

Abstract
Cells from patients with Fanconi anemia (FA), an inherited disorder that includes bone marrow failure and cancer predisposition, have increased sensitivity to oxidative stress through an unknown mechanism. We demonstrate that the FA group G (FANCG) protein is found in mitochondria. Wild-type but not G546R mutant FANCG physically interacts with the mitochondrial peroxidase peroxiredoxin-3 (PRDX3). PRDX3 is deregulated in FA cells, including cleavage by a calpainlike cysteine protease and mislocalization. FA-G cells demonstrate distorted mitochondrial structures, and mitochondrial extracts have a sevenfold decrease in thioredoxin-dependent peroxidase activity. Transient overexpression of PRDX3 suppresses the sensitivity of FA-G cells to H2O2, and decreased PRDX3 expression increases sensitivity to mitomycin C. Cells from the FA-A and -C subtypes also have PRDX3 cleavage and decreased peroxidase activity. This study demonstrates a role for the FA proteins in mitochondria witsh sensitivity to oxidative stress resulting from diminished peroxidase activity. These defects may lead to apoptosis and the accumulation of oxidative DNA damage in bone marrow precursors.
Introduction
The clinical hallmark of Fanconi anemia (FA) is the development of pancytopenia (loss of blood cells) in childhood (Schmid and Fanconi 1978; Butturini et al., 1994; Alter 1995). There is a progressive loss of hematopoietic stem cells by enhanced apoptosis, and it affects all blood lineages (for review see Grompe and D'Andrea 2001). Another consistent feature of FA is a high propensity toward both hematological and nonhematological malignancies, including myelodysplastic syndrome, acute myelogenous leukemia, and squamous cell carcinomas. A wide variety of birth defects, such as short stature, skeletal abnormalities, abnormal skin pigmentation, and developmental abnormalities of other organs, are also observed. Markedly reduced life expectancy has been observed in FA patients with death resulting from hematological complications and cancer. Thus, understanding the basis of the bone marrow failure is of critical importance to improve current treatment approaches for patients with FA.
The cellular phenotype of FA is characterized by the occurrence of spontaneous chromosomal aberrations and hypersensitivity to DNA cross-linking agents, such as mitomycin C (MMC) and diepoxybutane. There are 12 complementation groups (A–C, D1, D2, E–G, I, J, L, and M), and 11 of the FA genes have been cloned (D'Andrea 2003; Meetei et al., 2004; Thompson 2005). A key event in the FA pathway is the activation of FA subtype D2 protein (FANCD2) by monoubiquitination, which critically depends on the formation of a core complex of at least eight FA proteins (FANCA–C, E–G, L, and M) in the nucleus, in which FANCL is likely to function as the E3 ubiquitin ligase (Meetei et al., 2003, 2004; Thompson 2005).
Localization studies suggest that a high molecular weight FA complex is found in both the nucleus and cytoplasm (Thomashevski et al., 2004; Mi and Kupfer, 2005). For example, a microscopic study revealed that FANCA and FANCG are cytoplasmic in G1 and G2-M phase but are predominantly nuclear during S phase (Mi and Kupfer, 2005). In addition, the FANCC protein has been found to interact with several cytoplasmic proteins involved in redox metabolism, including GSTP1 and the molecular chaperones GRP94 and HSP70 (Hoshino et al., 1998; Cumming et al., 2001; Otsuki et al., 2002; Pang et al., 2002). FANCG interacts with CYP2E1, which is associated with the production of reactive oxygen species (ROS) and the bioactivation of carcinogens, possibly implicating FANCG in protection against oxidative damage (Futaki et al., 2001, 2002). These data suggest that the FA proteins may function in more than one cellular compartment.
Oxygen sensitivity of FA cells was first reported by Joenje et al. (1981), and further studies demonstrated abnormal oxygen metabolism of FA cells, suggesting a defective antioxidant mechanism (Schindler and Hoehn 1988; Pagano 2000). In particular, oxidative damage and a senescent phenotype in response to hypoxia followed by reoxygenation is accentuated in FA cells (Zhang et al., 2005). In FA fibroblasts, however, the activity of antioxidative enzymes, including catalase, superoxide dismutase, glutathione reductase, and phospholipid hydroperoxide glutathione peroxidase, are normal (Gille et al., 1987; Ruppitsch et al., 1997). Oxidative stress caused multimerization and an increased interaction of FANC proteins (Park et al., 2004). Antioxidants have been shown to be beneficial for DNA stability and survival of FA cells (Porfirio et al., 1989; Ruppitsch et al., 1997). Thioredoxin (Trx) is an intracellular antioxidant and regulator of redox-sensitive gene expression. The overexpression of Trx in FA fibroblasts prevents the cytotoxic and DNA-damaging effect of MMC and diepoxybutane (Ruppitsch et al., 1998), suggesting a direct association of oxidative stress with the primary genetic defect in FA (Pagano, 2000; Ahmad et al., 2002; for review see Pagano and Youssoufian 2003). This hypothesis is strengthened by the finding that Fancc and superoxide dismutase double knockout mice exhibit severe defects in hematopoiesis, including histological evidence of bone marrow hypoplasia (Hadjur et al., 2001). These biochemical and genetic data strongly suggest that FA cells, including subtypes A, C, and G, experience increased oxidative stress, which can alter multiple cellular processes such as apoptosis, senescence, and DNA damage.
Although Blom et al. (2004) identified evolutionally conserved tetratricopeptide repeat domains in the FANCG protein, little is known about the biochemical function of the FANCG protein. To determine potential functional roles of FANCG protein, we performed a yeast two-hybrid screen. Peroxiredoxin-3 (PRDX3), the major Trx-dependent mitochondrial peroxidase, was identified as interacting with FANCG.
Mitochondrial PRDX3 is an important cellular antioxidant that regulates physiological levels of H2O2, leading to decreased cell growth while protecting cells from the apoptosis-inducing effects of high levels of H2O2 (Nonn et al., 2003). PRDX3 depletion resulted in the acceleration of apoptosis, with increased rates of ΔΨm (mitochondrial membrane potential) collapse, cytochrome c release, and caspase activation (Chang et al., 2004). PRDX3 contains a mitochondrial localization sequence, is found exclusively in the mitochondrion, comprises 5% of the total mitochondrial matrix, and uses mitochondrial Trx2 as the electron donor for its peroxidase activity (Watabe et al., 1997; Gourlay et al., 2003). PRDX3 expression is induced by oxidants in the cardiovascular system to maintain oxidant homeostasis (Araki et al., 1999; Wonsey et al., 2002). PRDX3 expression is regulated by myc (Wonsey et al., 2002), and elevated levels of PRDX3 protein were detected in breast cancers (Noh et al., 2001) and hepatocellular carcinoma (Choi et al., 2002). Prdx1- and Prdx2-deficient mice have severe hematological disorders and hemolytic anemia. Prdx1 knockout mice also develop malignant cancers (Lee et al., 2003; Neumann et al., 2003).
In this study, we demonstrate that the FANCG protein interacts physically with PRDX3 and that the proteins colocalize in the mitochondria. In FA cells from patients with subtypes G, C, and A, PRDX3 undergoes cleavage by a calpainlike cysteine protease, and mitochondrial peroxidase activity is diminished. These defects are associated with distorted mitochondrial structures. The overexpression of PRDX3 can restore the resistance of FA cells to H2O2 exposure, and down-regulation increases MMC sensitivity, suggesting that diminished mitochondrial peroxidase activity underlies the sensitivity of FA cells to oxidative stress.
Results
FANCG interacts with the mitochondrial protein PRDX3
To identify interacting partners of the FANCG protein, we initially used both the N-terminal (aa 1–322) and C-terminal (aa 301–622) regions of the FANCG protein as baits for yeast two-hybrid analysis. A human B lymphocyte cDNA library cloned into the pACT vector was used as prey for the two-hybrid screen. The bait and prey were sequentially transformed into yeast, and positive colonies were selected by growth on the minimal synthetic plates containing 3-aminotriazole lacking adenine, tryptophan, and leucine. These clones were further tested for β-galactosidase activities, and positive plasmids were isolated and sequenced. A cDNA encoding aa 1–256 of the PRDX3 protein was identified in the screen using the C-terminal portion of FANCG as bait (Fig. 1, A and B). A bait construct containing a specific missense mutation (G546R) identified in an FA-G patient was used to further probe this interaction (Nakanishi et al., 2001). Quantitative β-galactosidase assays demonstrate that wild-type PRDX3 interacts with the FANCG bait, and this activity is diminished when the mutant FANCG bait is used (Fig. 1 B).
Figure 1.
