

Abstract
Action potential initiation and propagation requires clustered Na+ (voltage-gated Na+ [Nav]) channels at axon initial segments (AIS) and nodes of Ranvier. In addition to ion channels, these domains are characterized by cell adhesion molecules (CAMs; neurofascin-186 [NF-186] and neuron glia–related CAM [NrCAM]), cytoskeletal proteins (ankyrinG and βIV spectrin), and the extracellular chondroitin-sulfate proteoglycan brevican. Schwann cells initiate peripheral nervous system node formation by clustering NF-186, which then recruits ankyrinG and Nav channels. However, AIS assembly of this protein complex does not require glial contact. To determine the AIS assembly mechanism, we silenced expression of AIS proteins by RNA interference. AnkyrinG knockdown prevented AIS localization of all other AIS proteins. Loss of NF-186, NrCAM, Nav channels, or βIV spectrin did not affect other neuronal AIS proteins. However, loss of NF-186 blocked assembly of the brevican-based AIS extracellular matrix, and NF-186 overexpression caused somatodendritic brevican clustering. Thus, NF-186 assembles and links the specialized brevican-containing AIS extracellular matrix to the intracellular cytoskeleton.
Introduction
The nervous system is a highly integrated network of neurons and glia that work together to generate, propagate, and modulate action potentials. In most neurons, high densities of voltage- gated Na+ (Nav) channels initiate action potentials at the axon initial segment (AIS; Khaliq and Raman, 2006; Palmer and Stuart, 2006; Shu et al., 2007), whereas rapid and efficient action potential conduction along axons depends on high densities of Nav channels located at nodes of Ranvier. In cells that fire repetitively, the integration of synaptic inputs into trains of action potentials is determined by the AIS (Naundorf et al., 2006). However, despite its importance, the molecular mechanisms underlying AIS formation and maintenance are poorly understood. In contrast, the cellular and molecular mechanisms of nodal Nav channel clustering are better characterized. In the peripheral nervous system (PNS), the available data point to a model where channel clustering at nodes is initiated by interactions between gliomedin, secreted by Schwann cells, and the axonal cell adhesion molecule (CAM) neurofascin-186 (NF-186; Eshed et al., 2005, 2007; Sherman et al., 2005). Subsequently, the cytoskeletal and scaffolding proteins ankyrinG (ankG) and βIV spectrin are recruited to nodes. Finally, Nav channels bind to ankG and mediate the currents necessary for action potential conduction (Garrido et al., 2003). Thus, the clustering of ion channels at nodes of Ranvier is thought to depend on binding to cytoskeletal and scaffolding proteins that are positioned along axons by extracellular, heterophilic interactions between axonal and glial CAMs (Schafer and Rasband, 2006).
Intriguingly, the molecular compositions of the AIS and nodes are nearly identical, consistent with the fact that these axonal domains perform similar functions. These similarities suggest that the developmental mechanisms regulating their formation may be conserved (Hedstrom and Rasband, 2006). By analogy with nodes, the CAMs NF-186 and neuron glia–related CAM (NrCAM) may initiate ion channel clustering at the AIS through as-yet-unknown extrinsic mechanisms. One possibility is that the ECM contributes to formation of the AIS. Consistent with this idea, John et al. (2006) reported that in vitro a specialized brevican-containing matrix surrounds the AIS. An alternative model places ankG as the central intrinsic scaffold to which all other AIS components become tethered. The latter view has strong experimental support, as AIS localization of Nav and KCNQ2/3 Kv channels, NF-186, and βIV spectrin depend on binding to ankG (Garrido et al., 2003; Lemaillet et al., 2003; Pan et al., 2006; Yang et al., 2007). Further, mice lacking ankG in their Purkinje neurons fail to cluster ion channels or any other AIS proteins (Zhou et al., 1998; Jenkins and Bennett, 2001; Pan et al., 2006). However, loss of βIV spectrin has also been proposed to disrupt the proper assembly of the AIS (Komada and Soriano, 2002). Finally, ablation of Nav channels by RNAi blocked the accumulation of ankG, NF-186, and NrCAM in cultured motorneurons, indicating that ion channels themselves may play previously unappreciated roles in domain formation or stability (Xu and Shrager, 2005).
What are the molecular requirements for AIS formation? Is NF-186 necessary for Na+ channel clustering, as at nodes of Ranvier, or does it play some other unknown function at the AIS? To answer these questions, we eliminated each AIS protein and determined whether its loss affected the localization of other AIS proteins. Our results show that although nodes and the AIS share a common molecular organization, their mechanisms of assembly are unique. In particular, AIS NF-186 is dispensible for ion channel clustering but is required for the assembly of the specialized brevican-based ECM.
Results
Nav channels, NF-186, NrCAM, βIV spectrin, and ankG are all highly enriched at the AIS in vivo (Fig. 1 A; NrCAM, NF-186, and ankG not depicted). We used cultured hippocampal neurons to determine the role these proteins play in AIS assembly. This well-characterized model (Banker and Goslin, 1998) has been used to determine the mechanisms regulating protein sorting and localization in central nervous system (CNS) neurons (Lim et al., 2000; Garrido et al., 2003; Rivera et al., 2003; Wisco et al., 2003; Yang et al., 2007). In the experiments described here, we define the AIS by the presence of high density, restricted clustering of at least one of the following five protein components: NF-186, NrCAM, βIV spectrin, Nav channels, and ankG. Both in vivo and in cultured hippocampal neurons, NrCAM, NF-186, ankG, βIV spectrin, and Nav channels all colocalize within the first 20–40 μm of the axon (Fig. 1, B–D). Further, these proteins are excluded from somatodendritic domains defined by microtubule-associated protein 2 (MAP2; Fig. 1, B–D, blue) immunoreactivity.
Figure 1.

