± 12.5 s [n = 34], respectively; P = 1.7 × 10−3; Fig. 2 D). We found that in ∼50% of the analyzed cells, PGE1 generated a transient response in both the subplasma membrane compartment and the bulk cytosol, with the level of cAMP returning to baseline levels in 5.8 ± 0.84 and 7.6 ± 1.04 min, respectively, after application of the stimulus (Fig. 2 B). In the other 50% of the cells, the response reached a plateau value and remained sustained for at least 10 min (unpublished data). Application of the nonselective PDE inhibitor isobutyl-methyl-xanthine (IBMX; 100 μM) in the continuous presence of PGE1 raised the cAMP level in both compartments, indicating that termination of the cAMP response to PGE1 was caused by PDE activity. We excluded any overt role for receptor desensitization in the termination of the cAMP response to PGE1 based on the observation that it is possible to repeatedly stimulate the PGE1 receptor and obtain comparable rises in intracellular [cAMP] (supplemental material and Fig. S1; available at http://www.jcb.org/cgi/content/full/jcb.200605050/DC1). The application of 1 μM PGE1 also generated different cAMP levels in the two compartments, with a response in the subplasma membrane compartment of 8.2 ± 1.5% (n = 34) and of 3.3 ± 0.6% (n = 33) in the bulk cytosol (P = 9.5 × 10−4; Fig. 2 E).
Figure 2.

The cAMP response to PGE1 is higher under the plasma membrane than in the bulk cytosol. (A) Wide-field images of a representative HEK293 cell cotransfected with R-CFP (top left) and C-YFP (not depicted) and with mpR-CFP (bottom left) and C-YFP (not depicted). For the same cell, the pseudocolor images of the 480/545-nm emission ratio before (time = 0 s) and after the addition of 10 μM PGE1 (time = 200 s) and 100 μM IBMX (time = 800 s) are shown on the right. Bars, 10 μm. (B) Kinetics of cAMP changes recorded in the cells shown in A. Open circles represent kinetics recorded in the bulk cytosol with PKA-GFP, and closed circles represent kinetics recorded at the plasma membrane with mpPKA-GFP. (C) Summary of all the experiments performed in the same conditions as in A and B. (D) Time to reach half-maximal response (t/2) to 10 μM PGE1. (E) The summary of experiments performed by applying 1 μM PGE1. Error bars indicate SEM. *, 0.01 < P < 0.05; **, 0.005 < P < 0.01; ***, P < 0.005.
A unimolecular sensor for cAMP confirms the generation of discrete cAMP domains upon PGE1 stimulation
When using the mpPKA-GFP sensor, the binding of cAMP to mpR-CFP causes subunit dissociation, releasing the C-YFP subunits to diffuse away from the mpRII-CFP FRET partners anchored at the plasma membrane. Conversely, when cAMP binds to the R-CFP subunits of PKA-GFP in the bulk cytosol, the dissociating C-YFP subunits remain in close proximity to their cytosolic R-CFP FRET partners. In our experiments, FRET changes are measured as CFP intensity/YFP intensity (480/545-nm fluorescence emission; see FRET imaging section). Therefore, as a result of the diffusion of C-YFP away from the plasma membrane, the FRET changes measured upon probe dissociation in the subplasma membrane compartment may result in artifactually larger values being observed than the FRET changes measured in the cytosol. To exclude this possibility and confirm that the subplasma membrane compartment and the cytosolic compartments show a distinct cAMP response to PGE1, we used a unimolecular FRET sensor for cAMP based on Epac1 (H30; Fig. 3 A). As H30 is a single polypeptide chain, CFP and YFP do not diffuse apart upon cAMP binding. We modified H30 by fusing the plasma membrane–targeting mp sequence (mpH30; Fig. 3 B) to its N terminus, which, as with RII, allowed for the effective targeting of this unimolecular sensor to the plasma membrane (Fig. 3 B). Membrane targeting of H30 did not substantially modify its sensitivity to cAMP, as shown by the apparent dissociation constants measured for H30 and mpH30 (EC50 = 12.5 and 20 μM, respectively; Fig. 3 C). Using these unimolecular probes, we show in Fig. 3 (D and E) that the stimulation of HEK293 cells expressing H30 or mpH30 with 1 μM PGE1 generated a mean ΔR/R0 of 23.5 ± 1.4 (n = 40) and 31.4 ± 1.2% (n = 36), respectively (P = 10−4). These results confirm that PGE1 stimulation generates a compartmentalized cAMP response, with a higher level of cAMP being generated in the subplasma membrane compartment as compared with the bulk cytosol. Interestingly, the steepness of the cAMP gradient between the subplasma membrane and bulk cytosol compartments is smaller (35% higher cAMP response at the plasma membrane vs. the cytosol) as compared with the steepness of the gradient recorded with the PKA-based sensor (148% higher cAMP response at the plasma membrane vs. the cytosol; compare Fig. 2 E with Fig. 3 E).
Figure 3.

A unimolecular Epac-based sensor detects different [cAMP] at the plasma membrane and in the bulk cytosol. (A) Schematic representation of the fusion protein constituting the Epac-based cAMP sensors H30 and confocal micrographs showing its distribution in HEK293 cells. (B) Structure and localization of the membrane-targeted version of mpH30. Bars, 10 μm. (C) cAMP dose-response curves measured as the percent FRET changes of H30 (white circles), mpH30 (black circles), and nlsH30 (gray circles). EC50 are 12.5, 20, and 17.5 μM, respectively. (D) Representative kinetics of cAMP changes generated in the cytosol (white circles) and at the plasma membrane (black circles) upon stimulation with 1 μM PGE1 followed by 100 μM IBMX. (E) Summary of the experiments performed as in D. Error bars represent SEM. **, P = 0.002; ***, P = 10−4.