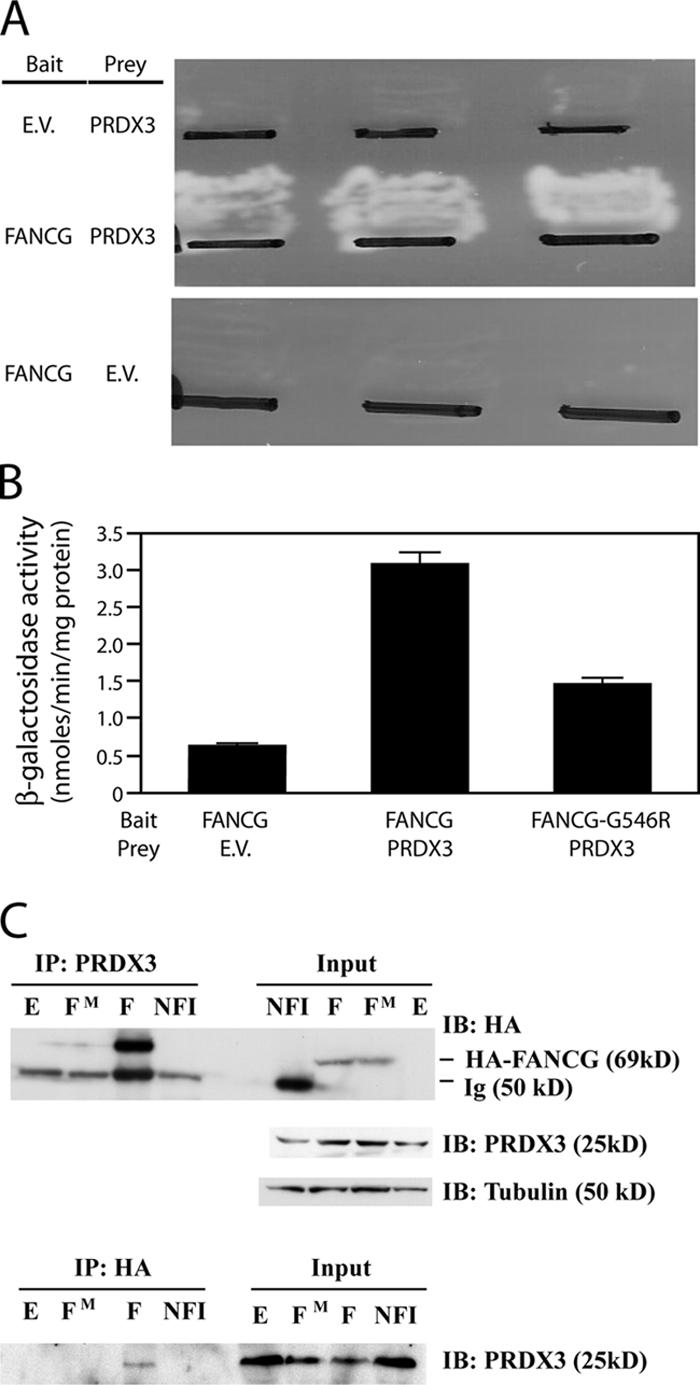
FANCG protein interacts with PRDX3. (A) Colonies of yeast strain PJ-69-4A transformed with the indicated bait and prey vectors were patched on selective synthetic medium plates lacking adenine, tryptophan, and leucine and grown for 2–3 d at 30°C. EV, empty vector. (B) β-galactosidase activity assays were measured on protein lysates from exponentially growing liquid yeast cultures containing the indicated plasmids. FANCG-G546R contains the FANCG cDNA with an introduced missense mutation in pAS1. Error bars represent the SEM of three independent determinations. (C) Total protein lysates from COS cells that were transfected with empty vector (E), HA-FANCG (F), HA-FANCG-G546R (FM), or HA-NF1 (NFI) constructs were analyzed. In the top panel, lysates were immunoprecipitated with PRDX3 C-terminal antibody (IP:PRDX3). 10% of the protein lysates used in the immunoprecipitation was electrophoresed on the right side of the blot (input). The membrane was serially probed with anti-HA, PRDX3, and tubulin antibodies. In the bottom panel, the same lysates were immunoprecipitated with the anti-HA antibody (IP:HA) and 10% of the lysate electrophoresed on the right (input). The blot was probed with the PRDX3 antibody.
To confirm the interaction of PRDX3 and FANCG in mammalian cells, we used a coimmunoprecipitation assay. The full-length wild-type and G546R mutant FANCG cDNAs were cloned into the pCDNA3 mammalian expression vector as a fusion with the HA epitope. An unrelated HA-tagged nuclear factor 1 (NF1) construct, pCH-HA-NF1, was used as a negative control (Mukhopadhyay et al., 2001). These expression constructs were transfected individually into COS-1 cells. 48 h after transfection, total cell lysates were precipitated with rabbit C-terminal PRDX3 antibody. The mutant and wild-type FANCG constructs were expressed equivalently when probed with the HA antibody (Fig. 1 C, input). Immunoprecipitated proteins were analyzed on SDS gel followed by immunoblotting with HA antibody (Fig. 1 C, top). The HA-FANCG protein is detected consistently with the endogenous PRDX3 protein interacting with the HA-FANCG protein. Similar to our two-hybrid result, there was little to no interaction between the endogenous PRDX3 protein and the HA-FANCG G546R mutant protein, and there was no evidence for interaction with an unrelated HA-NF1–tagged protein. This further demonstrates the specificity of the interaction between FANCG and PRDX3.
A reverse experiment was performed with the same lysates but with coimmunoprecipitation using the anti-HA epitope antibody. The immunoprecipitated proteins were visualized by Western blotting performed with PRDX3 antibody (Fig. 1 C, bottom). This antibody detects endogenous PRDX3 protein in the immunoprecipitate from cells transfected with wild-type but not G546R mutant HA-FANCG or HA-NF1. Therefore, by two-hybrid analysis and coimmunoprecipitation assays, the FANCG and PRDX3 proteins interact. This interaction is diminished in FANCG protein containing the patient-associated G546R mutation.
FANCG protein is localized to mitochondria
Previous studies have found that PRDX3 is exclusively a mitochondrial protein (Wonsey et al., 2002; Nonn et al., 2003). Therefore, we wanted to identify whether FANCG protein colocalized with PRDX3 within the mitochondria. The pDsRed2-Mito plasmid was used to identify mitochondria. We transfected the pCDNA3-HA FANCG and pDsRed2-Mito constructs into HeLa cells and used immunofluorescence to detect the localization of transfected proteins. C-terminal PRDX3 antibody was used to detect the endogenous PRDX3 protein. Inspection of asynchronous HeLa cells transfected with the HA-FANCG construct revealed cells with localization either in the nuclear or cytoplasmic compartments as previously reported (Mi and Kupfer 2005). Fig. 2 shows a deconvolution analysis of a representative cell with predominantly cytoplasmic staining of HA-FANCG. There was substantial colocalization of HA-FANCG with the mitochondrial marker (Fig. 2, A–C). As expected, PRDX3 colocalized with the mitochondrial marker (not depicted; see Fig. 4 A). Staining of both the HA-FANCG and endogenous PRDX3 demonstrates colocalization (Fig. 2, D–F). These immunofluorescent experiments suggest that both the FANCG and PRDX3 proteins are found within mitochondria.
Figure 2.

In vivo colocalization of FANCG and PRDX3 in mitochondria. (A–F) Fluorescent localization experiments were performed on HeLa cells transfected with the indicated plasmids, including pDsRed2-Mito (RFP-tagged mitochondrial marker; A–C) and HA-FANCG (A–F). Anti-HA monoclonal antibody and FITC-labeled secondary antibody were used to detect HA-FANCG. C-terminal PRDX3 polyclonal antibody and Texas red–labeled secondary antibody were used to detect the endogenous PRDX3. RFP was observed with the same red filter as used for Texas red. (G) HeLa cells transfected with the HA-FANCG wild type (left) or HA-FANCG G546R mutant (right) construct were fractionated into a nuclear fraction (N) and cytoplasmic fraction containing organelles (C/M). Mitochondria (M) were further purified. Lysates were electrophoresed and hybridized with anti-HA monoclonal antibody and the indicated polyclonal antibodies. Cytochrome c, tubulin, and lamin B represent mitochondrial, cytoplasmic, and nuclear markers, respectively. (H) Mitochondrial lysates were made from FA-G mutant (EUFA316) and corrected lymphoblasts and HeLa cells transiently transfected with HA-FANCG. The lysates were electrophoresed, transferred, and hybridized with FANCG and cytochrome c polyclonal antibodies.
Figure 4.

Mislocalization of PRDX3 and distorted mitochondrial structures in FA-G mutant fibroblasts. (A–C) Normal primary human fibroblasts (A), FA-G mutant (B), and FA-G–corrected (C) primary fibroblasts (PD352) were transfected with mitochondrial marker (pDsRed-Mito). The C-terminal PRDX3 antibody and FITC-labeled secondary antibody were used to detect the endogenous PRDX3 protein. Merge of the green and red images. Arrows (B) indicate the PRDX3 (green signals) that does not colocalize with the red mitochondrial marker in FA-G fibroblasts. (D and E) Transmission EM of FA-G mutant primary fibroblasts. Mitochondria from FA-G fibroblasts demonstrate frequent elongation and irregular shapes with acute and right angle branching (arrows). Some mitochondria have a z-shaped focal constriction of their diameters and bleblike extensions. (F) Transmission EM of FA-G–corrected primary fibroblasts with occasional mitochondria that have irregular outlines and branching pattern (top two arrowheads) and more normal structure (bottom arrowhead). Bars (D), 600 nm; (E and F) 400 nm.