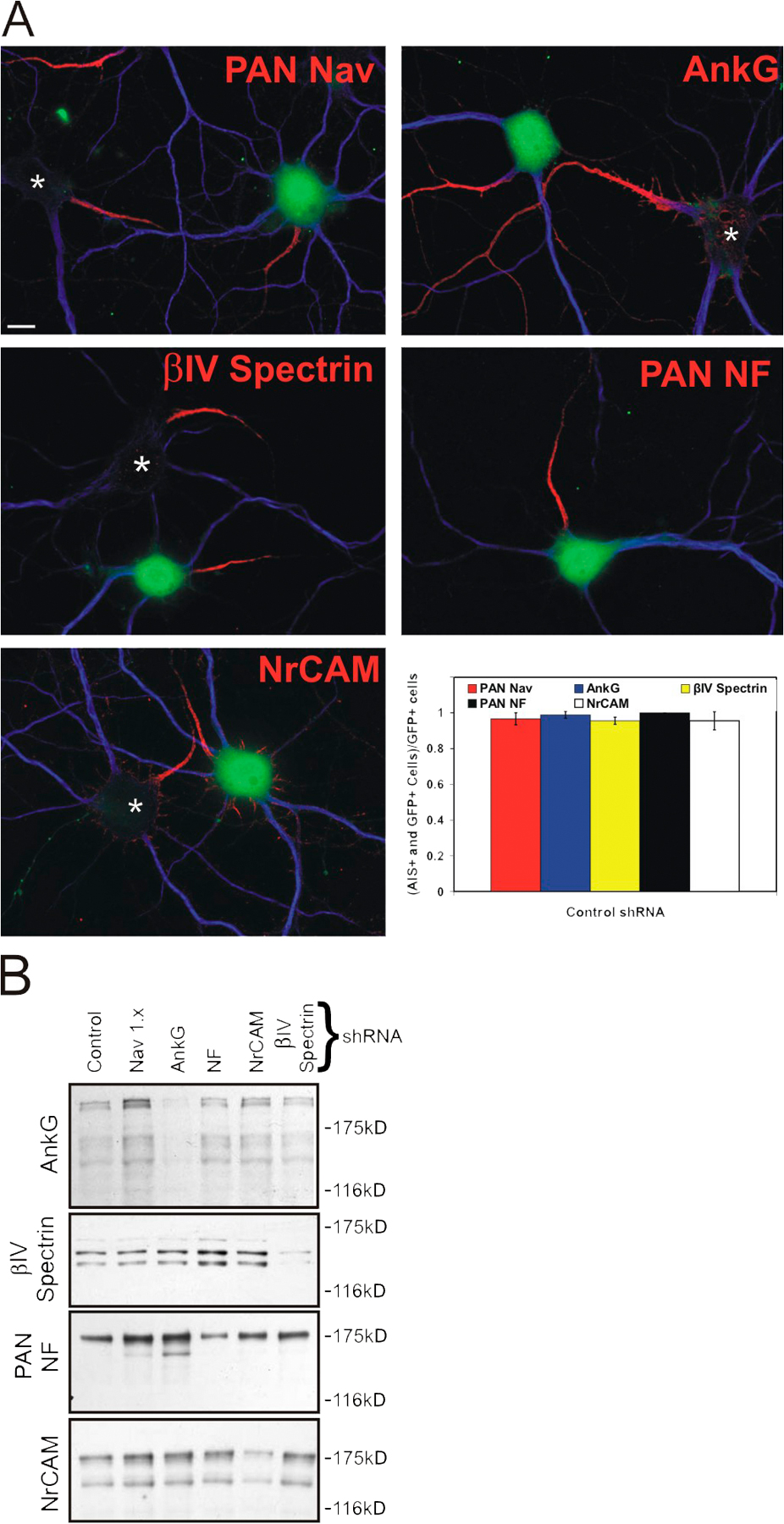
The molecular composition of AIS in vitro is identical to those in vivo, and shRNA expression plasmids effectively knock down their target proteins in hippocampal neurons. (A) Immunolabeling of cortical brain sections for Nav channels (green) and βIV spectrin (red). (B–D) Cultured hippocampal neurons labeled with antibodies against Nav channels (B; green), NrCAM (B; red), ankG (C; red), NF (D; green), and βIV spectrin (D; red). In B–D, somatodendritic domains were immunolabeled with antibodies against MAP2 (blue). (E) Hippocampal neurons were cotransfected with one shRNA and EGFP (GFP) 3 h after plating. After 17 DIV, neurons were immunolabeled for MAP2 (blue) to mark the somatodendritic domain and the targeted AIS protein (red). Control, untransfected cells are indicated by an asterisk. GFP fluorescence identifies cells that were transfected with the shRNA. Bars, 10 μm.
Which protein is the first to define the AIS? To answer this question, we immunostained hippocampal neurons at 2, 4, 6, 8, and 10 d in vitro (DIV) using antibodies against Nav channels, ankG, βIV spectrin, NrCAM, and NF (pan-NF). However, similar to Boiko et al. (2007), we were unable to clearly identify a single protein that clustered first at the AIS (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200705119/DC1), as we observed only small differences in the time course of protein clustering. These results likely reflect differences in antibody affinity rather than provide an accurate picture of the temporal regulation of AIS protein localization. Thus, other methods are necessary to determine the molecular events leading to AIS formation.
Short hairpin RNA (shRNA)–mediated ablation of AIS proteins
To determine the mechanisms underlying AIS formation, we used RNAi methods to silence expression of AIS proteins. Compared with mice lacking these proteins altogether, this strategy has several advantages, including the following: (1) all experimental manipulations of protein expression occur at the same time and in similar neurons, (2) mouse strain differences are avoided, and (3) by working with cells rather than the whole organism, the role of proteins essential for survival (e.g., Nav channels) or that may serve diverse functions in different cellular contexts (e.g., ankG) can be investigated. Therefore, we used shRNA expression plasmids for gene silencing of NF-186, NrCAM, Nav channels (Nav1.x), βIV spectrin, and ankG; we also constructed a control shRNA expression plasmid using a sequence directed against a nonmammalian protein. Cotransfection of the control shRNA and EGFP (to identify transfected neurons) showed that the assembly of the AIS was unaffected by transfection at both 10 and 17 DIV (Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200705119/DC1; 10 DIV not depicted).
To confirm the specificity of each shRNA, we cotransfected COS-7 cells with an expression plasmid for the target protein together with each shRNA. In transfected COS-7 cells, each shRNA only reduced expression of the target protein and did not affect other AIS proteins (Fig. S2 B).
As a direct measure of each shRNA's efficacy, we examined their ability to eliminate the endogenous target protein in cultured hippocampal neurons. 3 h after plating neurons, each shRNA expression plasmid was cotransfected into hippocampal neurons together with GFP. At 10 or 17 DIV, the transfected neurons were then immunostained with antibodies to MAP2 (to define the somatodendritic domain) and the targeted protein. Immunostaining for the target protein revealed that untransfected neurons (Fig. 1 E, asterisk) had the target protein at the AIS, but transfected neurons (GFP+) had no AIS immunoreactivity. The shRNA expression plasmids were found to be effective at eliminating all AIS immunoreactivity for the target protein in 50–90% of transfected hippocampal neurons (Fig. 2 B and Fig. 3 B; note that when the immunoreactivity for the targeted protein was reduced in transfected neurons, but not completely eliminated, the neuron was still counted as having an AIS). Thus, the shRNA expression plasmids are both specific and effective for knockdown of the targeted protein.
Figure 2.

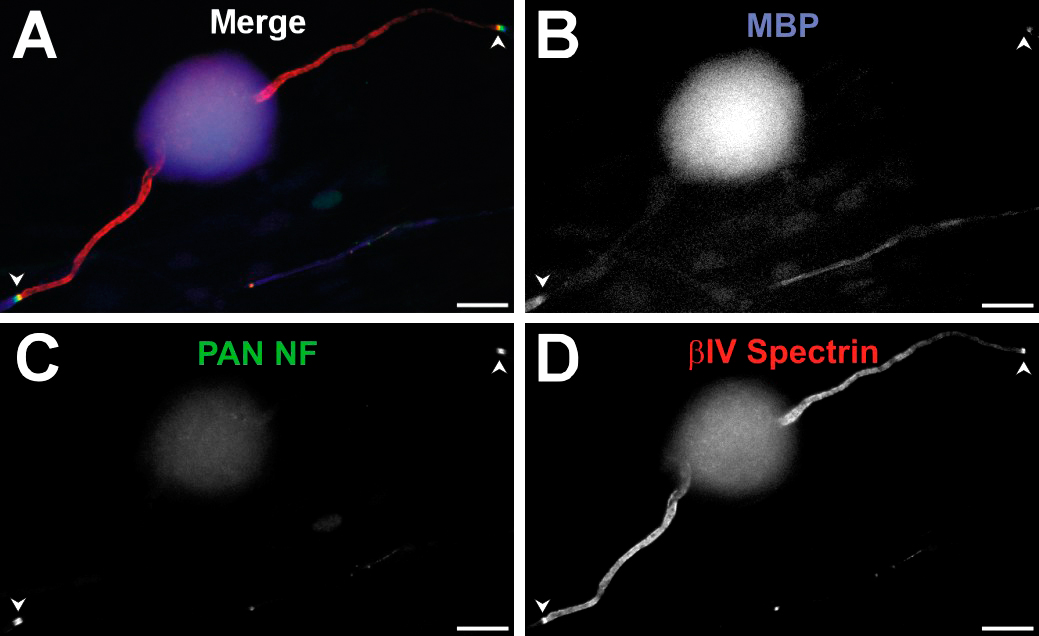
NF-186 is not required for Nav channel clustering or the molecular assembly of the AIS. (A) Immunolabeling of cultured hippocampal neurons transfected with either NF shRNA or NrCAM shRNA. At 10 DIV, cells were labeled for the nontargeted AIS proteins (red): Nav channels (pan-Nav), ankG, βIV spectrin, NF (pan-NF), or NrCAM (βIV spectrin, pan-NF, and NrCAM not depicted). GFP fluorescence indicates transfected cells, whereas MAP2 (blue) marks the somatodendritic domain. Asterisks indicate untransfected neurons. (B) The ratio of GFP+ neurons labeled at the AIS (for the protein indicated) to the total number of GFP+ neurons. In a minimum of three independent experiments, 25–30 GFP+ positive neurons were counted per shRNA. Error bars indicate ± SD. (C–E) Cultured DRG neurons in DRG/Schwann cell co-cultures immunolabeled for βIV spectrin (C [red] and D [green]), ankG (C and E, green), NF-186 (D, red), and NrCAM (E, red). MAP2 (blue) marks the soma in these neurons. (F) Nodes of Ranvier in PNS myelinated cultures have high densities of Nav channels, ankG, βIV spectrin, NF-186, and NrCAM (red). Paranodes are labeled with anti-Caspr (green). Myelin is labeled with anti-MBP (blue). Bars: (A–E) 10 μm; (F) 5 μm.