PKA enhances the cAMP gradient between the plasma membrane and the cytosol via PDE activation
The PKA-based and Epac1-based sensors both clearly reveal a gradient of cAMP between the plasma membrane and the cytosol upon PGE1 stimulation. However, such a gradient appears steeper when detected by the PKA-GFP probe as compared with H30. One possible explanation for this is that overexpression of the PKA-based sensor, the catalytic subunit of which is enzymatically active, may affect the steepness of the cAMP gradient. To test this hypothesis, we measured cAMP levels in HEK293 cells transfected with either H30 or mpH30 alone or in combination with untagged PKA. As shown in Fig. 4 (A and B), in the presence of overexpressed PKA, the FRET change recorded at the plasma membrane was ΔR/R0 = 9.5 ± 1% (n = 48), whereas the FRET change recorded in the bulk cytosol was ΔR/R0 = 5.8 ± 0.5% (n = 60; P = 0.0009), indicating that the level of cAMP is ∼62% higher at the plasma membrane as compared with the cytosol. Thus, PKA overexpression increases the steepness of the cAMP gradient between the two compartments around twofold. In further support of this, we found that the higher the level of PKA overexpression, the larger the effect is on the steepness of the cAMP gradient (supplemental material and Fig. S2; available at http://www.jcb.org/cgi/content/full/jcb.200605050/DC1).
Figure 4.

Role of PKA in shaping the cAMP gradient between the plasma membrane and the cytosol. (A and B) Representative cAMP kinetic (Romoser et al., 1996) and summary of experiments (B) performed in HEK293 cells cotransfected with PKA and either H30 or mpH30 and challenged with 1 μM PGE1 followed by either total PDE inhibition with 100 μM IBMX or PKA inhibition with 10 μM H89 as indicated. (C and D) Representative kinetics (C) and summary of experiments (D) showing the effect of endogenous PKA inhibition on the cAMP response induced by 10 nM PGE1 at the plasma membrane and bulk cytosol in the absence and presence of the PKA inhibitor H89 (10 μM). In all of the experiments, when the PKA inhibitor was used, cells were preincubated for 10 min with H89, and the inhibitor was present throughout the experiment. Error bars represent SEM. *, P = 0.02; **, P = 0.009; ***, P = 0.0009.
PKA is known to activate PDE3 and PDE4 families of PDEs, thereby stimulating the degradation of cAMP (Smith et al., 1996; MacKenzie et al., 2002). Interestingly, PDE4 and PDE3 are the major cAMP PDE activities represented in HEK293 cells, with PDE4 accounting for ∼68% of the total PDE activity and PDE3 accounting for a remaining 30% (Lynch et al., 2005). Therefore, we asked whether PDEs may be the effectors of PKA in modulating the steepness of the cAMP gradient. We found that the inhibition of PDEs with 100 μM of the nonselective PDE inhibitor IBMX completely abolished the effect of PKA overexpression on the cAMP gradient (cytosol ΔR/R0 = 26.03 ± 1.73% [n = 64] in the cytosol and 32.46 ± 1.72% [n = 56] at the plasma membrane; P = 0.009; Fig. 4 B), reestablishing a difference in the cAMP level present in the two compartments of ∼25% (compare Fig. 4 B with Fig. 3 E). These results confirm that PDEs mediate the effect of PKA on the cAMP gradient. In agreement with this finding and in support of the key role played by PKA in shaping the cAMP gradient, the inhibition of overexpressed PKA activity with 10 μM H89 completely abolished the cAMP gradient (ΔR/R0 = 12.7 ± 2.05% [n =35] in the cytosol and 16.18 + 2.7% [n =22] at the plasma membrane; P = NS; Fig. 4 B). Similarly, the inhibition of endogenous PKA with H89 was sufficient to completely dissipate the cAMP gradient generated upon PGE1 stimulation (Fig. 4 C). Interestingly, the cAMP response to PGE1 in the presence of H89 was invariably more sustained in time, with the level of cAMP being reduced by only ∼25% at 10 min after application of the stimulus.
To determine the contribution of different PDE families in shaping the cAMP gradient, we selectively inhibited either PDE4 with 10 μM rolipram (MacKenzie and Houslay, 2000) or PDE3 with 10 μM cilostamide (Manganiello and Degerman, 1999). We found that the sole inhibition of PDE4 reproduced the effect of total PDE inhibition with IBMX (ΔR/R0 = 30.88 ± 3.9% [n = 14] in the cytosol and 41.8 ± 2% [n = 16] at the plasma membrane; P = 0.01; compare Fig. 5 with Fig. 4 B). Rolipram showed a similar effect in cells transfected with H30 or mpH30 in the absence of overexpressed PKA (unpublished data). Conversely, selective PDE3 inhibition with 10 μM cilostamide did not substantially raise the cAMP level generated by PGE1 stimulation in either compartments both in the presence (ΔR/R0 = 5.05 ± 0.6% [n = 16] in the cytosol and 10.8 ± 1.7% [n = 12] at the plasma membrane; P = 0.00095; Fig. 5) and in the absence of overexpressed PKA (not depicted). These results strongly indicate that PDE4 is the key regulator of the intracellular cAMP gradient in HEK293 cells.
Figure 5.

Identification of the prevalent PDE affecting the cAMP response to PGE1. Summary of the effect of selective PDE3 (10 μM cilostamide) or PDE4 (10 μM rolipram) inhibition on the cAMP response in the cytosolic (H30) and subplasma membrane (mpH30) compartments upon 1 μM PGE1 stimulation of HEK cells overexpressing untagged PKA. Error bars represent SEM. *, P = 0.01; ***, P < 0.00095.
Tethered PDEs determine the direction of the cAMP gradient in HEK293 cells upon PGE1 stimulation
Our results are in agreement with studies indicating that PDEs may be responsible for the generation of intracellular cAMP gradients by acting as an enzymatic barrier that by rapidly degrading cAMP limits its diffusion from the site of synthesis (the plasma membrane) to the deep cytosol (Rich et al., 2000; Rochais et al., 2004; Mongillo et al., 2004). According to this view, the inhibition of PDE activity would abolish the barrier to cAMP diffusion and should result in a fast reequilibration of cAMP concentration within the cell. Surprisingly, however, our results show that PDE inhibition with either IBMX or rolipram does not dissipate the cAMP gradient between the plasma membrane and the bulk cytosol elicited by PGE1 stimulation (Figs. 2, B–E; 3 E, 4, and 5). We reasoned that as both IBMX and rolipram are competitive PDE inhibitors, in the high [cAMP] subplasma membrane compartment, they may be less efficient in inhibiting PDEs than in the bulk cytosol (lower [cAMP]), thus sustaining the cAMP gradient between the two compartments.