To confirm the localization of FANCG in the mitochondrial compartment, we performed immunoblotting of lysates from fractionated cells. After transfection with either the HA-FANCG wild-type or G546R mutant construct, HeLa cells were first fractionated into a nuclear and cytoplasmic lysate. The cytoplasmic lysate contains organelles, including mitochondria. Mitochondria were then further isolated from this extract. Immunoblots of the fractionated lysates from cells transfected with the wild-type construct are shown in the left panel in Fig. 2 G. Immunoblotting with appropriate antibodies demonstrates the expected pattern with cytoplasmic/mitochondrial lysates containing both tubulin and cytochrome c and the purified mitochondrial lysate containing only cytochrome c. Consistent with our immunofluorescence experiments, antibody to the HA epitope detects a single protein band with the expected size of HA-FANCG in both the mitochondrial and nuclear lysates. The pattern of localization of the G546R mutant protein is similar to the wild-type HA-FANCG (Fig. 2 G, right). Thus, the lack of coimmunoprecipitation of PRDX3 and mutant FANCG is not related to mislocalization of the mutant protein and is most likely caused by a direct impact of the mutation on FANCG–PRDX3 interaction.
Because transiently transfected proteins may be localized differently in comparison with the endogenous protein, we also isolated mitochondrial extracts from the FA-G mutant and stably corrected lymphoblasts and also from HeLa cells transiently transfected with HA-FANCG as a control (Fig. 2 H). An antibody to a FANCG peptide detects the FANCG band in the mitochondrial lysate of HeLa cells and FA-G–corrected cells but not in the FA-G mutant cells (Fig. 2 H). Thus, both the immunofluorescent and Western blot assays are consistent with a portion of FANCG protein localizing to the mitochondria.
A low molecular weight form of PRDX3 protein (PRDX3-S) is found in FA-G mutant lymphoblasts and in FANCG-corrected cells under oxidative stress
To understand the physiological relevance of the interaction between FANCG and PRDX3 in mitochondria, we determined the expression of PRDX3 in a FA-G mutant lymphoblast cell line compared with FA-G–corrected cells. We isolated total protein from both the mutant and corrected FA-G lymphoblasts, and equal amounts of protein were analyzed by Western blotting with C-terminal PRDX3 antibody (Fig. 3 A). In addition to the expected 25-kD protein, we found an additional smaller molecular mass band (named PRDX3-S) of ∼16 kD in the lysates from FA-G mutant cells (Fig. 3 A, lanes 3 and 4). This band was not seen in the FANCG-corrected FA-G cells (Fig. 3 A, lanes 1 and 2). To determine whether the PRDX3-S species is also a mitochondrial protein, we isolated the mitochondria from the mutant and corrected lymphoblasts. Similar to the whole cell lysates, PRDX3-S is found in the mitochondrial extracts of untreated FA-G mutant cells (Fig. 3 B, lane 4) but not the corrected cells (Fig. 3 B, lane 1). Previous investigators have reported that FA cells are sensitive to oxidative stress (for review see Pagano and Yousoufian, 2003). We treated the mutant and corrected FA-G cells with 100 μM and 1 mM of H2O2 for 90 min and isolated mitochondrial lysates for Western blotting. Interestingly, the FA-G–corrected cells exposed to 1 mM H2O2 demonstrate a band of the same mobility as the PRDX3-S band seen in unexposed FA-G mutant cells (Fig. 3 B, lanes 3 and 4). These results suggest that the PRDX3-S band may be generated by ongoing oxidative stress in the FA-G mutant lymphoblasts.
Figure 3.

Proteolytic cleavage of PRDX3 by a calpainlike cysteine protease in the FA-G mutant lymphoblasts and in FA-G–corrected cells after oxidative stress. (A) Total protein was isolated from FA-G mutant (EUFA316) and corrected lymphoblasts and immunoblotted with C-terminal PRDX3 antibody. The membrane was stripped and probed with tubulin antibody (bottom). Protein size markers are indicated on the right. (B) Mitochondrial proteins were isolated from corrected and mutant FA-G lymphoblasts (EUFA316) and hybridized with C-terminal PRDX3 antibody. Cells were treated with the indicated amounts of H2O2 for 90 min. The filter was stripped and probed with cytochrome c antibody (bottom). M represents the lane with protein size markers. (C) FA-G (mutant) lymphoblasts were treated without (lane 1) and with (lane 2) ALLN (cysteine protease inhibitor) for 4–5 h. Mitochondrial proteins were then isolated, and Western blotting was performed with the C-terminal PRDX3 antibody. The same membrane was stripped and probed with cytochrome c antibody (bottom). (D) A constant amount of in vitro–transcribed/translated 35S-labeled PRDX3 protein incubated without (lane 1) and with recombinant calpain II in increasing amounts (0.25–1.5 μg; lanes 2–5) in the presence of calpain buffer, which contains 20 mM CaCl2. In lane 6, the same amount of PRDX3 was incubated with 1.5 μg calpain II in the presence of 20 mM EGTA. (E) A constant amount of in vitro–transcribed/translated 35S-labeled PRDX3 incubated without (lane 1) and with recombinant calpain II (lane 2). 35S-labeled PRDX3 incubated with mitochondrial extract from FA-G–corrected lymphoblasts (lane 3), FA-G mutant lymphoblasts (lane 4), and FA-G–corrected lymphoblasts treated with 1 mm H2O2 for 90 min before lysis. (F) A constant amount of in vitro–transcribed/translated 35S-labeled PRDX3 was incubated with 10 μl calpain II (lanes 2–6) in the presence of in vitro–translated 35S-labeled FANCG in increasing amounts (10, 20, 40, and 50 μl; lanes 3–6).
Calpainlike cysteine protease–mediated cleavage of PRDX3 in the FA-G lymphoblasts
Several proteases are activated by oxidative stress, including members of the cysteine protease family. To determine whether PRDX3-S is generated by the cleavage of full-length PRDX3 with a cysteine protease, we added a cysteine protease inhibitor, ALLN, to the medium 4–5 h before harvesting mitochondria from the FA-G mutant lymphoblasts. Immunoblotting demonstrates that the ALLN treatment blocks generation of the PRDX3-S band (Fig. 3 C). This experiment suggests that a cysteine residue in PRDX3 is cleaved by a calpainlike cysteine protease active in the FA-G mutant cells. We tested this hypothesis directly in vitro. We produced 35S-labeled PRDX3 protein using a wheat germ in vitro transcription/translation assay and incubated the protein with recombinant calpain II enzyme. As demonstrated in Fig. 3 D, the addition of increasing amounts of calpain II (0.25–1.5 μg; lanes 2–5) to the in vitro–translated protein generates a band of the same molecular weight as PRDX3-S. Calpain II cleavage activity requires high calcium (20 mM), and the addition of 20 mM EGTA blocks the production of PRDX3-S by 1.5 μg calpain II (Fig. 3 D, lane 6). These data are consistent with the endogenous PRDX3-S band seen in Western blots being a cleavage product of full-length PRDX3.
The finding of PRDX3-S in Western blots of lysates from isolated mitochondria (Fig. 3 B) suggests that the protease activity is mitochondrial in origin. To test this directly, we incubated in vitro–translated 35S-labeled PRDX3 protein with mitochondrial extracts isolated from untreated FA-G mutant and FA-G–corrected cells before and after exposure to 1 mM H2O2. The cleavage product PRDX3-S was generated by incubation with the untreated FA-G mutant mitochondrial extract, but only from FA-G–corrected cells after exposure to oxidative stress (Fig. 3 E). The addition of in vitro–translated FANCG protein protects PRDX3 from calpain-mediated cleavage (Fig. 3 F). Collectively, these results suggest that PRDX3 is cleaved by a mitochondrial calpainlike cysteine protease in vivo to produce PRDX3-S. This protease activity is continuously active in FA-G mutant cells and can be induced in FA-G–corrected cells by acute exposure to H2O2.
Mislocalization of PRDX3 and distorted mitochondrial structure in FA-G fibroblasts
PRDX3 has been reported as an exclusively mitochondrial protein that reduces the level of H2O2 continuously generated in mitochondria (Araki et al., 1999; Wonsey et al., 2002; Nonn et al., 2003). To determine whether the localization of PRDX3 is altered in cells from FA-G patients, we performed immunofluorescence of PRDX3 in FA-G mutant (PD352-F1°) and corrected (PD352-F2) primary fibroblasts and normal human primary fibroblasts. The endogenous PRDX3 was hybridized with C-terminal PRDX3 antibody and was detected by FITC-labeled anti–rabbit IgG secondary antibody. Localization of the FITC-stained PRDX3 with pDsRed2-Mito was studied by deconvolution microscopy. As expected from prior studies (Wonsey et al., 2002; Nonn et al., 2003), in normal human fibroblasts, there is nearly 100% colocalization of the mitochondrial and PRDX3 signals (Fig. 4 A). In contrast, in FA-G mutant cells, most of the PRDX3 (FITC signal) does not colocalize with the pDsRed2-Mito RFP marker (Fig. 4 B). Interestingly, in the FA- G–corrected cells, there is an intermediate pattern, with the amount of colocalization (yellow signal) intermediate between the FA-G mutant and normal fibroblasts (Fig. 4 C).