Figure 3.

AnkG is required for AIS formation. (A) 10-DIV hippocampal neurons transfected with Nav channel shRNA (Nav1.x shRNA), ankG shRNA, or βIV spectrin shRNA, and immunolabeled for Nav channels (pan-Nav), ankG, βIV spectrin, NF (pan-NF), or NrCAM (red), and MAP2 (blue). GFP fluorescence indicates transfected neurons. Asterisks indicate untransfected neurons. (B) The ratio of GFP+ neurons labeled at the AIS (for the protein indicated) to the total number of GFP+ neurons. In a minimum of three independent experiments, 25–30 GFP+ positive neurons were counted per shRNA. Error bars indicate ± SD. Bar, 10 μm.
Neither NF-186 nor NrCAM is required for Nav channel clustering at the AIS of CNS neurons
Because NF-186 is essential for Nav channel and ankG clustering at nodes of Ranvier (Sherman et al., 2005), we tested whether the CAMs NF-186 or NrCAM are also necessary for the molecular assembly of the AIS (Fig. 2). We eliminated NF-186 using NF shRNA and then immunostained the cultures at 8–10 DIV with anti-MAP2 (Fig. 2, blue) and antibodies to Nav channels, ankG, βIV spectrin, or NrCAM (red). Cells transfected with the NrCAM shRNA were immunostained with the same set of antibodies, except anti–pan-NF was substituted for anti-NrCAM. In neurons transfected with NF or NrCAM shRNA expression plasmids, ∼80% of neurons had no detectable staining for the targeted protein (Fig. 1 E and Fig. 2 B; transfected neurons are GFP+). However, elimination of these CAMs did not affect Nav channel, ankG, or βIV spectrin (not depicted) localization and clustering at the AIS (Fig. 2, A and B; untransfected neurons are identified by an asterisk). We measured ∼30% reduction in NrCAM+ AIS in neurons transfected with NF shRNA, and ∼20% reduction in NF-186+ AIS when neurons were transfected with NrCAM shRNA (Fig. 2 B). This observation is interesting because Sherman et al. (2005) reported that NFasc-null mice lack NrCAM at PNS nodes of Ranvier, and NrCAM-null mice have delayed PNS node formation (Custer et al., 2003). Thus, although there may be some interdependence between NrCAM and NF-186, these results demonstrate that neither of these CAMs is required for the clustering of Nav channels at the AIS of CNS neurons.
NF-186 and NrCAM are not enriched at the AIS of dorsal root ganglion (DRG) neurons
As NF-186 has been shown to be essential for node formation in the PNS, we considered the possibility that NF-186 or NrCAM may contribute to AIS formation in the PNS rather than the CNS. To test this possibility, we used a DRG neuron–Schwann cell co-culture system, where Schwann cells make many myelin segments and form nodes of Ranvier. After 21 DIV and after induction of myelination by the addition of ascorbic acid, we determined that DRG neurons had ankG, βIV spectrin, and Nav channels at the AIS (Fig. 2 C; Nav channels not depicted). However, we were unable to detect any enrichment for NF-186 or NrCAM (Fig. 2, D and E, red). Even cultures that were 5 mo old did not have high densities of NrCAM or NF-186 at the DRG neuron AIS (unpublished data). In contrast, nodes of Ranvier along these same axons, defined by Caspr-labeled paranodes (Fig. 2, D and E, green), had high levels of clustered NF-186, NrCAM, Nav channels, ankG, and βIV spectrin (Fig. 2 F, red; and Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200705119/DC1). These results indicate that NF-186 and NrCAM are not required for Nav channel clustering at the AIS, and that the molecular requirements for AIS assembly are distinct from nodes of Ranvier. These results parallel those recently reported by Dzhashiashvili et al. (2007).
AnkG is required for Nav channel clustering and the molecular assembly of the AIS
If NrCAM and NF-186 are not required for AIS assembly, what protein is essential? As ankG, βIV spectrin, and Nav channels have each been reported to contribute to AIS formation (Zhou et al., 1998; Komada and Soriano, 2002; Xu and Shrager, 2005), we introduced shRNA expression plasmids directed against these proteins into cultured hippocampal neurons. After 8–10 DIV, knockdown of Nav channels or βIV spectrin using the shRNA expression plasmids had no effect on the clustering of other AIS components (Fig. 3). However, when ankG shRNA expression plasmids were transfected into hippocampal neurons, ∼80% of cells had no detectable clustering of any other AIS protein (Fig. 3, A and B, middle). Thus, ankG is required for the clustering of Nav channels and the molecular assembly of the AIS.
Because loss of βIV spectrin or Nav channels has also been reported to disrupt the AIS (Komada and Soriano, 2002; Xu and Shrager, 2005), we considered the possibility that NF-186, NrCAM, Nav channels, or βIV spectrin contribute to maintenance and long-term stability of the AIS. To test this, we transfected the various shRNA expression plasmids as described and then immunostained neurons at 17 DIV to determine whether loss of any one AIS component influenced the clustering of any other AIS protein at later time points. We found that at 17 DIV, the shRNA expression plasmids (with the exception of βIV spectrin shRNA) were effective at eliminating their target proteins in ∼70% of neurons (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200705119/DC1). However, only the ankG shRNA disrupted localization of the other AIS protein components.
To confirm further that ankG is essential for the molecular assembly of the AIS, we electroporated neurons with either NF-186 or ankG shRNA at embryonic day (E) 16 in utero. The shRNA expression plasmids also expressed a membrane-bound form of GFP to permit identification of transfected neurons and their axons. This method results in transfection of neurons that subsequently migrate into the cortex and become layer II–III pyramidal neurons. 6 wk after birth, we cut coronal brain sections and immunostained these sections to detect ankG, βIV spectrin, NF-186, and Nav channels. The axons of GFP+ neurons were easily identified (Fig. 4, arrowheads). 80% (n = 33) of NF-186 shRNA–transfected neurons lacked NF-186 at the AIS (Fig. 4 A, arrowheads), but untransfected neurons had robust NF-186 immunoreactivity (Fig. 4 A, arrows). Further, other components of the AIS, such as βIV spectrin (Fig. 4 B, arrowheads), were still appropriately clustered in neurons transfected with the NF-186 shRNA (22/22 NF-186 shRNA–transfected neurons had βIV spectrin immunoreactivity). In contrast, 70% (n = 27) of neurons electroporated with ankG shRNA lacked ankG at the AIS (Fig. 4 C, arrowheads; n = 27), but untransfected neurons had robust AIS ankG immunoreactivity (Fig. 4 C, arrows). As with cultured hippocampal neurons, silencing of ankG expression blocked the AIS clustering of βIV spectrin, NF-186, and Nav1.6 (Fig. 4, D and E; Nav1.6 not depicted); 1/21, 0/24, and 0/12 neurons had AIS immunoreactivity for βIV spectrin, NF-186, and Nav1.6, respectively. Together, these results confirm ankG's essential role in vivo for Nav channel clustering and AIS assembly.