To test the effect of noncompetitive inhibition of PDE4 on the intracellular cAMP gradient, we performed genetic ablation of PDEs by an RNA silencing approach to reduce enzyme concentration. The most represented PDE4 subfamilies in HEK293 cells are PDE4B and PDE4D, with PDE4B representing ∼30% of the total PDE4 activity and PDE4D representing ∼65% in these cells (Lynch et al., 2005). Therefore, we focused on these two subfamilies. The design of siRNA oligonucleotides as well as the time and means of transfection had been previously optimized by us to achieve ∼95% selective knockdown of each PDE subfamily in HEK293 cells (Lynch et al., 2005). We found that the genetic knockdown of PDE4B did not affect the cAMP gradient (ΔR/R0 = 13.9 ± 2.2 [n = 27] and 8.5 ± 1.2% [n = 23] in the subplasma membrane and bulk cytosol, respectively; P = 0.04; Fig. 6 B). Surprisingly, when we cotransfected the cAMP sensor with siRNA sequences that selectively knock down PDE4D, we found that the cAMP response was higher in the cytosol as compared with the subplasma membrane compartment (ΔR/R0 = 22.8 ± 2.4 [n = 25] and 14.6 ± 2.5% [n = 23], respectively; P = 0.04; Fig. 6 C). The finding that the ablation of PDE4D results in the inversion of the cAMP gradient suggests that a PDE different from PDE4D is active in the subplasma membrane compartment and maintains the cAMP concentration low in that domain. In fact, when we ablated both PDE4B and PDE4D, we found that the cAMP gradient was completely abolished (ΔR/R0 = 22.8 ± 2.2% [n = 20] in the cytosol and 20.3 ± 2.4% [n = 22] in the subplasma membrane compartment; Fig. 6 D). Overall, our results suggest that PDE4B mainly regulates the subplasma membrane compartment, whereas PDE4D mainly regulates the cytosolic pool.
Figure 6.

Effect of genetic ablation of selected PDEs on the cAMP gradient between the plasma membrane and bulk cytosol. (A–D) Summary of the effect on the cAMP response generated by 1 μM PGE1 either in the bulk cytosol (open bars) or at the plasma membrane (closed bars) in HEK293 cells expressing either H30 or mpH30 in control cells (A; Romoser et al., 1996) or cells transfected with siRNA oligonucleotides for the selective genetic knockdown of PDE4B (B), PDE4D (C), or both (D). For each experimental group, n ≥ 25. In each panel, the schematics on the right show the observed distribution of the cAMP gradient (in red) and the predicted distribution of endogenous PDEs upon the selective knockdown of specific PDE4 subfamilies. As shown, a lower concentration of cAMP corresponds to a region with higher PDE4 activity. Error bars represent SEM. *, P = 0.04; ***, P = 10−4.
The ability of individual PDE4 subfamilies to control cAMP concentration independently in defined compartments implies that they are selectively tethered within such compartments. To test this, we overexpressed catalytically inactive point mutants of PDE4B and PDE4D in HEK293 cells. Such mutants have been shown to exert a dominant-negative (dn) effect by displacing the cognate endogenous active PDE4 isoforms from their functionally relevant anchor sites (Baillie et al., 2003). As shown in Fig. 7 A, upon the overexpression of either dnPDE4B1 or dnPDE4B2, the cAMP response to PGE1 was higher in the subplasma membrane compartment than in the cytosol. This finding indicates that the displacement of PDE4B from its anchor sites and its consequent distribution inside the cell do not affect the gradient dictated by the prevalent PDE4D activity localized in the cytoplasm. On the contrary, the overexpression of dnPDE4D3 or dnPDE4D5 completely abolished the cAMP gradient (Fig. 7 B), as expected upon displacement of the prevalent PDE4D activity from its anchor sites within the cytoplasm. Overall, these results confirm that the different cAMP concentrations in these two compartments are dependent on compartmentalized PDE4 isoforms.
Figure 7.

Effect of the selective displacement of PDE4 subfamilies on the cAMP gradient. (A and B) Response to 1 μM PGE1 in HEK293 cells expressing the dn variants of PDE4B1 or PDE4B2 (A; Romoser et al., 1996) and expressing the dn variants of PDE4D3 or PDE4D5 (B). For each experimental group, n ≥ 17. Schematics on the right of each panel show the observed cAMP gradient (in red) and the predicted redistribution of endogenous, active PDE4 subfamilies upon overexpression of the cognate, inactive dn proteins (not depicted). Error bars represent SEM. *, P = 0.01; **, P = 0.004.
PDE4B and PDE4D localize in distinct compartments in HEK293 cells
The aforementioned effect of the genetic manipulation of PDE4B and PDE4D implies a distinct localization of the two enzyme subfamilies. To confirm this, we performed immunocytochemistry by using monoclonal antibodies specific for either subfamilies and found that, as expected, PDE4B shows a prevalent localization at the plasma membrane, whereas PDE4D localizes mainly in the bulk cytosol (Fig. 8). Such membrane sequestration of PDE4B is consistent with a previous study showing that in HEK293 cells, PDE4B2 is unavailable to interact with cytosolic β-arrestin (Lynch et al., 2005).
Figure 8.
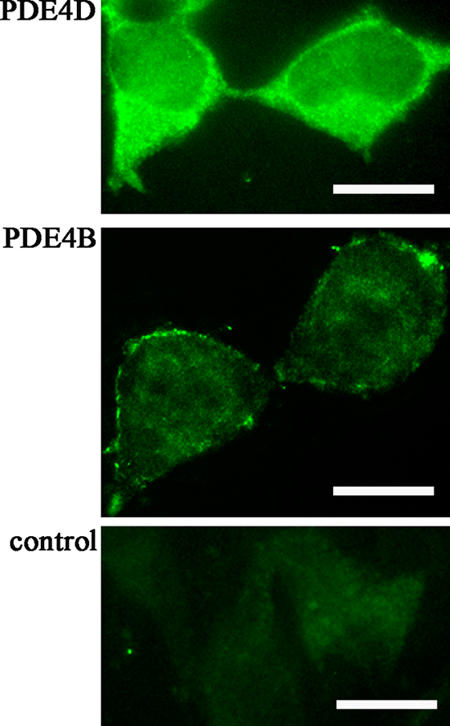
Subcellular localization of PDE4B and PDE4D. Confocal images of HEK293 cells stained with monoclonal antibodies specific for either PDE4B or PDE4D. The control is the secondary antibody alone. Bars, 10 μm.