These immunolocalization experiments suggested that there might be a distortion of mitochondrial structures in FA-G cells, which is only partially restored when the FANCG cDNA is reintroduced (FA-G–corrected cells). To study mitochondrial structure further, we performed EM on the same cell lines used for the immunolocalization studies. As shown in Fig. 4 (D and E), the mitochondrial structures are quite abnormal in FA-G cells. The FA-G mutant cells possess mitochondria with markedly irregular outlines and sizes. There was noticeable branching, irregular angulation, and elongated shapes, including mitochondria with acute and right angle outlines, as well as some with a z shape (Fig. 4 E, arrowheads). There were also areas with constriction of the mitochondria, with decreased diameters and elongation of the mitochondria with bleblike extensions. The crista and internal structures of the mitochondria from both FA-G mutant and corrected cells did not show alterations. Mitochondria from the FA-G–corrected cells (Fig. 4 F, top arrowheads) had occasional mitochondria with irregular outlines and less noticeable branching. The majority of the mitochondria from corrected cells had more typical mitochondrial morphology (Fig. 4 F, bottom arrowhead). Overall, the total number of abnormally shaped mitochondria in FA-G mutant fibroblast cells (Fig. 4, D and E) is higher compared with the corrected cells (Fig. 4 F). These immunofluorescent and EM structural experiments suggest that there are defects in both PRDX3 localization and mitochondrial structure in cells from FA-G patients, implicating defective mitochondrial function in FA patients. The lack of complete rescue in the FA-G cells after correction with the FANCG cDNA may result from inefficient turnover of the abnormal mitochondria already formed in FA-G mutant cells.
PRDX3-S is also found in lymphoblasts from patients with FA subtypes C and A
Several lines of evidence argue that the proteins encoded by the different FA genes function coordinately. This includes the similar clinical phenotypes of patients from the different subtypes as well as biochemical experiments demonstrating that many FA proteins form a complex required for FANCD2 monoubiquitination. Therefore, we tested whether the expression of PRDX3 protein is altered in FA-C and -A mutants and in corrected lymphoblasts, as these represent the most common FA subtypes. Isolation of mitochondrial protein and immunoblotting with C-terminal anti-PRDX3 antibody is shown in Fig. 5 A. PRDX3-S was found in FA-A and -C mutant lymphoblasts (Fig. 5 A, lanes 2 and 4) but not in the corresponding corrected lymphoblasts (Fig. 5 A, lanes 1 and 2). This result suggests that the loss of any one of these three FA proteins results in the cleavage of PRDX3 in untreated cells.
Figure 5.

PRDX3 cleavage product is present and peroxidase activity is diminished in lymphoblasts from FA subgroups C, A, and G. (A) Mitochondrial proteins were isolated from FA-A (HSC72) and -C (HSC536) mutant and corrected lymphoblasts and hybridized with C-terminal PRDX3 antibody. The blot was stripped and reprobed with cytochrome c antibody. (B) Mitochondrial lysates from HeLa cells transiently transfected with HA-FANCG (lane 1), FA-G–corrected (lane 2), FA-A mutant (lane 3), FA-C mutant (lane 4), and FA-G mutant (lane 5) lymphoblasts were probed with FANCG antibody. The membrane was stripped and reprobed with cytochrome c antibody. (C and D) Peroxidase activity of mitochondrial extracts isolated from FA mutant and corrected cells. Recombinant PRDX1 and a buffer sample alone were used as controls. All samples contained equal amounts of TR, Trx, NADPH, and H2O2. Each bar represents the mean initial activity of the mitochondrial extract isolated from three different aliquots of the same cell line. Error bars represent the SEM.
The aforementioned result could be an indirect effect secondary to diminished FANCG protein in these other subtypes or a direct effect of loss of the FANCA or FANCC function. Therefore, we determined endogenous FANCG expression in mitochondrial extracts of FA-A, -C, and -G mutant cells (Fig. 5 D). As expected, FANCG protein is absent from FA-G mutant cells. FANCG is present in the FA-C mitochondrial extract and is somewhat diminished in the FA-A extract.
Previous studies demonstrated diminished FANCG levels in FA-A cells (Waisfisz et al., 1999; Medhurst et al., 2006). The presence of FANCG protein in the mitochondria from FA-C cells suggests that PRDX3 cleavage is not simply caused by the loss of FANCG and that multiple FA proteins are required for the maintenance of PRDX3 integrity. This model is consistent with the fact that FA cells from multiple subtypes demonstrate increased sensitivity to oxidative stress (for review see Pagano and Youssoufian, 2003).
Mitochondrial peroxidase enzymatic activity is diminished in lymphoblasts from FA patient types G, C, and A
PRDX3 reduces the H2O2 level in mitochondria by catalyzing the conversion of H2O2 into H2O using Trx reductase (TR) and Trx as electron donors (Kang et al., 1998). An important question is whether the cleavage and mislocalization of PRDX3 observed in the previous experiments is associated with alteration in mitochondrial peroxidase activity. We performed Trx-dependent peroxidase activity assays in mitochondrial lysates from mutant and corrected FA cells. We used recombinant PRDX1 as a positive control for these assays. In Fig. 5 B, the addition of 0.6 μg of recombinant PRDX1 shows a ninefold higher peroxidase activity than the negative control, which contains all of the reagents except the enzyme. 0.6 μg mitochondrial lysate from the FA-G–corrected cells supplies a similar level of peroxidase activity. The mitochondrial extract from FA-G mutant cells showed almost a sevenfold lower peroxidase activity than the FA-G–corrected cells. We excluded the possibility of an inhibitor of peroxidase activity in the mutant cells by assaying a 1:1 mixture of lysates from FA-G mutant and corrected cells. This activity was comparable with the FA-G–corrected lysates alone.
Similar to the finding of the PRDX3-S cleavage product in FA-A and -C cells, mitochondrial extracts from FA-A and -C mutant (but not corrected) lymphoblasts also had decreased mitochondrial peroxidase activity similar to the FA-G mutant cells (Fig. 5 C). Given that PRDX3 is the major Trx-dependent mitochondrial peroxidase, these data suggest that the PRDX3-mediated peroxidase activity is diminished in all three FA subtype mutant cells compared with corrected cells. Thus, we see both the cleavage of PRDX3 and diminished mitochondrial peroxidase activity in multiple FA subtypes.
Overexpression of PRDX3 suppresses the sensitivity of FA-G cells to H2O2
Our data are consistent with the decreased mitochondrial peroxidase activity of PRDX3 in cells from FA-G, -A, and -C patients as the basis of increased sensitivity to ROS. To test this hypothesis, we determined whether the transient overexpression of PRDX3 in FA-G mutant cells increases resistance to treatment with H2O2. FA-G mutant and corrected fibroblasts (PD352-F1° and PD352-F2) were transfected with either the pEF6/His6 empty vector or PRDX3 expression construct pEF6-PRDX3 using a protocol that transfects ∼80% of the cells. 48 h after transfection, cells were exposed to varying concentrations of H2O2 for 90 min, and the number of viable cells was determined using the 3-(4,5-dimethyl-thiazol-2yl)-2,5-diphenyl-tetrazolium bromide (MTT) colorimetric assay. As previously reported (Park et al., 2004), FA-G mutant cells have increased sensitivity to H2O2 compared with FA-G–corrected cells (Fig. 6). Transient transfection of PRDX3 restores the survival of FA-G mutant cells to similar levels as the FA-G–corrected cells transfected with the empty vector.
Figure 6.