Figure 4.

ShRNA knockdown of ankG in vivo, but not NF-186, inhibits AIS assembly. (A and B) NF shRNA-gapEGFP plasmid was electroporated into rat brain in utero at E16. Transfected cortical neurons were immunolabeled with anti–pan-NF (A) or βIV spectrin (B). (C–E) AnkG shRNA-gapEGFP plasmid was electroporated into rat brain in utero at E16. Layer II–III cortical neurons immunolabeled with antibodies against ankG (C), pan-NF (D), and βIV spectrin (E). Arrowheads indicate axons of each transfected (GFP+) neuron. Arrows indicate the AIS of nontransfected neurons. Bar, 10 μm.
Brevican, an ECM molecule, is enriched at the AIS and nodes of Ranvier
If NF-186 and NrCAM are not required for Nav channel clustering at the AIS, what are their functions? As both of these CAMs have large extracellular immunoglobulin domains, they might be important for mediating interactions with other cells or proteins. Consistent with this idea, John et al. (2006) recently reported that the chondroitin sulfate proteoglycan brevican, mainly secreted by glia, is highly enriched at the AIS of cultured neurons. Brevican is a member of a family of ECM molecules called lecticans that assemble into perineuronal nets surrounding neurons (Yamaguchi, 2000). Other lecticans include aggrecan and neurocan. Immunostaining of cultured hippocampal neurons with antibodies against aggrecan and neurocan at 16 DIV revealed prominent somatic staining (Fig. 5, A–D). In ∼40% of neurons, there was punctate aggrecan and neurocan immunoreactivity along the AIS (Fig. 5, A and C, insets). In contrast, 93% of cultured hippocampal neurons had robust brevican immunoreactivity that was restricted to the AIS, where it colocalized with other AIS markers, such as βIV spectrin (Fig. 5 E). Brevican immunoreactivity was also enriched at the AIS of cortical neurons in vivo (Fig. 5 F), but neither aggrecan nor neurocan could be detected at these sites in vivo (not depicted). As the molecular composition of nodes is nearly identical to that at the AIS, we immunostained optic nerve with antibodies against aggrecan, neurocan, and brevican. Among these lecticans, only brevican could be detected at nodes (Fig. 5, G–I, arrowheads). These results indicate that brevican is highly enriched at sites where NF-186 and NrCAM are located.
Figure 5.

Brevican is enriched at the AIS and node of Ranvier. (A–D) Double immunostaining for aggrecan or neurocan (red) and pan-NF (green) in cultured hippocampal neurons. Insets show punctate aggrecan (A) or neurocan (C) staining at the AIS of some neurons. (E) Brevican (red) and βIV spectrin (green) immunostaining of cultured hippocampal neurons. Brevican is enriched only at the AIS (see inset). In A–E, MAP2 (blue) defines the somatodendritic domain. (F) Double immunostaining for brevican (red) and βIV spectrin (green) in vivo; brevican immunoreactivity colocalizes with βIV spectrin. Inset shows brevican immunoreactivity alone. The soma of the neuron is indicated by an asterisk. (G–I) Immunostaining of optic nerve using antibodies against neurocan (G, red), aggrecan (H, green), or brevican (I, green). Nodes of Ranvier were identified by anti–βIV spectrin (G [green] and I [blue]) or anti-Caspr to label paranodes (H and I; red). Arrowheads indicate nodes. Bars: (A–F) 10 μm; (G–I) 5 μm.
The brevican-based ECM interacts with NF-186
To determine if NF-186 and/or NrCAM interact with brevican, we expressed each CAM in COS cells and then added soluble GFP-brevican to determine if it could bind to the transfected cells. We found punctate GFP-brevican bound to the surface of NF-186–transfected cells (Fig. 6, A and B), but not to untransfected (Fig. 6, A and B) or NrCAM-transfected cells (Fig. 6, C and D), suggesting that brevican interacts with NF-186.
Figure 6.

Brevican interacts with NF-186. (A–D) Soluble GFP-brevican (green) binds to COS-7 cells transfected with HA-tagged NF-186 (A [red] and B) but not cells transfected with NrCAM (C [red] and D). Nuclei are labeled using Hoechst (blue). (E–H) Transfected neurons overexpressing HA-tagged NF-186 labeled with anti-HA (green; E and G) and anti-brevican (red; E and H). MAP2 (blue; E and F) was used to mark the somatodendritic domain. (I and J) Intensity plots measuring fluorescence along the white lines shown in E for the AIS (I) and a dendrite (J). Bar, 10 μm.
To further understand the mechanisms underlying recruitment of brevican to the AIS, we examined the effect of ectopically expressed NF-186 on brevican localization. We expressed an HA-tagged NF-186 in hippocampal neurons. Low levels of HA–NF-186 expression resulted in HA–NF-186 (Lemaillet et al., 2003) and brevican being restricted to the AIS (Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200705119/DC1). In contrast, overexpressed HA–NF-186 was found both at the AIS and somatodendritic domains (Fig. 6 E). Overexpression did not interfere with the normal recruitment of brevican to the AIS (Fig. 6, E and I). However, whenever HA–NF-186 was ectopically localized to somatodendritic domains, brevican was recruited to these same sites (Fig. 6, F–H). Nontransfected neurons never had somatodendritic brevican (Fig. S5). The redistribution of brevican along dendrites of cells overexpressing HA–NF-186 can be seen in line scans through the AIS and dendrites (Fig. 6, I and J; these line scans correspond to the white lines crossing the AIS and a dendrite in Fig. 6 E). Thus, ectopic expression of NF-186 is sufficient to recruit the brevican-based ECM to somatodendritic domains.
AIS clustering of brevican depends on NF-186
Because ankG knockdown eliminates clustering of all neuronal AIS proteins, including NF-186 and NrCAM (Fig. 2, Fig. 3, and Fig. S4), we first tested whether loss of ankG also blocked brevican from clustering at the AIS. Cotransfection of the ankG shRNA together with GFP in cultured hippocampal neurons showed that 84% of GFP+ neurons had no detectable brevican at the AIS (Fig. 7, A and D). However, untransfected neurons (Fig. 7 A, asterisk) had robust brevican immunoreactivity at the AIS. To directly determine whether AIS brevican clustering depends on the CAMs NF-186 and/or NrCAM, we silenced expression of these proteins in cultured hippocampal neurons using shRNA expression plasmids. Although untransfected neurons had robust brevican immunoreactivity at the AIS (Fig. 7 B, asterisk), knockdown of NF-186, but not NrCAM, completely blocked the AIS clustering of brevican (Fig. 7, B–D).
Figure 7.