PDE4s define local drains for cAMP and generate multiple contiguous, intracellular compartments with different concentrations of the second messenger
The results presented in Figs. 6 and 7 indicate that PDE4 enzymes, rather than acting as a barrier to cAMP diffusion from the site of synthesis to the bulk cytosol, function as a local drain to degrade freely diffusible cAMP and maintain the low concentration of the second messenger in discrete compartments. Thus, rather than a continuous gradient from the plasma membrane (higher [cAMP]) to the deep cytosol (lower [cAMP]), we may expect to find contiguous compartments with higher or lower [cAMP] independent of their distance from the site of synthesis but depending on the localization and activity of PDEs. According to this hypothesis, a subcellular compartment deeper inside the cell as compared with the bulk cytosol and in which PDE activity is low should show a higher [cAMP] as compared with the bulk cytosol. The nucleus is such a compartment.
To monitor [cAMP] inside the nucleus, we fused a nuclear localization sequence (nls) to the C terminus of the H30 sensor (Fig. 9 A) that selectively targets the probe to the nucleus (Fig. 9 A). When we challenged HEK293 cells transfected with nlsH30 with 1 μM PGE1, we found that the FRET change was higher in the nucleus as compared with the bulk cytosol (ΔR/R0 = 28.5 ± 1.7 [n = 25] and 23.5 ± 1.4% [n = 28], respectively; P = 0.03) and was similar to the FRET change found in the subplasma membrane compartment (Fig. 9, B and C). The different response recorded in the nucleus was not caused by a higher sensitivity of the nlsH30 sensor to cAMP changes, as indicated by similar EC50 (17.5 μM for nlsH30 vs. 12.5 μM for H30; Fig. 3 C). In apparent contrast with our data showing that there is a delay in the cAMP changes in the bulk cytosol as compared with the subplasma membrane compartment (Fig. 2 D), the kinetics of the nuclear cAMP changes appear to be as fast as those occurring at the plasma membrane (Fig. 9 B). This is caused by differences in probe kinetics in that the velocity of nlsH30 FRET change upon [cAMP] rise is significantly faster than the velocity of H30 (P = 0.004) and mpH30 (P = 0.0001) FRET changes at the same [cAMP] (supplemental material). The difference in the FRET change between the nucleus and the bulk cytosol was completely abolished when PDE4D was ablated by siRNA duplex cotransfection (ΔR/R0 = 17.7 ± 1.5% [n = 26] in the nucleus and 22.8 ± 2.5% [n = 28] in the cytosol; P = 0.12; Fig. 6 D). These results confirm that multiple contiguous, subcellular domains with diverse [cAMP] may coexist within HEK293 cells irrespective of their distance from the site of cAMP synthesis and depending on PDE4B and PDE4D activity and localization.
Figure 9.

PGE1 stimulation generates a larger cAMP response in the nucleus as compared with the bulk cytosol. (A) Schematic representation of the sensor H30 targeted to the nucleus (nlsH30) and its distribution in HEK293 cells. Bars, 10 μm. (B) Representative kinetics of cAMP changes in response to 1 μM PGE1 as detected in the bulk cytosol in cells transfected with H30 (white circles), at the plasma membrane in cells transfected with mpH30 (black circles), and in the nucleus in cells transfected with nlsH30 (gray circles). (C) Summary of the experiments performed in the aforementioned conditions. (D) Summary of the FRET changes recorded in the bulk cytosol (white bar) and in the nucleus (gray bar) of HEK293 cells cotransfected with either H30 or nlsH30 and siRNA oligonucleotides for PDE4D and stimulated with 1 μM PGE1. Error bars represent SEM. **, P = 0.03; ***, P = 10−4.
Discussion
The pleiotropic effects of cAMP pose the pressing question of how signaling specificity is achieved. In the past few years, compartmentalization of the cAMP signal transduction pathway has emerged as an important mechanism to ensure the necessary specificity of response (Tasken and Aandahl, 2004; Wong and Scott, 2004). A particular focus has been placed on the organization of macromolecular complexes, including receptors, effectors, modulators, and targets effectively organized in restricted domains, within which the molecular components of the complex only affect each other appropriately. Some of these domains are localized at the plasma membrane, one example of which is the assembly of the β2 adrenergic receptor, heterotrimeric G proteins, AC, and PKA and its target, the L-type Ca2+ channel Cav1.2 (Davare et al., 2001). Within the complex, activation of the receptor leads to the synthesis of cAMP and activation of PKA, which, in turn, can regulate the activity of the channel in a highly localized manner. AKAP/PKA signaling domains have also been found to be located deep in the cytosol and away from the site of cAMP synthesis, as is the case, for example, for the centrosome-associated AKAPs (Witczak et al., 1999) or the nuclear membrane-associated muscle AKAP (Dodge et al., 2001). Specific activation of the PKA pools, which are spatially segregated deep inside the cell, requires that cAMP is made available selectively in such compartments. How the highly hydrophilic, freely diffusible cAMP molecule can selectively activate deep intracellular targets without affecting PKA enzymes located closer to the site of cAMP synthesis remains to be defined.
One example of the limited diffusion of cAMP is the generation of a subplasma membrane pool of the second messenger in response to PGE1 stimulation of HEK293 cells. To explain such compartmentalization, the hypothesis was formulated of a physical barrier that is possibly formed by elements of the endoplasmic reticulum and localizes underneath the plasma membrane, thereby limiting cAMP diffusion from the site of synthesis to the deep cytosol (Rich et al., 2000). However, an important contribution to cAMP compartmentalization appears to derive from the activity of PDEs, as pointed out by a large body of indirect evidence based on altered cell functioning after selective PDE inhibition (Rich et al., 2001; Zaccolo and Pozzan, 2002; Jurevicius et al., 2003; Abrahamsen et al., 2004; Mongillo et al., 2004, 2006). PDEs can be targeted to subcellular compartments, including the plasma membrane, and can be recruited into multiprotein signaling complexes (Perry et al., 2002), thereby providing a means to terminate the cAMP signal in a spatially restricted manner (Baillie et al., 2005). Such features suggested a role for PDEs as an enzymatic barrier to cAMP diffusion (for review see Brunton, 2003; Barnes et al., 2005; Willoughby et al., 2006).