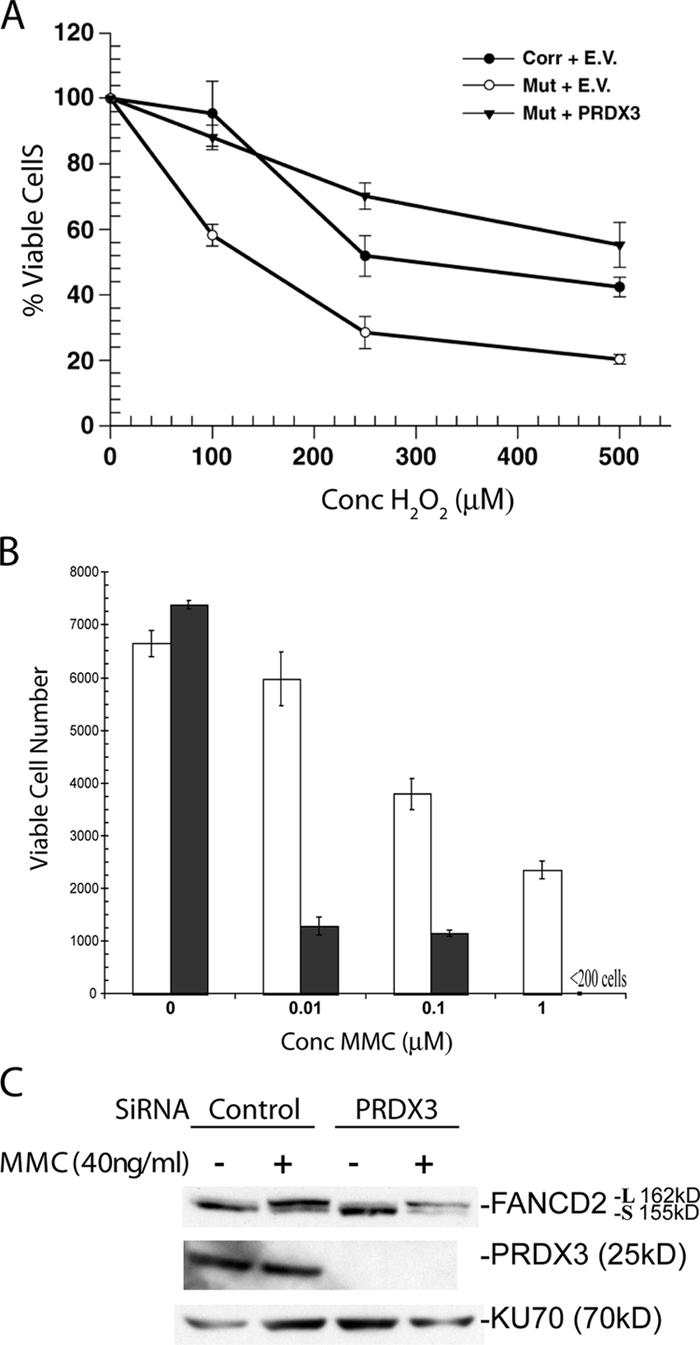
PRDX3 expression impacts sensitivity to oxidative stress and MMC independent of FANCD2 monoubiquitination. (A) FA-G mutant (PD352-F1°) and corrected (PD352-F2) fibroblasts were transfected with the pEF6/His6 vector (empty vector [EV]) or pEF6-PRDX3 (PRDX3). 24 h later, cells were exposed to H2O2 for 90 min, and their viability was assessed with an MTT assay. A representative experiment is shown with the line going through the mean and error bars showing the minimum and maximum value for each cell line. (B) HeLa cells were transfected with control siRNA (white bars) or PRDX3 siRNA (black bars). 24 h later, cells were exposed to MMC for an additional 24 h, and the number of viable cells was determined by MTT assay. (C) HeLa cells were transfected with control or PRDX3 siRNA and subsequently treated with 40 ng/ml MMC for 24 h. Protein lysates were made, electrophoresed, and immunoblotted with antibodies to PRDX3, FAND2, and KU70. The long (L) and short (S) FANCD2 species are noted.
Increase in MMC sensitivity but maintenance of FANCD2 monoubiquitination in cells with diminished PRDX3 expression
We next determined whether decreasing PRDX3 expression in cells with a normal FA pathway would alter the response to MMC. HeLa cells were transfected with either a control siRNA or PRDX3 siRNA (as previously developed by Chang et al., 2004) to reduce PRDX3 expression. Cells with diminished PRDX3 expression showed dose-dependent increases in cell death after 24 h of MMC treatment (Fig. 6 B), which is consistent with a previous study of the impact of Trx levels on MMC sensitivity (Ruppitsch et al., 1998). The increase in MMC sensitivity occurred despite the maintenance of FANCD2 monoubiquitination in the PRDX3 knockdown cells (Fig. 6 C).
Discussion
Localization of FANCG in the mitochondria
FANC proteins, including G, A, and C, have been reported to be present both in the nucleus and cytoplasm. However, the methods used in previous studies did not distinguish between cytoplasmic and mitochondrial proteins (Thomashevski et al., 2004; Mi and Kupfer 2005; Medhurst et al., 2006). In this study, we present data based on two-hybrid interaction, coimmunoprecipitation, and immunolocalization that the FANCG protein binds and colocalizes with PRDX3 in mitochondria. The interaction of these two proteins is lost in the patient-associated G546R-FANCG mutant protein. We specifically demonstrated that a portion of FANCG localizes with mitochondria in HeLa and FA-G–corrected cells. These results suggest that the FANCG protein can translocate into mitochondria and interact with PRDX3.
Proteolytic cleavage of PRDX3 in the FA lymphoblasts
The finding of FANCG interacting with PRDX3 is further strengthened by the characterization of defects in the structure and function of PRDX3 in FA cells from subtypes A, C, and G. PRDX3 is one of the major proteins that controls the level of ROS in the mitochondria (Nonn et al., 2003), and the level of ROS impacts proliferation and apoptosis. Therefore, we focused our experiments on determining whether cells from FA patients have intrinsic defects in regulation of the PRDX3 protein and enzymatic activity. We find a proteolytic cleavage product, PRDX3-S, in FA cells that is also seen in corrected cells exposed to high levels of H2O2. Importantly, the PRDX3-S product is found in three different FA subtypes (G, C, and A), suggesting that multiple FA proteins are required to maintain the integrity of PRDX3 in the mitochondria.
We propose that the cleavage of PRDX3 in FA mutant cells or normal cells exposed to oxidative stress is caused by a calpainlike cysteine protease based on inhibition by ALLN in vivo and by in vitro cleavage assays using mitochondrial extracts from FA cells and recombinant calpains. Calpains, a family of Ca2+-activated neutral cysteine proteases, are involved in cell death in a variety of models. For example, p35 (a neuron-specific activator) is cleaved by calpain into p25, which accumulates in the brains of Alzheimer's patients (Lee et al., 2000). Although there are some reports of mitochondrial calpains, they are not well characterized (Chen et al., 2002; Daniel et al., 2003). Oxidative stress can release Ca2+ from the mitochondrial Ca2+ pool (Pariente et al., 2001), which may then activate mitochondrial calpains. Our results suggest that this activation occurs constitutively in FA cells. Further research to characterize mitochondrial calpains and to understand their activation by oxidative stress is needed.
Mitochondrial peroxidase activity is diminished in FA cells
PRDX3 mediates the level of ROS through its peroxidase activity. The endogenous level of mitochondrial peroxidase activity is substantially reduced in FA-G, -A, and -C mutant cells, suggesting that a basic function of FA proteins is to maintain the integrity and biochemical activity of PRDX3. Consistent with this model, the transient overexpression of PRDX3 restores the resistance of FA-G cells to acute oxidative stress. The loss of peroxidase activity in mitochondria from FA cells may result in ongoing oxidative stress with an increase in ROS and ROS-mediated DNA damage (Chang et al., 2004).
The mechanism by which the FA proteins impact the structure and function of PRDX3 is not clear. Based on our finding that the FANCG protein can bind PRDX3 protein directly, can prevent PRDX3 cleavage in an in vitro assay, and is localized to the mitochondria, FANCG may have a direct protective effect. This binding is lost in the G546R mutant protein even though the mutant protein is still present in mitochondria. Given the relative cellular abundance of PRDX3 compared with FA proteins, it is possible that the FA proteins play a more transient role in the folding of PRDX3 or its proper translocation into the inner mitochondrial membrane. Further research is required to discern this protective mechanism.
Collectively, our data suggest that FA proteins have important roles outside the nucleus in maintaining the structure and peroxidase activity of the mitochondrial PRDX3 protein. This is similar to the finding that the p53 protein translocates to both the nucleus and mitochondria after DNA damage (Marchenko et al., 2000; Zhao et al., 2005). Others have also suggested the importance of PRDX3 in modulating levels of ROS and apoptosis in non-FA cells (Nonn et al., 2003; Chang et al., 2004). We propose that the loss of PRDX3 activity results in a negative feedback loop (Fig. 7). With decreased peroxidase activity, ROS can build up within mitochondria, triggering apoptosis as well as further activating calpainlike proteases to further cleave PRDX3, which is consistent with the constitutive PRDX3 cleavage activity demonstrated in mitochondrial extracts of FA-G cells. Apart from cleavage, ROS can also directly interfere with PRDX3 activity by overoxidation of the active site cysteine to cysteine sulfinic acid. This form of inactivation may be particularly important for PRDX3 because the rate of regeneration of active PRDX3 by sulfinylation is slower than for PRDX1 and PRDX2 (Woo et al., 2003).
Figure 7.

Schematic model of the regulation of PRDX3 activity and the impact of decreased PRDX3 activity on oxidative stress–induced apoptosis and DNA damage in FA cells.
This model suggests that cells from FA patients would accumulate mitochondrial lesions from constitutive oxidative stress and may result in the distorted mitochondrial structures seen in EM experiments. In addition, ROS can leak out of the mitochondria and may cause nuclear DNA damage and stimulate apoptosis or the senescent phenotype described in bone marrow precursors of Fancc −/− mice (Zhang et al., 2005). The reduction in PRDX3 expression in non-FA cells results in increased sensitivity to MMC with an intact FANCD2 monoubiquitination pathway. In FA cells, the increased ROS-mediated DNA damage would be coupled with impaired DNA repair processes and could also increase genomic instability and cancer susceptibility. This model is consistent with the recent finding in Saccharomyces cerevisiae that the loss of PRDX function increases the mutator phenotype of strains deficient in DNA repair functions (Huang and Kolodner 2005). Thus, minimizing the production of ROS, inhibiting calpainlike protease activity, or using treatments that activate other ROS scavenger pathways may achieve improvements in the FA phenotype.