NF-186, but not NrCAM, is required for brevican clustering at the AIS. (A and B) Knockdown of ankG or NF-186 via shRNA plasmids in hippocampal neurons (GFP+) blocks AIS localization of brevican (red). (C) Hippocampal neurons lacking NrCAM still cluster brevican (red) at the AIS. MAP2 immunoreactivity (blue) marks the somatodendritic domain. Asterisks indicate untransfected neurons. (D) The ratio of GFP+ neurons labeled at the AIS (for the protein indicated) to the total number of GFP+ neurons. In a minimum of three independent experiments, 25–30 GFP+ positive neurons were counted per shRNA. (E and F) NF shRNA-gapEGFP plasmid was electroporated into rat brain in utero at E16. Transfected cortical neurons were visualized by GFP fluorescence (green), and sections were immunostained with anti-brevican (E [red] and F). The axon of the transfected neuron is indicated by the arrowheads, and the anti-brevican–labeled initial segments of untransfected neurons are indicated by the arrow. (G and H) NF-186 and ankG are both normally clustered at the AIS in brevican−/− brain. Note the neurons shown in G and H are not the same neurons but come from different brain sections, and NF-186 immunostaining was performed using a rabbit polyclonal antibody. Error bars indicate ± SD. Bars, 10 μm.
Finally, using the same in utero electroporation strategy as described, we knocked down expression of NF-186 in vivo using NF shRNA expression plasmids. We then immunolabeled postnatal day 10 brain sections using anti-brevican antibodies. The axons of electroporated neurons were easily identified by GFP fluorescence (Fig. 7 E, arrowheads). Although untransfected neurons had brevican immunoreactivity at the AIS (Fig. 7, E and F, arrows), we could not detect brevican immunoreactivity at the AIS of electroporated neurons (Fig. 7, E and F, arrowheads). Thus, NF-186 is required for the enrichment of brevican at the AIS. In contrast, NF-186 and ankG were both normally clustered at the AIS of brevican-null mice (Fig. 7, G and H).
Discussion
Neuronal excitability depends not only on the kinds of ion channels and receptors expressed by a cell but also on their specific localization. In axons, ion channels are clustered in high densities at the nodes of Ranvier and the AIS. Analysis of βIV spectrin mutant mice suggested that βIV spectrin could be responsible for the assembly of the AIS and nodes by directing the recruitment of ankG to these domains (Komada and Soriano, 2002). More recent results indicate that βIV spectrin localization depends on ankG binding. Further, βIV spectrin by itself cannot recruit other interacting proteins to the AIS (Yang et al., 2007). Instead, βIV spectrin contributes to maintenance of these domains and the overall plasma membrane structure of the node and the AIS (Yang et al., 2004). Consistent with these ideas, we observed no impairment in ankG, Nav channel, or CAM clustering in hippocampal neurons lacking βIV spectrin at the AIS.
Nav channels themselves have also been proposed to be required for AIS assembly (Xu and Shrager, 2005). This result was based on the observation that knockdown of Nav channels in spinal motor neurons (using the same shRNA expression construct used here) blocked the clustering of ankG at the AIS. This result is different from the one we report for hippocampal neurons. We do not have a clear explanation for these differences, although one notable distinction between the two models is that the AIS assembles much more rapidly in motorneurons than hippocampal neurons.
Although nodes and the AIS share a common molecular organization, the data presented here show that the initial events leading to their assembly are unique: NF-186 is dispensible for Nav channel clustering at the AIS but not at nodes (Sherman et al., 2005). Despite this clear difference, there are also common elements to the mechanism of Nav channel clustering in these domains. For example, we show that ankG is essential for AIS formation. This conclusion is consistent with analyses of mutant mice lacking ankG in their Purkinje neurons (Jenkins and Bennett, 2001). AnkG is also required for node of Ranvier formation based on its simultaneous interactions between NF-186, Nav channels, and KCNQ2/3 channels (Garver et al., 1997; Garrido et al., 2003; Pan et al., 2006). Furthermore, one recent report demonstrated that knockdown of ankG in myelinated DRG Schwann cells results in failure to cluster Na+ channels (Dzhashiashvili et al., 2007). Thus, ankG functioning as a scaffold to facilitate clustering of Nav channels is common to both nodes and AIS.
How does ankG become localized to the AIS? The answer to this question remains unknown and will likely depend on a more complete description of the proteins located at the AIS and the developmental mechanisms that underlie axon specification (Arimura and Kaibuchi, 2007). It is interesting to note that other ankyrin repeat–containing proteins are also found at the AIS, including IκBα, a component of the NFκB signaling pathway (Schultz et al., 2006). However, as ankyrinB is restricted to distal axons rather than the AIS (Boiko et al., 2007), it is not likely that ankyrin repeats themselves determine AIS localization. Indeed, structure function analyses of ankG's targeting mechanism indicated that multiple protein domains in ankG are necessary for its restriction to the AIS (Zhang and Bennett, 1998).
The role of CAMs at the AIS
At nodes of Ranvier CAMs are positioned along axons through heterophilic interactions with glia, or molecules secreted by glia (Schafer and Rasband, 2006; Eshed et al., 2007). In contrast, NF-186 and NrCAM AIS clustering depends on an FIGQY ankG-interacting motif conserved among all L1 CAMs (Garver et al., 1997). The roles of NF-186 and NrCAM at the AIS have not been investigated in Nfasc- or NrCAM-null mice, respectively. This analysis may be difficult though, as Nfasc-null mice die at about postnatal day 6 (Sherman et al., 2005), and NrCAM-null mice do not exhibit a phenotype suggesting altered neuronal excitability (Custer et al., 2003). Ango et al. (2004) proposed that NF-186 contributes to appropriate GABA-ergic innervation between basket cells and the AIS of Purkinje neurons. However, this conclusion was based, in part, on an analysis of the localization of NF-186 in mice lacking ankG in Purkinje neurons. As we, and others, have shown (Jenkins and Bennett, 2001), loss of ankG blocks the recruitment of all AIS proteins, not just NF-186. AIS localization of NF-186 may lead to GABA-ergic synapse stabilization through brevican, rather than a direct cell– cell interaction between NF-186 and some other axonal CAM or receptor.
The data presented here provide the first direct evidence for the function of NF-186 at the AIS: to recruit and assemble a specialized brevican-containing ECM. We showed that loss of NF-186, but not NrCAM, results in failure to assemble this matrix. We also showed that overexpression of NF-186 in somatodendritic domains caused brevican to be recruited to these same sites. These data strongly suggest that NF-186 interacts directly with brevican, although we cannot rule out the possibility that there is some as-yet-unidentified intermediary linking brevican and NF-186. The view that brevican is a ligand for NF-186 is consistent with the fact that brevican binds strongly to fibronectin type III domains, of which NF-186 has three (Davis et al., 1993; Yamaguchi, 2000).
The role of brevican
Brevican is only one component of the perineuronal net that surrounds most CNS neurons. These nets form only during postnatal development, suggesting that they are structures integral to mature neurons. Proposed functions of the perineuronal net include stabilization of synapses and cellular contacts, ionic buffering, and serving as a link to the intracellular cytoskeleton (Celio et al., 1998). Within this net, there exists a specialized brevican-containing matrix at the AIS. Because chandelier and basket cells make axo-axonic synapses on cortical pyramidal neuron AIS (Inda et al., 2006), brevican may be important for stabilizing these contacts. Alternatively, brevican may function at the AIS and nodes of Ranvier as an “exoskeleton” to stabilize NF-186, which may then function as a link between this exoskeleton and the cytoskeleton to stabilize not only NF-186 but also its interacting proteins. The AIS and nodes, which function as highly specialized axonal compartments responsible for action potential initiation and propagation, may organize their own specific brevican-based ECM as a microenvironment for optimized Nav channel function at these sites. Although electrophysiological studies of brevican-null mice revealed a clear reduction in hippocampal long-term potentiation, the physiological significance is still obscure (Brakebusch et al., 2002).