The model of a barrier to cAMP diffusion implies the generation of a gradient of cAMP with a higher concentration of the second messenger at the plasma membrane and a progressively lower concentration toward the deep cytosol. However, the direction of such a gradient is incompatible with the selective activation of a subset of PKA anchored deep inside the cytosol without concomitant activation of those PKA enzymes that are localized at the plasma membrane and closer to the site of cAMP synthesis.
In this study, we set out to study the cAMP response to GPCR stimulation of the transmembrane AC with PGE1 in HEK293 cells with the aim of defining the role of PDEs in the generation of a subplasma membrane pool of high cAMP. We applied an imaging approach for real-time monitoring of cAMP dynamics in single living cells and used FRET-based biosensors selectively targeted to different subcellular domains. We found that the cAMP response in the subplasma membrane compartment is higher as compared with the bulk cytosol because of a compartmentalized PDE4D located in the cytosol that keeps the level of the second messenger low in this compartment. Indeed, genetic ablation of PDE4D inverted the direction of the cAMP gradient, resulting in a higher concentration of the second messenger in the cytosol as compared with the subplasma membrane compartment. On the contrary, genetic ablation of PDE4B did not affect the direction of the gradient. The cAMP gradient was completely abolished only when the expression of both PDE4B and PDE4D was blocked. These results demonstrate that the concerted activity of PDE4B and PDE4D is sufficient to generate an intracellular cAMP gradient in response to PGE1 in HEK293 cells. We also found that dislocation of PDE4D from its anchoring sites by means of the overexpression of catalytically inactive enzymes dissipated the cAMP gradient, indicating that the compartmentalization of PDE4D is responsible for shaping such a gradient.
Intriguingly, we found that the inhibition of endogenous PKA with H89 completely abolished the cAMP gradient between the plasma membrane and bulk cytosol. On the contrary, the overexpression of PKA via PDE activation increased the steepness of the subplasma membrane cAMP gradient generated by PGE1. These results indicate that PKA not only has a role in shaping the intracellular cAMP gradient upon receptor stimulation but also that PKA itself can reinforce the boundaries between the intracellular cAMP compartments and can self-regulate the specificity of its own activity.
In this study, we show for the first time that a compartment deep inside the cell may accumulate higher levels of cAMP as compared with the bulk cytosol. These findings demonstrate that multiple and contiguous domains with different concentrations of cAMP can be generated simultaneously inside the cell irrespective of their distance from the site of synthesis of the second messenger. To explain such findings, we must assume that PDEs, rather than acting as enzymatic barriers to cAMP diffusion from the plasma membrane to the bulk cytosol, are organized to generate local drains that dump cAMP in defined compartments. Such a mechanism of control of cAMP diffusion may be more general and may also apply to the production of nonuniform intracellular cAMP domains in response to soluble AC activation. Therefore, we propose a new model whereby cAMP is free to diffuse from the site of synthesis and to accumulate in the cell to levels effective for PKA activation except in those domains in which localized PDEs degrade it to protect sensitive targets from inappropriate activation.
Materials and methods
Reagents
DME, Opti-MEM, FBS, l-glutamine, penicillin, trypsin/EDTA, PBS, and LipofectAMINE 2000 were purchased from Invitrogen. PGE1 and IBMX were obtained from Sigma-Aldrich. Restriction enzymes, T4 ligase, and shrimp AP were purchased from New England Biolabs, Inc. FuGENE-6 transfection reagent was obtained from Roche.
Construct generation
The membrane-targeted version of the R-CFP subunit of the PKA-based cAMP sensor (Lissandron et al., 2005) was generated by inserting the N-terminal targeting signal MGCIKSKRKDNLNDD (mp) from Lyn kinase at the N terminus of R-CFP. This becomes posttranslationally myristoylated and palmitoylated, resulting in its targeting to membrane rafts. The cAMP Epac1-based sensor, called H30, was provided by K. Jalink (The Netherlands Cancer Institute, Amsterdam, Netherlands) and corresponds to the CFP-Epac(δDEP-CD)-YFP sensor (Ponsioen et al., 2004). The membrane-targeted version of H30 was generated by fusion at the N terminus of the MGCIKSKRKDNLNDD plasma membrane–targeting sequence. The nuclear-targeted version of H30 was generated by fusion of the nuclear localization signal PKKKRKVEDA (nls) at the C terminus (DiPilato et al., 2004).
Untagged PKA was generated by cloning both the RIIβ subunit of PKA and the Cα subunit of PKA into pCDNA3.1 (Invitrogen). Constructs for the expression of PKA subunits in Escherichia coli were generated by cloning RIIβ, mpRIIβ, and Cα into pRSETB (Invitrogen).
Activation constants and apparent dissociation constants determination
To determine apparent activation constants for PKA-GFP and mpPKA-GFP, the pRSETB vectors carrying the RIIβ, mpRIIβ, or Cα subunits were introduced into BL21(DE3) (Stratagene) for heterologous protein expression in bacteria. Individual subunits were subsequently purified with Ni–nitrilotriacetic acid resin according to the manufacturer's instructions (QIAGEN). Purified proteins were buffer changed by gel filtration to 20 mM MOPS, 150 mM NaCl, 5 mM MgCl2, 100 μM ATP, and 1 mM β-mercaptoethanol, pH 7.0 (dialysis buffer), to remove imidazole. For PKA holoenzyme formation, PKA-R (PKA-GFP and mpPKA-GFP) and PKA Cα subunits (Slice et al., 1989; Herberg et al., 1993) were mixed in a molar ratio of 1.5:1.0, and dialysis was performed overnight with three buffer changes against dialysis buffer (Herberg et al., 1993). For the determination of activation constants, a coupled spectrophotometric assay (Cook et al., 1982) was performed using the peptide kemptide (LRRASLG; Biosynthan) as the substrate. Phosphotransferase activity was measured in an assay mixture consisting of 100 mM MOPS, pH 7.0, 10 mM MgCl2, 1 mM phosphoenol pyruvate, 1 mM ATP, 200 μM NADH, 1 mM DTT, 15 U/ml lactate dehydrogenase (Roche), and 70 U/ml pyruvate kinase (Roche). For the reaction, 100 μl of assay mixture, 20 nM PKA holoenzyme, and 1 μl kemptide peptide (200 μM) were mixed in a quartz cuvette. The enzymatic activity (ΔE340nm × min−1) was monitored at room temperature using a spectrophotometer (Lambda Bio UV/Vis; PerkinElmer). EC50 values for activation were determined by preincubating the PKA holoenzyme with increasing concentrations of cAMP for 1 min in assay mixture before starting the reaction with kemptide peptide. Data points were determined in duplicates, and experiments were repeated at least twice with similar results. Statistical analyses were performed using Prism 4.0 software (GraphPad Software).