Materials and methods
Yeast two-hybrid screen
The N-terminal (aa 135–1,093) and C-terminal (aa 1,044–2,000) regions of FANCG cDNA were cloned into pAS2-1 bait vector (CLONTECH Laboratories, Inc.). A human B lymphocyte cDNA library fused with the GAL4 activation domain (gift from S. Elledge, Harvard University, Boston, MA) was screened. A total of 800,000 transformants was screened in the yeast strain PJ-69-4A (Gietz et al., 1997). Colonies capable of growth on minimal synthetic plates containing 3-aminotriazole and lacking adenine, tryptophan, and leucine were selected. The liquid β-galactosidase activity assay was performed (Rose et al., 1981) to quantify the interaction of specific bait-prey combinations.
Plasmids
Full-length FANCG cDNA was subcloned into the HindIII and NotI sites of pcDNA3 to encode an N-terminal HA epitope-tagged FANCG protein. PRDX3 cDNA was cloned into the BamH1 and Xba1 sites of the pEF6/His6 mammalian expression vector (Invitrogen). pCH-HA-NF1 has been previously described (Mukhopadhyay et al., 2001). The construct pDsRed2-Mito encoding RFP fused to the mitochondrial targeting sequence from subunit VII of human cytochrome c oxidase was purchased from BD Biosciences.
Cell culture
HeLa and COS-1 cells were grown in DME and 10% FBS (American Type Culture Collection). The mutant and corrected FA-A (HSC72) and -C (HSC536) lymphoblasts were obtained from G. Bagby (Oregon Health Sciences University, Portland, OR) and H. Joenje (VU University Medical Center, Amsterdam, Netherlands), EUFA316 (FA-G corrected and mutant) cells were obtained from A. D'Andrea (Dana Farber Cancer Institute, Boston, MA), and PD352 cells were obtained from the Fanconi Anemia Research Fund. Normal human fibroblasts were obtained from the American Type Culture Collection (cell line GM08398). All FA cells were grown in RPMI and 15% FBS except that the PD352-F fibroblasts were grown in α-MEM and 15% FBS. All cells were grown in a 37°C incubator with 5% CO2. Transfections were performed with Effectene (QIAGEN) according to the manufacturer's protocol.
Immunoblotting
Polyclonal rabbit antibodies to PRDX3 were generated against peptides from N-terminal (GEFKELSLDDFKGKY) and C-terminal (SPTASKEYFEKYHQ) regions of mouse PRDX3 (Zymed Laboratories). Rabbit polyclonal antibody against a FANCG peptide was obtained from S.-H. Lee (Indiana University School of Medicine, Indianapolis, IN; Park et al., 2004). Sources of commercial monoclonal antibodies were as follows: anti-HA (HA.11) antibody (Covance), cytochrome c antibody (BD Biosciences), α-tubulin (Oncogene Research Products), and FANCD2 (Novus).
Cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EGTA, 1 mM PMSF, 1 mM sodium orthovanadate, 10 mM sodium fluoride, and Complete Protease Inhibitor Cocktail tablet [1 tablet/10 ml of buffer; Roche]). Protein concentration was determined by detergent-compatible reagent (Bio-Rad Laboratories). Samples were boiled with sample buffer (125 mM Tris-HCl, pH 6.8, 86 mM 2-mercaptoethanol, 4% SDS, 10% glycerol [vol/vol], and 0.2 mg/ml bromophenol blue) for 5 min and were subjected to 10–13% SDS-PAGE. After electrophoresis, proteins were transferred to polyvinylidene difluoride. Immobilon-P membrane (Millipore) using a submerged transfer apparatus (Bio-Rad Laboratories) was filled with 25 mM Tris base, 200 mM glycine, and 20% methanol. After blocking with 5% nonfat dried milk in TBS-T (200 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween 20), the membrane was incubated with the primary antibody diluted in TBS-T with 5% milk (1:1,000 dilution for both the HA and PRDX3 antibody). Other antibodies were used according to the manufacturer's recommendations. Appropriate HRP-linked secondary antibodies were used for detection by ECL (GE Healthcare).
Coimmunoprecipitation
Cells were lysed with RIPA buffer, and ∼500 μg of lysate was incubated with 4–5 μl of antibody (HA) for 30 min on ice. 20 μl of protein A plus/protein G agarose beads (Oncogene Research Products) was added and incubated overnight at 4°C with constant shaking. Beads were washed with either PBS or RIPA three to four times and boiled with the sample buffer for 5 min followed by 10% SDS-PAGE and immunoblotting as described in the previous section.
Immunofluorescent microscopy
Cells were plated onto poly-l-lysine–coated coverslips in 60-mm dishes and transfected the next day with the indicated constructs using Effectene. 48 h later, the cells were fixed for 30 min at RT in a 4% PFA-buffered solution and permeabilized with 0.5% Triton X-100 for 10 min at RT. Staining with either the polyclonal PRDX3 C-terminal antibody and/or monoclonal HA antibody at a 1:300 dilution was performed for 1 h at RT in TBS-T containing 5% nonfat dairy milk. The species-specific fluorescein (FITC)- or Texas red–conjugated secondary antibodies (Abcam) at a 1:400 dilution were applied for 1 h at RT followed by counterstaining with DAPI and mounting together with Vectashield mounting medium (Vector Laboratories) for 10–15 min at RT in the dark. The cells were analyzed on a microscope (Axiovert 200M; Carl Zeiss MicroImaging, Inc.) equipped with a digital CCD camera (C4742-95-12ERG; Hamamatsu) and the Openlab 3.1.5 and Volocity 2.1d19 software packages (Improvision) using a plan-Apochromat 63× NA 1.40 oil objective (Carl Zeiss MicroImaging, Inc.).
EM studies for mitochondrial structure of the FA cells
Primary fibroblasts of FA-G mutant (PD352-F1°) and corrected (PD352-F2) cells at approximately passage 8 were harvested and washed three to four times with 1× PBS. The cell pellets were fixed in 3% glutaraldehyde and underwent graded dehydration and infiltration with plastic to create plastic tissue blocks. Semithin sections stained with toluidine blue were created to determine the suitability for ultrastructural examination. Thin sections were prepared, placed on copper grids, and stained with uranyl acetate and lead citrate. The section was then examined with transmission EM to assess the ultrastructure of the mitochondria using an electron microscope (model 1210; JEOL).
Preparation of mitochondrial protein
Cells were harvested, washed once with ice-cold PBS, pH 7.4, resuspended, and incubated in a hypotonic buffer (10 mM Tris, 10 mM NaCl, 3 mM MgCl2, 1 mM EDTA, and 1 mM EGTA) for 30 min on ice. Cells were lysed by 15 strokes with a tight-fitting Dounce homogenizer, and nuclei and unbroken cells were pelleted by centrifugation for 15 min at 600 g and 4°C. The supernatant was centrifuged for 20 min at 12,000 g and 4°C to pellet mitochondria. The pellet was resuspended in lysis buffer (100 mM Tris-HCl, pH 7.4, and 0.1% Triton X-100 supplemented with protease and phosphatase inhibitors as described for RIPA buffer) and broken by sonification for 15 s. Membranes were removed by centrifugation at 12,000 g for 15 min at 4°C (Nonn et al., 2003). Mitochondrial protein was quantified using protein reagent (Bio-Rad Laboratories).
Peroxidase assay
NADPH, recombinant human PRDX1, human Trx, and TR from rat liver were purchased from Sigma-Aldrich. The initial rate of NADPH oxidation was monitored spectrophotometrically (Ultraspec2100 Pro; GE Healthcare) at A340. The 150-μl reaction mixture contained 50 mM Hepes-NaOH, pH 7.0, 250 μM NADPH, 46 nM TR, 2.2 μM Trx, and 0.6 μg of recombinant human PRDX1 or mitochondrial extract of the indicated cells. The reaction was initiated by the addition of 0.5 mM H2O2 (Kang et al., 1998).
In vitro translation and calpain digestion
35S-labeled PRDX3 protein was produced by in vitro transcription-translation using the TNT-coupled wheat germ extract lysate (Promega). 10 μl of labeled PRDX3 protein was incubated with increasing amounts (0.25–1.5 μg) of rat recombinant calpain II (Calbiochem) or mitochondrial lysates and the activation buffer (20 mM CaCl2) for 10–15 min at 37°C. Calpain was diluted with assay buffer (50 mM Tris-HCl, pH 8.0). After digestion, the proteins were separated on a 10% SDS polyacrylamide gel and transferred to polyvinylidene difluoride membrane to decrease background. The membranes were exposed to phosphorimager screen (Molecular Dynamics) for quantitative analysis as well as x-ray film (Kodak).