Brevican is also enriched at mature and developing nodes (unpublished data), suggesting that it may contribute to node formation and/or maintenance. Other ECM molecules located at CNS nodes include OMGP, versican, and tenascin, some of which have been suggested to be important for CNS node formation (Huang et al., 2005; Melendez-Vasquez et al., 2005; Nie et al., 2006). In the PNS, gliomedin, secreted from Schwann cells, interacts with heparin sulfate proteoglycans in the ECM and axonal NF-186 to initiate Nav channel clustering (Eshed et al., 2007). We speculate that brevican and other ECM proteins may function in a similar way to initiate CNS node formation. ECM proteins may bind to and stabilize NF-186 at axonal membranes not covered by myelin (i.e., putative nodes). The nodal CAMs may then function as attachment sites for ankG, followed by accumulation and clustering of ion channels. The diversity of CNS nodal ECM proteins may explain why loss of any single component (e.g., tenascin, OMGP; Weber et al., 1999; Nie et al., 2006) does not inhibit channel clustering. Some data suggest that soluble, glial-derived factors initiate Nav channel clustering in cultured CNS neurons (Kaplan et al., 1997). Future experiments will test the model described and help to determine whether these soluble factors include brevican and other nodal ECM proteins.
The results presented here clearly demonstrate that ankG, rather than βIV spectrin, CAMs, or Nav channels, is essential for the molecular assembly of the AIS. Thus, in contrast to nodes, NF-186 is dispensible for Nav channel clustering at the AIS. However, similar to nodes, NF-186 interacts with and facilitates the assembly of a specialized ECM. NF-186 interactions with brevican and the subsequent assembly of a specialized AIS and nodal ECM may be necessary for a variety of physiological functions critical for integrating neurons into a complex network of communicating cells.
Materials and methods
Animals
Animals were housed at the University of Connecticut Health Center, and all experiments involving animals were approved by the institutional animal care and use committee in accordance with all National Institutes of Health guidelines for the humane treatment of animals. Brevican−/− brains were generously provided by R. Faessler (Max Planck Institute of Biochemistry, Martinsried, Germany).
Antibodies
The mouse monoclonal pan-NF and pan-Nav channel antibodies and the polyclonal Caspr antibodies were previously described (Schafer et al., 2004). The polyclonal βIV spectrin antibody was previously described (Ogawa et al., 2006). The polyclonal ankG antibody was provided by V. Bennett (Duke University, Durham, NC). A mouse monoclonal ankG antibody was purchased from Zymed Laboratories. The rat monoclonal MBP and the rabbit polyclonal anti-Aggrecan antibodies were purchased from Chemicon International. The chicken MAP2 antibody was purchased from EnCor Biotechnology, Inc. The rabbit polyclonal NrCAM antibody was purchased from Abcam. The rabbit polyclonal anti–NF-186 antibody was provided by A. Gow (Wayne State University, Detroit, MI). The anti-Neurocan antibody 1F6 developed by R.U. Margolis and R.K. Margolis was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development by the University of Iowa (Iowa City, IA). The guinea pig anti-brevican antibodies were previously described (John et al., 2006). The rabbit and mouse anti-GFP antibodies were purchased from Invitrogen and CLONTECH Laboratories, Inc., respectively. All fluorescent secondary antibodies were purchased from Invitrogen except for the AMCA-conjugated anti-chicken antibody, which was obtained from Accurate Chemical.
cDNA constructs
The shRNA constructs were made as previously described (Xu and Shrager, 2005). The sense sequences for each oligonucleotide are as follows: Nav1.x, 5′-GTTCGACCCTGACGCCACT-3′; ankG, 5′-GCCGTCAGTACCATCTTCT-3′; βIV spectrin, 5′-CACTGGATAGCCGAGAAGG-3′; NF, 5′-TGCCTTCGTCAGCGTATTA-3′; NrCAM, 5′-CCAATAATCCTCCGAAGTG-3′; control, 5′-CTACTGAGAACTAAGAGAG-3′. The ankG-GFP and NrCAM constructs were gifts from V. Bennett. The HA-tagged NF-186 cDNA was provided by S. Lambert (University of Massachusetts Medical School, Worcester, MA). The myc-tagged βIV spectrin construct was a gift from M. Komada (Tokyo Institute of Technology, Yokohama, Japan). pEGFP-N1 was provided by J. Hewett (University of Connecticut Health Center, Farmington, CT). shRNAs used in electroporation experiments were constructed using pENTR 11. The H1 promoter driving shRNA expression and the shRNA sequence were inserted to pENTR 11 via the EcoRI and HindIII sites. The CAG promoter driving expression of membrane-bound gap-EGFP was inserted using the HindIII and XhoI sites. In the brevican-EGFP fusion construct, EGFP was fused to the C terminus of the full-length cDNA of rat brevican. We introduced an EcoRI and a BamHI restriction site into the brevican cDNA and cloned the entire open reading frame into the pEGFP-N1 vector.
Immunostaining
Optic nerves and brains were dissected rapidly and immediately fixed with 4% PFA at 4°C for 30 min (optic nerves) or 1 h (brains). Optic nerves and brains were then transferred to ice-cold 20% sucrose (wt/vol) in 0.1 M PB until equilibrated. The tissue was sectioned and immunolabeled as previously described (Schafer et al., 2004). For cultured neurons, cells to be immunostained using pan-Nav or ankG antibodies were fixed with 1 or 2% PFA, respectively, whereas cells to be immunostained using βIV spectrin, pan-NF, or NrCAM antibodies were fixed with 4% PFA. Cells were permeabilized using 0.3% Triton X-100 (Sigma-Aldrich) in milk.
Image acquisition
Fluorescence images were collected on an Axiovert 200M (Carl Zeiss MicroImaging, Inc.) fitted with an apotome for optical sectioning, and a digital camera (AxioCam; Carl Zeiss MicroImaging, Inc.). Images were taken using a 63× Plan-APOCHROMAT (1.4 NA) objective. AxioVision (Carl Zeiss MicroImaging, Inc.) acquisition software was used for collection of images. In some cases, stacks of images were acquired and volume reconstructions were generated using AxioVision software. In some images, contrast and brightness were subsequently adjusted using Photoshop (Adobe). No other processing of the images was performed. All figures were assembled using CorelDraw.
Tissue culture
Primary hippocampal neurons were prepared from E18 rat embryos as described previously (Ogawa et al., 2006). Neurons were plated at a density of 200 cells/mm2. Primary myelinating co-cultures were prepared as described by Svenningsen et al. (2003) with minor alterations. In brief, DRG were dissected from E16 Wistar rats and collected in HBSS without calcium and magnesium (Invitrogen). Cells were mechanically dissociated after 15 min of trypsin (0.25% in HBSS; Invitrogen) digestion at 37°C. Trypsinization was stopped by adding 10% FBS (Invitrogen) in Neurobasal medium (Invitrogen). Cells were centrifuged, washed, and resuspended in growth medium (Neurobasal; 2% B27), penicillin/streptomycin, Glutamax, and 100 ng/ml 7S NGF (all from Invitrogen). Cells were plated at a density of 230 cells/mm2 on glass (Carolina Biological) or Permanox slides (Nunc) coated with 0.4 mg/ml matrigel (BD Biosciences) and10 μg/ml poly-l-lysine (Invitrogen). Cells were maintained and myelinated with 50 μg/ml ascorbic acid (wt/vol; Sigma-Aldrich) as described by Svenningsen et al. (2003).