To determine in vivo apparent dissociation constants for H30, mpH30, and nlsH30, HeLa cells expressing the cAMP sensor were injected with known concentrations of cAMP via a patch pipette, and FRET changes were recorded as described in the FRET imaging section. Cells were continuously superfused at 2 ml/min in a standard extracellular solution, ECS-1, which contained 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM Hepes, 2 mM CaCl2, 2 mM pyruvate, and 5 mM glucose, pH 7.4. Patch pipettes were filled with an intracellular solution, ICS-1, containing 120 mM K- aspartate, 10 mM TEA-Cl, 1 mM MgCl2, 10 mM Hepes, 10 mM CsCl, 0.3 mM GTP-Na, 3 mM ATP-K (adjusted to pH 7.2 with KOH), 5 mM BAPTA, 1 mM thapsigargin, 0.1 mM IBMX, and 0.007–0.18 mM cAMP as indicated and filtered through 0.22-μm pores (Millipore). Only cells that displayed a seal resistance of >2 GΩ before achieving the whole cell configuration were retained. Cells admitted to the analysis after achieving the whole cell configuration were further characterized by an access resistance of <12 MΩ and an apparent membrane resistance of >300MΩ. At least six cells were analyzed for each concentration of cAMP.
Cell culture and transfection
HEK293 cells were grown in DME containing 10% FBS supplemented with 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere containing 5% CO2. For transient expression, cells were seeded onto 24-mm diameter round glass coverslips, and transfections were performed at 50–70% confluence with FuGENE-6 transfection reagent according to the manufacturer's instructions using 1–2 μg DNA per coverslip. Imaging experiments were performed after 24–48 h.
To achieve the selective knockdown of PDE4B or PDE4D subfamilies, we used double-stranded 21-mer RNA duplexes (Dharmacon) targeted at regions of sequence that are unique to each of these subfamilies as described previously (Lynch et al., 2005). Each siRNA duplex was delivered into target cells via the reagent LipofectAMINE 2000 (Invitrogen). Specifically, 5 μl LipofectAMINE 2000 (1 mg/ml) was diluted in 100 μl Opti-MEM, and, separately, 125 pmol of each siRNA sample and 1 μg cAMP sensor DNA were diluted in 100 μl Opti-MEM. 200 μl siRNA–DNA transfection complexes were added to each well, and the plates were incubated for 3–4 h at 37°C (5% CO2). These complexes were then removed and replaced with DME. Imaging experiments were performed after 48 h.
FRET imaging
FRET imaging experiments were performed 24–48 h after transfection. Cells were maintained in Hepes-buffered Ringer-modified saline containing 125 mM NaCl, 5 mM KCl, 1 mM Na3PO4, 1 mM MgS04, 5.5 mM glucose, 1 mM CaCl2, and 20 mM Hepes, pH 7.5, at room temperature (20–22°C) and imaged on an inverted microscope (IX50; Olympus) with a 60× NA 1.4 oil immersion objective (Olympus). The microscope was equipped with a CCD camera (Sensicam QE; PCO), a software-controlled monochromator (Polychrome IV; TILL Photonics), and a beam-splitter optical device (Multispec Microimager; Optical Insights). Images were acquired using custom-made software and processed using ImageJ (National Institutes of Health). FRET changes were measured as changes in the background-subtracted 480/545-nm fluorescence emission intensities on excitation at 430 nm and expressed as either R/R0, where R is the ratio at time t and R0 is the ratio at time = 0 s, or ΔR/R0, where ΔR = R – R0.
Confocal imaging
Cells were stained with anti-PDE4B and -PDE4D monoclonal antibodies (FabGennix). AlexaFluor488-conjugated anti–mouse antibody was used as the secondary antibody. Confocal images were acquired with an inverted microscope (Eclipse TE300; Nikon) equipped with a spinning disk confocal system (Ultraview LCI; PerkinElmer) and a 12-bit CCD camera (Hamamatsu; Orca ER). Cells were excited using the 488-nm line of a krypton-argon laser (643-Ryb-A02; Melles Griot) for imaging YFP and the AlexaFluor488 fluorophore and using the 405-nm diode laser (iFlex2000; Point Source) for imaging CFP.
Online supplemental material
Fig. S1 shows an evaluation of PGE1 receptor desensitization. Fig. S2 shows an evaluation of the dose-response relationship between PKA overexpression and the steepness of the cAMP gradient. Video 1 shows the dynamics interactions between mpRII-CFP and C-YFP upon cAMP binding and release. Supplemental material also provides information about determination of the velocity of FRET change for the cAMP sensors. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200605050/DC1.
Supplementary Material
Acknowledgments
We thank Prof. F. Mammano for technical advice.
This work was supported by Telethon Italy (grants TCP00089 and GGP05113), the Italian Cystic Fibrosis Research Foundation, the Fondazione Compagnia di San Paolo, the Human Frontier Science Program Organization (grant RGP0001/2005-C to M. Zaccolo), the Medical Research Council of the United Kingdom (grant G8604010 to M.D. Houslay), the European Union (grant QLK3-CT-2002-02149 to M. Zaccolo, F.W. Herberg, and M.D. Houslay), and the BMBF (grant 01 GR0441 [NGFN] to F.W. Herberg).