H2O2 sensitivity assay
Approximately 104 FA-G primary fibroblasts (mutant, PD352F1°, and corrected PD352-F2) were split into 96-well plates. 24 h later, cells were transfected with the indicated construct using Effectene. 48 h after transfection, cells were treated with H202 for 90 min. Cells were washed several times with PBS, and 15 μl MTT (5 mg/ml in PBS) was added to each well (Denizot and Lang 1986). Plates were incubated for 4 h, 150 μl DMSO was added to each well, and the optical density at 550 nm was determined with a plate reader. Varying numbers of each cell line were plated, and, 24 h later, the MTT assay was performed. These data were used for log-linear regression of cell number versus OD550 to determine the number of viable cells remaining after treatment with H2O2.
MMC sensitivity assay
The PRDX3 and control siRNAs (as described in Chang et al., 2004) were introduced into HeLa cells by transfection with the use of a Nucleofactor instrument (Amaxa Biosystems). 24 h later, cells were treated with the indicated dose of MMC for 24 h. MTT assay was performed as described in the previous paragraph for cell viability assay. For immunoblotting, cells were lysed with RIPA buffer and separated by 8% SDS-PAGE followed by immunoblotting with FANCD2 antibody (Novus Biologicals).
Online supplemental material
Fig. S1 shows the mislocalization of PRDX3 in FA-G mutant fibroblasts. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200607061/DC1.
Supplementary Material
Acknowledgments
We thank Steve Elledge for the human cDNA library; Maureen E. Hoatlin and Barbara A. Cox of the Fanconi Anemia Research Fund; Allan D'Andrea, Hans Joenje, and Grover Bagby for FA cell lines; and Suk-Hee Lee for FANCG antibody. We acknowledge Heather Walker and Linna Zhang for technical help and Kathyrn Poland for the β-galactosidase assays. Deborah Shardy, Debananda Pati, and James Huang provided advice on the manuscript.
This work was supported by National Institutes of Health grant HL52138.
S.S. Mukhopadhyay's present address is The University of Texas MD Anderson Cancer Center, Houston, TX 77030.
H. Youssoufian's present address is ImClone Systems, Inc., New York, NY 10014.
Abbreviations used in this paper: FA, Fanconi anemia; MMC, mitomycin C; MTT, 3-(4,5-dimethyl-thiazol-2yl)-2,5-diphenyl-tetrazolium bromide; NF1, nuclear factor 1; PRDX, peroxiredoxin; RIPA, radioimmunoprecipitation assay; ROS, reactive oxygen species; TR, Trx reductase; Trx, thioredoxin.
References
- Ahmad, S.I., F. Hanaoka, and S.H. Kirk. 2002. Molecular biology of Fanconi anaemia–an old problem, a new insight. Bioessays. 24:439–448. [DOI] [PubMed] [Google Scholar]
- Alter, B.P. 1995. Hematologic abnormalities in Fanconi anemia. Blood. 85:1148–1149. [PubMed] [Google Scholar]
- Araki, M., H. Nanri, K. Ejima, Y. Murasato, T. Fujiwara, Y. Nakashima, and M. Ikeda. 1999. Antioxidant function of the mitochondrial protein SP-22 in the cardiovascular system. J. Biol. Chem. 274:2271–2278. [DOI] [PubMed] [Google Scholar]
- Blom, E., H.J. van de Vrugt, Y. de Vries, J.P. de Winter, F. Arwert, and H. Joenje. 2004. Multiple TPR motifs characterize the Fanconi anemia FANCG protein. DNA Repair (Amst.). 3:77–84. [DOI] [PubMed] [Google Scholar]
- Butturini, A., R.P. Gale, P.C. Verlander, B. Adler-Brecher, A.P. Gillio, and A.D. Auerbach. 1994. Hematologic abnormalities in Fanconi anemia: an International Fanconi Anemia Registry study. Blood. 84:1650–1655. [PubMed] [Google Scholar]
- Chang, T.S., C.S. Cho, S. Park, S. Yu, S.W. Kang, and S.G. Rhee. 2004. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J. Biol. Chem. 279:41975–41984. [DOI] [PubMed] [Google Scholar]
- Chen, M., D.J. Won, S. Krajewski, and R.A. Gottlieb. 2002. Calpain and mitochondria in ischemia/reperfusion injury. J. Biol. Chem. 277:29181–29186. [DOI] [PubMed] [Google Scholar]
- Choi, J.H., T.N. Kim, S. Kim, S.H. Baek, J.H. Kim, S.R. Lee, and J.R. Kim. 2002. Overexpression of mitochondrial thioredoxin reductase and peroxiredoxin III in hepatocellular carcinomas. Anticancer Res. 22:3331–3335. [PubMed] [Google Scholar]
- Cumming, R.C., J. Lightfoot, K. Beard, H. Youssoufian, P.J. O'Brien, and M. Buchwald. 2001. Fanconi anemia group C protein prevents apoptosis in hematopoietic cells through redox regulation of GSTP1. Nat. Med. 7:814–820. [DOI] [PubMed] [Google Scholar]
- D'Andrea, A. 2003. Fanconi anemia. Curr. Biol. 13:R546. [DOI] [PubMed] [Google Scholar]
- Daniel, K.G., J.S. Anderson, Q. Zhong, A. Kazi, P. Gupta, and Q.P. Dou. 2003. Association of mitochondrial calpain activation with increased expression and autolysis of calpain small subunit in an early stage of apoptosis. Int. J. Mol. Med. 12:247–252. [PubMed] [Google Scholar]
- Denizot, F., and R. Lang. 1986. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods. 89:271–277. [DOI] [PubMed] [Google Scholar]
- Futaki, M., S. Watanabe, S. Kajigaya, and J.M. Liu. 2001. Fanconi anemia protein, FANCG, is a phosphoprotein and is upregulated with FANCA after TNF-alpha treatment. Biochem. Biophys. Res. Commun. 281:347–351. [DOI] [PubMed] [Google Scholar]
- Futaki, M., T. Igarashi, S. Watanabe, S. Kajigaya, A. Tatsuguchi, J. Wang, and J.M. Liu. 2002. The FANCG Fanconi anemia protein interacts with CYP2E1: possible role in protection against oxidative DNA damage. Carcinogenesis. 23:67–72. [DOI] [PubMed] [Google Scholar]
- Gietz, R.D., B. Triggs-Raine, A. Robbins, K.C. Graham, and R.A. Woods. 1997. Identification of proteins that interact with a protein of interest: applications of the yeast two-hybrid system. Mol. Cell. Biochem. 172:67–79. [PubMed] [Google Scholar]
- Gille, J.J., H.M. Wortelboer, and H. Joenje. 1987. Antioxidant status of Fanconi anemia fibroblasts. Hum. Genet. 77:28–31. [DOI] [PubMed] [Google Scholar]
- Gourlay, L.J., D. Bhella, S.M. Kelly, N.C. Price, and J.G. Lindsay. 2003. Structure-function analysis of recombinant substrate protein 22 kDa (SP-22). A mitochondrial 2-CYS peroxiredoxin organized as a decameric toroid. J. Biol. Chem. 278:32631–32637. [DOI] [PubMed] [Google Scholar]
- Grompe, M., and A. D'Andrea. 2001. Fanconi anemia and DNA repair. Hum. Mol. Genet. 10:2253–2259. [DOI] [PubMed] [Google Scholar]
- Hadjur, S., K. Ung, L. Wadsworth, J. Dimmick, E. Rajcan-Separovic, R.W. Scott, M. Buchwald, and F.R. Jirik. 2001. Defective hematopoiesis and hepatic steatosis in mice with combined deficiencies of the genes encoding Fancc and Cu/Zn superoxide dismutase. Blood. 98:1003–1011. [DOI] [PubMed] [Google Scholar]
- Hoshino, T., J. Wang, M.P. Devetten, N. Iwata, S. Kajigaya, R.J. Wise, J.M. Liu, and H. Youssoufian. 1998. Molecular chaperone GRP94 binds to the Fanconi anemia group C protein and regulates its intracellular expression. Blood. 91:4379–4386. [PubMed] [Google Scholar]
- Huang, M.E., and R.D. Kolodner. 2005. A biological network in Saccharomyces cerevisiae prevents the deleterious effects of endogenous oxidative DNA damage. Mol. Cell. 17:709–720. [DOI] [PubMed] [Google Scholar]
- Joenje, H., F. Arwert, A.W. Eriksson, H. de Koning, and A.B. Oostra. 1981. Oxygen-dependence of chromosomal aberrations in Fanconi's anaemia. Nature. 290:142–143. [DOI] [PubMed] [Google Scholar]
- Kang, S.W., H.Z. Chae, M.S. Seo, K. Kim, I.C. Baines, and S.G. Rhee. 1998. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 273:6297–6302. [DOI] [PubMed] [Google Scholar]
- Lee, M.S., Y.T. Kwon, M. Li, J. Peng, R.M. Friedlander, and L.H. Tsai. 2000. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 405:360–364. [DOI] [PubMed] [Google Scholar]