Cell transfections and quantification
Primary hippocampal neurons were transfected 3 h after plating using Lipofectamine 2000 (Invitrogen). Each shRNA construct was cotransfected into hippocampal neurons with pEGFP-N1, permitting GFP to serve as a marker for transfection. Cells were transfected using 1 μg of shRNA vector plus 1 μg pEGFP-N1 combined with 3 μg of Lipofectamine 2000 per ml of OptiMem (Invitrogen). Transfections were allowed to proceed for 4 h, after which cells were returned to growth medium. After 10 or 17 DIV, cells were fixed and immunostained as described. All experiments were performed in triplicate with independent dissections. Cells transfected with each shRNA separately were immunolabeled with ankG, pan-NAV, βIV spectrin, pan-NF, or NrCAM antibodies. 25–30 MAP2- and GFP-positive cells were counted per dissection for each shRNA and AIS antibody combination (∼90 transfected cells for each condition). Cells containing any positive AIS clustering of the immunolabeled protein were counted as having properly formed AIS with respect to that protein, even if the immunofluorescence intensity was clearly reduced compared with untransfected neurons. Only GFP-positive cells without any detectable AIS immunoreactivity were counted as negatives. For validation of shRNAs, COS-7 cells were cotransfected as above with a combination of each shRNA and ankG-GFP, myc-βIV spectrin, HA–NF-186, or NrCAM cDNAs. Cells were lysed, and proteins were size fractionated by SDS-PAGE (Schafer et al., 2004). Proteins were then electrophoretically transferred to nitrocellulose membranes and immunoblotted for each protein of interest as described previously (Schafer et al., 2004).
In vivo electroporation
In utero electroporation was performed as described previously (Saito, 2006), except that we introduced shRNA plasmids into embryonic rats at E16 rather than mice. We used rats because the pan-NF antibody only recognizes rat NF-186.
COS binding assay
COS-7 cells were transfected with 2 μg/ml brevican-EGFP cDNA using 3 μg/ml Lipofectamine 2000 (Invitrogen) in OptiMem. Serum-free media (VP-SFM; Invitrogen), supplemented with Glutamax replaced the transfection solution, and the culture supernatant was collected 3 d after transfection. For NF-186 and NrCAM transfections, COS-7 cells were plated on 25-mm glass coverslips (Fisher Scientific) and transfected with either 1 μg/ml of HA-tagged NF-186 or NrCAM cDNAs. 2 d after transfection, cells were incubated in serum-free media for 2 h before treatment. Brevican-EGFP was pre–cross-linked using anti-GFP antibodies for 1 h. Cells were treated with pre–cross-linked brevican-EGFP for 2 h. Cells were fixed with 4% PFA and immunolabeled with either pan-NF or NrCAM antibodies. Hoechst (Sigma-Aldrich) was used to identify nuclei.
Online supplemental material
Fig. S1 shows immunostaining of cultured hippocampal neurons immunostained for Nav channels, NrCAM, ankG, βIV spectrin, and NF-186 as a function of DIV. Fig. S2 shows that control shRNA does not affect AIS clustering of Nav channels, ankG, βIV spectrin, NrCAM, or NF-186 and that the shRNAs are specific and do not affect the expression of other AIS proteins. Fig. S3 shows a myelinated DRG neuron triple labeled with anti-MBP, anti–pan-NF, and anti–βIV spectrin. Fig. S4 shows the effect of shRNA-mediated protein knockdown on the localization of other AIS proteins at 17 DIV. Fig. S5 shows an untransfected neuron and a transfected neuron expressing low levels of HA–NF-186. In both neurons, brevican is restricted to the axon. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200705119/DC1.
Supplementary Material
Acknowledgments
We thank Dr. Reinhard Faessler for kindly providing brevican−/− brains.
This work was supported by National Institutes of Health grants NS044916 (M.N. Rasband) and NS17965 (P. Shrager), New York State grant CO19772-3784 (P. Shrager), and Deutsche Forschungsgemeinschaft grant Gu-230/5-2 (C.I. Seidenbecher). M.N. Rasband is a Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society. Y. Ogawa is supported by a postdoctoral fellowship from the National Multiple Sclerosis Society. X. Xu was a recipient of grants from the Christopher Reeve Foundation and the Schmitt Program on Integrative Brain Research.
The authors declare that they have no conflicts of interest.
M.N. Rasband's present address is Department of Neuroscience, Baylor College of Medicine, Houston, TX 77030.
Abbreviations used in this paper: AIS, axon initial segment; ankG, ankyrinG; CAM, cell adhesion molecule; CNS, central nervous system; DIV, days in vitro; DRG, dorsal root ganglion; E, embryonic day; MAP2, microtubule-associated protein 2; Nav, voltage-gated Na+; NF, neurofascin; NrCAM, neuron glia–related CAM; PNS, peripheral nervous system; shRNA, short hairpin RNA.
References
- Ango, F., G. di Cristo, H. Higashiyama, V. Bennett, P. Wu, and Z.J. Huang. 2004. Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at purkinje axon initial segment. Cell. 119:257–272. [DOI] [PubMed] [Google Scholar]
- Arimura, N., and K. Kaibuchi. 2007. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat. Rev. Neurosci. 8:194–205. [DOI] [PubMed] [Google Scholar]
- Banker, G., and K. Goslin. 1998. Culturing Nerve Cells. The MIT Press, Cambridge, England. 666 pp.
- Boiko, T., M. Vakulenko, H. Ewers, C.C. Yap, C. Norden, and B. Winckler. 2007. Ankyrin-dependent and -independent mechanisms orchestrate axonal compartmentalization of L1 family members neurofascin and L1/neuron-glia cell adhesion molecule. J. Neurosci. 27:590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brakebusch, C., C.I. Seidenbecher, F. Asztely, U. Rauch, H. Matthies, H. Meyer, M. Krug, T.M. Bockers, X. Zhou, M.R. Kreutz, et al. 2002. Brevican-deficient mice display impaired hippocampal CA1 long-term potentiation but show no obvious deficits in learning and memory. Mol. Cell. Biol. 22:7417–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celio, M.R., R. Spreafico, S. De Biasi, and L. Vitellaro-Zuccarello. 1998. Perineuronal nets: past and present. Trends Neurosci. 21:510–515. [DOI] [PubMed] [Google Scholar]
- Custer, A.W., K. Kazarinova-Noyes, T. Sakurai, X. Xu, W. Simon, M. Grumet, and P. Shrager. 2003. The role of the ankyrin-binding protein NrCAM in node of Ranvier formation. J. Neurosci. 23:10032–10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, J.Q., T. McLaughlin, and V. Bennett. 1993. Ankyrin-binding proteins related to nervous system cell adhesion molecules: candidates to provide transmembrane and intercellular connections in adult brain. J. Cell Biol. 121:121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhashiashvili, Y., Y. Zhang, J. Galinska, I. Lam, M. Grumet, and J.L. Salzer. 2007. Nodes of Ranvier and axon initial segments are ankyrin G-dependent domains that assemble by distinct mechanisms. J. Cell Biol. 177:857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshed, Y., K. Feinberg, S. Poliak, H. Sabanay, O. Sarig-Nadir, I. Spiegel, J.R. Bermingham Jr., and E. Peles. 2005. Gliomedin mediates schwann cell-axon interaction and the molecular assembly of the nodes of ranvier. Neuron. 47:215–229. [DOI] [PubMed] [Google Scholar]
- Eshed, Y., K. Feinberg, D.J. Carey, and E. Peles. 2007. Secreted gliomedin is a perinodal matrix component of peripheral nerves. J. Cell Biol. 177:551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido, J.J., P. Giraud, E. Carlier, F. Fernandes, A. Moussif, M.P. Fache, D. Debanne, and B. Dargent. 2003. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 300:2091–2094. [DOI] [PubMed] [Google Scholar]