A. Terrin and G. Di Benedetto contributed equally to this paper.
Abbreviations used in this paper: AC, adenylyl cyclase; AKAP, A kinase–anchoring protein; dn, dominant negative; FRET, fluorescence resonance energy transfer; GPCR, Gs protein–coupled receptor; HEK, human embryonic kidney; IBMX, isobutyl-methyl-xanthine; PDE, phosphodiesterase; PGE1, prostaglandin 1.
References
- Abrahamsen, H., G. Baillie, J. Ngai, T. Vang, K. Nika, A. Ruppelt, T. Mustelin, M. Zaccolo, M. Houslay, and K. Tasken. 2004. TCR- and CD28-mediated recruitment of phosphodiesterase 4 to lipid rafts potentiates TCR signaling. J. Immunol. 173:4847–4858. [DOI] [PubMed] [Google Scholar]
- Bacskai, B.J., B. Hochner, M. Mahaut-Smith, S.R. Adams, B.K. Kaang, E.R. Kandel, and R.Y. Tsien. 1993. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science. 260:222–226. [DOI] [PubMed] [Google Scholar]
- Baillie, G.S., A. Sood, I. McPhee, I. Gall, S.J. Perry, R.J. Lefkowitz, and M.D. Houslay. 2003. beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc. Natl. Acad. Sci. USA. 100:940–945. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Baillie, G.S., J.D. Scott, and M.D. Houslay. 2005. Compartmentalisation of phosphodiesterases and protein kinase A: opposites attract. FEBS Lett. 579:3264–3270. [DOI] [PubMed] [Google Scholar]
- Barnes, A.P., G. Livera, P. Huang, C. Sun, W.K. O'Neal, M. Conti, M.J. Stutts, and S.L. Milgram. 2005. Phosphodiesterase 4D forms a cAMP diffusion barrier at the apical membrane of the airway epithelium. J. Biol. Chem. 280:7997–8003. [DOI] [PubMed] [Google Scholar]
- Beavo, J.A., and L.L. Brunton. 2002. Cyclic nucleotide research–still expanding after half a century. Nat. Rev. Mol. Cell Biol. 3:710–718. [DOI] [PubMed] [Google Scholar]
- Biel, M., X. Zong, A. Ludwig, A. Sautter, and F. Hofmann. 1999. Structure and function of cyclic nucleotide-gated channels. Rev. Physiol. Biochem. Pharmacol. 135:151–171. [DOI] [PubMed] [Google Scholar]
- Bos, J.L. 2003. Epac: a new cAMP target and new avenues in cAMP research. Nat. Rev. Mol. Cell Biol. 4:733–738. [DOI] [PubMed] [Google Scholar]
- Brunton, L.L. 2003. PDE4: arrested at the border. Sci. STKE. 10.1126/stke.2003.204.pe44. [DOI] [PubMed]
- Buxton, I.L., and L.L. Brunton. 1983. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem. 258:10233–10239. [PubMed] [Google Scholar]
- Cook, P.F., M.E. Neville Jr., K.E. Vrana, F.T. Hartl, and R. Roskoski Jr. 1982. Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry. 21:5794–5799. [DOI] [PubMed] [Google Scholar]
- Cooper, D.M. 2003. Regulation and organization of adenylyl cyclases and cAMP. Biochem. J. 375:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, D.M. 2005. Compartmentalization of adenylate cyclase and cAMP signalling. Biochem. Soc. Trans. 33:1319–1322. [DOI] [PubMed] [Google Scholar]
- Davare, M.A., V. Avdonin, D.D. Hall, E.M. Peden, A. Burette, R.J. Weinberg, M.C. Horne, T. Hoshi, and J.W. Hell. 2001. A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science. 293:98–101. [DOI] [PubMed] [Google Scholar]
- DiPilato, L.M., X. Cheng, and J. Zhang. 2004. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signalling within discrete subcellular compartments. Proc. Natl. Acad. Sci. USA. 101:16513–16518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge, K.L., S. Khouangsathiene, M.S. Kapiloff, R. Mouton, E.V. Hill, M.D. Houslay, L.K. Langeberg, and J.D. Scott. 2001. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 20:1921–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge-Kafka, K.L., J. Soughayer, G.C. Pare, J.J. Carlisle Michel, L.K. Langeberg, M.S. Kapiloff, and J.D. Scott. 2005. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature. 437:574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, S.H., I.V. Turko, and J.D. Corbin. 2001. Cyclic nucleotide phosphodiesterases: relating structure and function. Prog. Nucleic Acid Res. Mol. Biol. 65:1–52. [DOI] [PubMed] [Google Scholar]
- Herberg, F.W., S.M. Bell, and S.S. Taylor. 1993. Expression of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli: multiple isozymes reflect different phosphorylation states. Protein Eng. 6:771–777. [DOI] [PubMed] [Google Scholar]
- Houslay, M.D., and G. Milligan. 1997. Tailoring cAMP-signalling responses through isoform multiplicity. Trends Biochem. Sci. 22:217–224. [DOI] [PubMed] [Google Scholar]
- Houslay, M.D., and D.R. Adams. 2003. PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem. J. 370:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurevicius, J., and R. Fischmeister. 1996. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc. Natl. Acad. Sci. USA. 93:295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurevicius, J., V.A. Skeberdis, and R. Fischmeister. 2003. Role of cyclic nucleotide phosphodiesterase isoforms in cAMP compartmentation following beta2-adrenergic stimulation of ICa,L in frog ventricular myocytes. J. Physiol. 551:239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissandron, V., A. Terrin, M. Collini, L. D'Alfonso, G. Chirico, S. Pantano, and M. Zaccolo. 2005. Improvement of a FRET-based indicator for cAMP by linker design and stabilization of donor-acceptor interaction. J. Mol. Biol. 354:546–555. [DOI] [PubMed] [Google Scholar]
- Lugnier, C. 2006. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol. Ther. 109:366–398. [DOI] [PubMed] [Google Scholar]
- Lynch, M.J., G.S. Baillie, A. Mohamed, X. Li, C. Maisonneuve, E. Klussmann, G. van Heeke, and M.D. Houslay. 2005. RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with beta arrestin to control the protein kinase A/AKAP79-mediated switching of the beta2-adrenergic receptor to activation of ERK in HEK293B2 cells. J. Biol. Chem. 280:33178–33189. [DOI] [PubMed] [Google Scholar]