- Lee, T.H., S.U. Kim, S.L. Yu, S.H. Kim, D.S. Park, H.B. Moon, S.H. Dho, K.S. Kwon, H.J. Kwon, Y.H. Han, et al. 2003. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood. 101:5033–5038. [DOI] [PubMed] [Google Scholar]
- Marchenko, N.D., A. Zaika, and U.M. Moll. 2000. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J. Biol. Chem. 275:16202–16212. [DOI] [PubMed] [Google Scholar]
- Medhurst, A.L., E.H. Laghmani, J. Steltenpool, M. Ferrer, C. Fontaine, J. Groot, M.A. Rooimans, R.J. Scheper, A.R. Meetei, W. Wang, H. Joenje and J.P. Winter. 2006. Evidence for subcomplexes in the Fanconi anemia pathway. Blood. 108:2072–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meetei, A.R., J.P. de Winter, A.L. Medhurst, M. Wallisch, Q. Waisfisz, H.J. van de Vrugt, A.B. Oostra, Z. Yan, C. Ling, C.E. Bishop, et al. 2003. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet. 35:165–170. [DOI] [PubMed] [Google Scholar]
- Meetei, A.R., M. Levitus, Y. Xue, A.L. Medhurst, M. Zwaan, C. Ling, M.A. Rooimans, P. Bier, M. Hoatlin, G. Pals, et al. 2004. X-linked inheritance of Fanconi anemia complementation group B. Nat. Genet. 36:1219–1224. [DOI] [PubMed] [Google Scholar]
- Mi, J., and G.M. Kupfer. 2005. The Fanconi anemia core complex associates with chromatin during S phase. Blood. 105:759–766. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, S.S., S.L. Wyszomierski, R.M. Gronostajski, and J.M. Rosen. 2001. Differential interactions of specific nuclear factor I isoforms with the glucocorticoid receptor and STAT5 in the cooperative regulation of WAP gene transcription. Mol. Cell. Biol. 21:6859–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi, K., A. Moran, T. Hays, Y. Kuang, E. Fox, D. Garneau, R.M. de Oca, M. Grompe, and A.D. D'Andrea. 2001. Functional analysis of patient-derived mutations in the Fanconi anemia gene, FANCG/XRCC9. Exp. Hematol. 29:842–849. [DOI] [PubMed] [Google Scholar]
- Neumann, C.A., D.S. Krause, C.V. Carman, S. Das, D.P. Dubey, J.L. Abraham, R.T. Bronson, Y. Fujiwara, S.H. Orkin, and R.A. Van Etten. 2003. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 424:561–565. [DOI] [PubMed] [Google Scholar]
- Noh, D.Y., S.J. Ahn, R.A. Lee, S.W. Kim, I.A. Park, and H.Z. Chae. 2001. Overexpression of peroxiredoxin in human breast cancer. Anticancer Res. 21:2085–2090. [PubMed] [Google Scholar]
- Nonn, L., M. Berggren, and G. Powis. 2003. Increased expression of mitochondrial peroxiredoxin-3 (thioredoxin peroxidase-2) protects cancer cells against hypoxia and drug-induced hydrogen peroxide-dependent apoptosis. Mol. Cancer Res. 1:682–689. [PubMed] [Google Scholar]
- Otsuki, T., D.B. Young, D.T. Sasaki, M.P. Pando, J. Li, A. Manning, M. Hoekstra, M.E. Hoatlin, F. Mercurio, and J.M. Liu. 2002. Fanconi anemia protein complex is a novel target of the IKK signalsome. J. Cell. Biochem. 86:613–623. [DOI] [PubMed] [Google Scholar]
- Pagano, G. 2000. Mitomycin C and diepoxybutane action mechanisms and FANCC protein functions: further insights into the role for oxidative stress in Fanconi's anaemia phenotype. Carcinogenesis. 21:1067–1068. [DOI] [PubMed] [Google Scholar]
- Pagano, G., and H. Youssoufian. 2003. Fanconi anaemia proteins: major roles in cell protection against oxidative damage. Bioessays. 25:589–595. [DOI] [PubMed] [Google Scholar]
- Pang, Q., T.A. Christianson, W. Keeble, T. Koretsky, and G.C. Bagby. 2002. The anti-apoptotic function of Hsp70 in the interferon-inducible double-stranded RNA-dependent protein kinase-mediated death signaling pathway requires the Fanconi anemia protein, FANCC. J. Biol. Chem. 277:49638–49643. [DOI] [PubMed] [Google Scholar]
- Pariente, J.A., C. Camello, P.J. Camello, and G.M. Salido. 2001. Release of calcium from mitochondrial and nonmitochondrial intracellular stores in mouse pancreatic acinar cells by hydrogen peroxide. J. Membr. Biol. 179:27–35. [DOI] [PubMed] [Google Scholar]
- Park, S.J., S.L. Ciccone, B.D. Beck, B. Hwang, B. Freie, D.W. Clapp, and S.H. Lee. 2004. Oxidative stress/damage induces multimerization and interaction of Fanconi anemia proteins. J. Biol. Chem. 279:30053–30059. [DOI] [PubMed] [Google Scholar]
- Porfirio, B., G. Ambroso, G. Giannella, G. Isacchi, and B. Dallapiccola. 1989. Partial correction of chromosome instability in Fanconi anemia by desferrioxamine. Hum. Genet. 83:49–51. [DOI] [PubMed] [Google Scholar]
- Rose, M., M.J. Casadaban, and D. Botstein. 1981. Yeast genes fused to beta-galactosidase in Escherichia coli can be expressed normally in yeast. Proc. Natl. Acad. Sci. USA. 78:2460–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppitsch, W., C. Meisslitzer, H. Weirich-Schwaiger, H. Klocker, C. Scheidereit, M. Schweiger, and M. Hirsch-Kauffmann. 1997. The role of oxygen metabolism for the pathological phenotype of Fanconi anemia. Hum. Genet. 99:710–719. [DOI] [PubMed] [Google Scholar]
- Ruppitsch, W., C. Meisslitzer, M. Hirsch-Kauffmann, and M. Schweiger. 1998. Overexpression of thioredoxin in Fanconi anemia fibroblasts prevents the cytotoxic and DNA damaging effect of mitomycin C and diepoxybutane. FEBS Lett. 422:99–102. [DOI] [PubMed] [Google Scholar]
- Schindler, D., and H. Hoehn. 1988. Fanconi anemia mutation causes cellular susceptibility to ambient oxygen. Am. J. Hum. Genet. 43:429–435. [PMC free article] [PubMed] [Google Scholar]
- Schmid, W., and G. Fanconi. 1978. Fragility and spiralization anomalies of the chromosomes in three cases, including fraternal twins, with Fanconi's anemia, type Estren-Dameshek. Cytogenet. Cell Genet. 20:141–149. [DOI] [PubMed] [Google Scholar]
- Thomashevski, A., A.A. High, M. Drozd, J. Shabanowitz, D.F. Hunt, P.A. Grant, and G.M. Kupfer. 2004. The Fanconi anemia core complex forms 4 different sized complexes in different subcellular compartments. J. Biol. Chem. 279:26201–26209. [DOI] [PubMed] [Google Scholar]
- Thompson, L.H. 2005. Unraveling the Fanconi Anemia - DNA repair connection. Nat. Genet. 37:921–922. [DOI] [PubMed] [Google Scholar]
- Waisfisz, Q., J.P. de Winter, F.A. Kruyt, J. de Groot, L. van der Weel, L.M. Dijkmans, Y. Zhi, F. Arwert, R.J. Scheper, H. Youssoufian, M.E. Hoatlin, and H. Joenje. 1999. A physical complex of the Fanconi anemia proteins FANCG/XRCC9 and FANCA. Proc. Natl. Acad. Sci. USA. 96:10320–10325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe, S., T. Hiroi, Y. Yamamoto, Y. Fujioka, H. Hasegawa, N. Yago, and S.Y. Takahashi. 1997. SP-22 is a thioredoxin-dependent peroxide reductase in mitochondria. Eur. J. Biochem. 249:52–60. [DOI] [PubMed] [Google Scholar]
- Wonsey, D.R., K.I. Zeller, and C.V. Dang. 2002. The c-Myc target gene PRDX3 is required for mitochondrial homeostasis and neoplastic transformation. Proc. Natl. Acad. Sci. USA. 99:6649–6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, H.A., H.Z. Chae, S.C. Hwang, K.S. Yang, S.W. Kang, K. Kim, and S.G. Rhee. 2003. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 300:653–656. [DOI] [PubMed] [Google Scholar]
- Zhang, X., J. Li, D.P. Sejas, and Q. Pang. 2005. Hypoxia-reoxygenation induces premature senescence in FA bone marrow hematopoietic cells. Blood. 106:75–85. [DOI] [PubMed] [Google Scholar]
- Zhao, Y., L. Chaiswing, J.M. Velez, I. Batinic-Haberle, N.H. Colburn, T.D. Oberley, and D.K. St Clair. 2005. p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res. 65:3745–3750. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.