- Garver, T.D., Q. Ren, S. Tuvia, and V. Bennett. 1997. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J. Cell Biol. 137:703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom, K.L., and M.N. Rasband. 2006. Intrinsic and extrinsic determinants of ion channel localization in neurons. J. Neurochem. 98:1345–1352. [DOI] [PubMed] [Google Scholar]
- Huang, J.K., G.R. Phillips, A.D. Roth, L. Pedraza, W. Shan, W. Belkaid, S. Mi, A. Fex-Svenningsen, L. Florens, J.R. Yates III, and D.R. Colman. 2005. Glial membranes at the node of Ranvier prevent neurite outgrowth. Science. 310:1813–1817. [DOI] [PubMed] [Google Scholar]
- Inda, M.C., J. Defelipe, and A. Munoz. 2006. Voltage-gated ion channels in the axon initial segment of human cortical pyramidal cells and their relationship with chandelier cells. Proc. Natl. Acad. Sci. USA. 103:2920–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins, S.M., and V. Bennett. 2001. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J. Cell Biol. 155:739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John, N., H. Krugel, R. Frischknecht, K.H. Smalla, C. Schultz, M.R. Kreutz, E.D. Gundelfinger, and C.I. Seidenbecher. 2006. Brevican-containing perineuronal nets of extracellular matrix in dissociated hippocampal primary cultures. Mol. Cell. Neurosci. 31:774–784. [DOI] [PubMed] [Google Scholar]
- Kaplan, M.R., A. Meyer-Franke, S. Lambert, V. Bennett, I.D. Duncan, S.R. Levinson, and B.A. Barres. 1997. Induction of sodium channel clustering by oligodendrocytes. Nature. 386:724–728. [DOI] [PubMed] [Google Scholar]
- Khaliq, Z.M., and I.M. Raman. 2006. Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J. Neurosci. 26:1935–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komada, M., and P. Soriano. 2002. βIV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J. Cell Biol. 156:337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaillet, G., B. Walker, and S. Lambert. 2003. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J. Biol. Chem. 278:27333–27339. [DOI] [PubMed] [Google Scholar]
- Lim, S.T., D.E. Antonucci, R.H. Scannevin, and J.S. Trimmer. 2000. A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron. 25:385–397. [DOI] [PubMed] [Google Scholar]
- Melendez-Vasquez, C., D.J. Carey, G. Zanazzi, O. Reizes, P. Maurel, and J.L. Salzer. 2005. Differential expression of proteoglycans at central and peripheral nodes of Ranvier. Glia. 52:301–308. [DOI] [PubMed] [Google Scholar]
- Naundorf, B., F. Wolf, and M. Volgushev. 2006. Unique features of action potential initiation in cortical neurons. Nature. 440:1060–1063. [DOI] [PubMed] [Google Scholar]
- Nie, D.Y., Q.H. Ma, J.W.S. Law, C.P. Chia, N.K. Dhingra, Y. Shimoda, W.L. Yang, N. Gong, Q.W. Chen, G. Xu, et al. 2006. Oligodendrocytes regulate formation of nodes of Ranvier via the recognition molecule OMgp. Neuron Glia Biol. 2:151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa, Y., D.P. Schafer, I. Horresh, V. Bar, K. Hales, Y. Yang, K. Susuki, E. Peles, M.C. Stankewich, and M.N. Rasband. 2006. Spectrins and ankyrinB constitute a specialized paranodal cytoskeleton. J. Neurosci. 26:5230–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, L.M., and G.J. Stuart. 2006. Site of action potential initiation in layer 5 pyramidal neurons. J. Neurosci. 26:1854–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Z., T. Kao, Z. Horvath, J. Lemos, J.-Y. Sul, S.D. Cranstoun, M.V. Bennett, S.S. Scherer, and E.C. Cooper. 2006. A common ankyrin-G-based mechanism retains KCNQ and Nav channels at electrically active domains of the axon. J. Neurosci. 26:2599–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera, J.F., S. Ahmad, M.W. Quick, E.R. Liman, and D.B. Arnold. 2003. An evolutionarily conserved dileucine motif in Shal K+ channels mediates dendritic targeting. Nat. Neurosci. 6:243–250. [DOI] [PubMed] [Google Scholar]
- Saito, T. 2006. In vivo electroporation in the embryonic mouse central nervous system. Nat Protoc. 1:1552–1558. [DOI] [PubMed] [Google Scholar]
- Schafer, D.P., and M.N. Rasband. 2006. Glial regulation of the axonal membrane at nodes of Ranvier. Curr. Opin. Neurobiol. 16:508–514. [DOI] [PubMed] [Google Scholar]
- Schafer, D.P., R. Bansal, K.L. Hedstrom, S.E. Pfeiffer, and M.N. Rasband. 2004. Does paranode formation and maintenance require partitioning of neurofascin 155 into lipid rafts? J. Neurosci. 24:3176–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, C., H.G. Konig, D. Del Turco, C. Politi, G.P. Eckert, E. Ghebremedhin, J.H. Prehn, D. Kogel, and T. Deller. 2006. Coincident enrichment of phosphorylated IkappaBalpha, activated IKK, and phosphorylated p65 in the axon initial segment of neurons. Mol. Cell. Neurosci. 33:68–80. [DOI] [PubMed] [Google Scholar]
- Sherman, D.L., S. Tait, S. Melrose, R. Johnson, B. Zonta, F.A. Court, W.B. Macklin, S. Meek, A.J. Smith, D.F. Cottrell, and P.J. Brophy. 2005. Neurofascins are required to establish axonal domains for saltatory conduction. Neuron. 48:737–742. [DOI] [PubMed] [Google Scholar]
- Shu, Y., A. Duque, Y. Yu, B. Haider, and D.A. McCormick. 2007. Properties of action potential initiation in neocortical pyramidal cells: evidence from whole cell axon recordings. J. Neurophysiol. 97:746–760. [DOI] [PubMed] [Google Scholar]
- Svenningsen, A.F., W.S. Shan, D.R. Colman, and L. Pedraza. 2003. Rapid method for culturing embryonic neuron-glial cell cocultures. J. Neurosci. Res. 72:565–573. [DOI] [PubMed] [Google Scholar]
- Weber, P., U. Bartsch, M.N. Rasband, R. Czaniera, Y. Lang, H. Bluethmann, R.U. Margolis, S.R. Levinson, P. Shrager, D. Montag, and M. Schachner. 1999. Mice deficient for tenascin-R display alterations of the extracellular matrix and decreased axonal conduction velocities in the CNS. J. Neurosci. 19:4245–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisco, D., E.D. Anderson, M.C. Chang, C. Norden, T. Boiko, H. Folsch, and B. Winckler. 2003. Uncovering multiple axonal targeting pathways in hippocampal neurons. J. Cell Biol. 162:1317–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X., and P. Shrager. 2005. Dependence of axon initial segment formation on Na+ channel expression. J. Neurosci. Res. 79:428–441. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, Y. 2000. Lecticans: organizers of the brain extracellular matrix. Cell. Mol. Life Sci. 57:276–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., S. Lacas-Gervais, D.K. Morest, M. Solimena, and M.N. Rasband. 2004. BetaIV spectrins are essential for membrane stability and the molecular organization of nodes of Ranvier. J. Neurosci. 24:7230–7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., Y. Ogawa, K.L. Hedstrom, and M.N. Rasband. 2007. βIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J. Cell Biol. 176:509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X., and V. Bennett. 1998. Restriction of 480/270-kD ankyrin G to axon proximal segments requires multiple ankyrin G-specific domains. J. Cell Biol. 142:1571–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, D., S. Lambert, P.L. Malen, S. Carpenter, L.M. Boland, and V. Bennett. 1998. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J. Cell Biol. 143:1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}