- MacKenzie, S.J., and M.D. Houslay. 2000. Action of rolipram on specific PDE4 cAMP phosphodiesterase isoforms and on the phosphorylation of cAMP-response-element-binding protein (CREB) and p38 mitogen-activated protein (MAP) kinase in U937 monocytic cells. Biochem. J. 347:571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie, S.J., G.S. Baillie, I. McPhee, C. MacKenzie, R. Seamons, T. McSorley, J. Millen, M.B. Beard, G. van Heeke, and M.D. Houslay. 2002. Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in Upstream Conserved Region 1 (UCR1). Br. J. Pharmacol. 136:421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganiello, V.C., and E. Degerman. 1999. Cyclic nucleotide phosphodiesterases (PDEs): diverse regulators of cyclic nucleotide signals and inviting molecular targets for novel therapeutic agents. Thromb. Haemost. 82:407–411. [PubMed] [Google Scholar]
- Mongillo, M., T. McSorley, S. Evellin, A. Sood, V. Lissandron, A. Terrin, E. Huston, A. Hannawacker, M.J. Lohse, T. Pozzan, et al. 2004. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ. Res. 95:67–75. [DOI] [PubMed] [Google Scholar]
- Mongillo, M., C.G. Tocchetti, A. Terrin, V. Lissandron, Y.F. Cheung, W.R. Dostmann, T. Pozzan, D.A. Kass, N. Paolocci, M.D. Houslay, and M. Zaccolo. 2006. Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ. Res. 98:226–234. [DOI] [PubMed] [Google Scholar]
- Nikolaev, V.O., M. Bunemann, L. Hein, A. Hannawacker, and M.J. Lohse. 2004. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 279:37215–37218. [DOI] [PubMed] [Google Scholar]
- Perry, S.J., G.S. Baillie, T.A. Kohout, I. McPhee, M.M. Magiera, K.L. Ang, W.E. Miller, A.J. McLean, M. Conti, M.D. Houslay, and R.J. Lefkowitz. 2002. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science. 298:834–836. [DOI] [PubMed] [Google Scholar]
- Ponsioen, B., J. Zhao, J. Riedl, F. Zwartkruis, G. van der Krogt, M. Zaccolo, W.H. Moolenaar, J.L. Bos, and K. Jalink. 2004. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 5:1176–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resh, M.D. 1999. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta. 1451:1–16. [DOI] [PubMed] [Google Scholar]
- Rich, T.C., K.A. Fagan, H. Nakata, J. Schaack, D.M. Cooper, and J.W. Karpen. 2000. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J. Gen. Physiol. 116:147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich, T.C., K.A. Fagan, T.E. Tse, J. Schaack, D.M. Cooper, and J.W. Karpen. 2001. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc. Natl. Acad. Sci. USA. 98:13049–13054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochais, F., G. Vandecasteele, F. Lefebvre, C. Lugnier, H. Lum, J.L. Mazet, D.M. Cooper, and R. Fischmeister. 2004. Negative feedback exerted by cAMP-dependent protein kinase and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes: an in vivo study using adenovirus-mediated expression of CNG channels. J. Biol. Chem. 279:52095–52105. [DOI] [PubMed] [Google Scholar]
- Romoser, V., R. Ball, and A.V. Smrcka. 1996. Phospholipase C beta2 association with phospholipid interfaces assessed by fluorescence resonance energy transfer. G protein betagamma subunit-mediated translocation is not required for enzyme activation. J. Biol. Chem. 271:25071–25078. [DOI] [PubMed] [Google Scholar]
- Slice, L.W., and S.S. Taylor. 1989. Expression of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli. J. Biol. Chem. 264:20940–20946. [PubMed] [Google Scholar]
- Smith, K.J., G. Scotland, J. Beattie, I.P. Trayer, and M.D. Houslay. 1996. Determination of the structure of the N-terminal splice region of the cyclic AMP-specific phosphodiesterase RD1 (RNPDE4A1) by 1H NMR and identification of the membrane association domain using chimeric constructs. J. Biol. Chem. 271:16703–16711. [DOI] [PubMed] [Google Scholar]
- Soderling, S.H., and J.A. Beavo. 2000. Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions. Curr. Opin. Cell Biol. 12:174–179. [DOI] [PubMed] [Google Scholar]
- Tasken, K., and E.M. Aandahl. 2004. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol. Rev. 84:137–167. [DOI] [PubMed] [Google Scholar]
- Willoughby, D., W. Wong, J. Schaack, J.D. Scott, and D.M. Cooper. 2006. An anchored PKA and PDE4 complex regulates subplasmalemmal cAMP dynamics. EMBO J. 25:2051–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witczak, O., B.S. Skalhegg, G. Keryer, M. Bornens, K. Tasken, T. Jahnsen, and S. Orstavik. 1999. Cloning and characterization of a cDNA encoding an A-kinase anchoring protein located in the centrosome, AKAP450. EMBO J. 18:1858–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, W., and J.D. Scott. 2004. AKAP signalling complexes: focal points in space and time. Nat. Rev. Mol. Cell Biol. 5:959–970. [DOI] [PubMed] [Google Scholar]
- Zaccolo, M., and T. Pozzan. 2002. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 295:1711–1715. [DOI] [PubMed] [Google Scholar]
- Zaccolo, M., F. De Giorgi, C.Y. Cho, L. Feng, T. Knapp, P.A. Negulescu, S.S. Taylor, R.Y. Tsien, and T. Pozzan. 2000. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat. Cell Biol. 2:25–29. [DOI] [PubMed] [Google Scholar]
- Zaccolo, M., P. Magalhaes, and T. Pozzan. 2002. Compartmentalisation of cAMP and Ca(2+) signals. Curr. Opin. Cell Biol. 14:160–166. [DOI] [PubMed] [Google Scholar]
- Zippin, J.H., Y. Chen, P. Nahirney, M. Kamenetsky, M.S. Wuttke, D.A. Fischman, L.R. Levin, and J. Buck. 2003. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J. 17:82–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.