

Abstract
Actin filaments in cells depolymerize rapidly despite the presence of high concentrations of polymerizable G actin. Cofilin is recognized as a key regulator that promotes actin depolymerization. In this study, we show that although pure cofilin can disassemble Listeria monocytogenes actin comet tails, it cannot efficiently disassemble comet tails in the presence of polymerizable actin. Thymus extracts also rapidly disassemble comet tails, and this reaction is more efficient than pure cofilin when normalized to cofilin concentration. By biochemical fractionation, we identify Aip1 and coronin as two proteins present in thymus extract that facilitate the cofilin-mediated disassembly of Listeria comet tails. Together, coronin and Aip1 lower the amount of cofilin required to disassemble the comet tail and permit even low concentrations of cofilin to depolymerize actin in the presence of polymerizable G actin. The cooperative activities of cofilin, coronin, and Aip1 should provide a biochemical basis for understanding how actin filaments can grow in some places in the cell while shrinking in others.
Introduction
Actin filaments rapidly polymerize in some regions within cells while rapidly depolymerizing in others. In some cases, such as at the tips and bases of filopodia, polymerization and depolymerization are spatially separated (Mallavarapu and Mitchison, 1999). In others, such as lamellipodia, they are close together (Ponti et al., 2003). Simultaneous polymerization and depolymerization of a protein polymer in the same solution is a nonequilibrium behavior that requires an energy source. The most likely source of this energy is ATP hydrolysis by actin during polymerization, but how this energy is used in cells to promote actin dynamics is poorly understood (Mitchison, 1992).
Listeria monocytogenes propels itself through the host cell's cytoplasm by assembling an actin filament–based comet tail whose characteristic morphology depends on spatially separating actin assembly reactions from disassembly reactions. Comet tail assembly is restricted to the interface between the bacterial surface and the comet tail, where local activation of Arp2/3 rapidly nucleates a dendritic network of actin filaments (Theriot et al., 1992; Welch et al., 1997; Cameron et al., 2001; Brieher et al., 2004). The fast rate of Listeria propulsion indicates that mammalian cytoplasm contains a high concentration of available actin monomer that drives fast actin polymerization. Everywhere else in the tail, actin depolymerizes rapidly. The rate of subunit loss is proportional to the local concentration of actin filaments, leading to the exponential decay of filament density with a time constant of ∼25 s, suggesting that disassembly is a first-order reaction that proceeds through a single rate-limiting step (Theriot et al., 1992). However, the pool of actin monomer presents a challenge to the depolymerization reaction. How can filaments in the comet tail depolymerize rapidly in an environment that favors fast assembly? Providing a mechanistic answer to this question requires identification of the cellular factors that perform the reaction.
Members of the cofilin family of actin-binding proteins have been identified as key factors accelerating actin disassembly in all eukaryotic cells. Cofilin is necessary for the rapid turnover of actin arrays in cells (Bamburg, 1999), including the disassembly of Listeria actin comet tails (Rosenblatt et al., 1997). Whether cofilin alone is sufficient for disassembling actin arrays in cells is not known. Cofilin alone seems unlikely to explain the disassembly behavior of Listeria actin comet tails. Cofilin can sever actin filaments, creating new ends that grow in the presence of G actin (Ichetovkin et al., 2002), but filament growth is not detectable in comet tails. Furthermore, the experimentally triggered activation of cofilin in cells results in a burst of net actin assembly, not disassembly (Ghosh et al., 2004). Therefore, the question of what, in addition to cofilin, is required to depolymerize the Listeria actin comet tail in the presence of high concentrations of polymerizable actin remains open. In this study, we use the Listeria actin comet tail as a model substrate to identify cellular factors capable of rapidly disassembling it.
Results
To simplify the analysis of actin comet tail disassembly, we experimentally separated it from the assembly reaction. High speed supernatants of detergent extracts from HeLa cells readily assemble comet tails, but they have no detectable disassembly activity (unpublished data). To assay comet tail disassembly, comet tails were first assembled on Listeria adhered to coverslips in perfusion chambers by flowing in HeLa cell extract mixed with rhodamine-actin to mark the comet tails. Disassembly was then induced by replacing this solution with solutions containing actin-depolymerizing factors. The reaction was followed by time-lapse imaging, and disassembly was quantified by measuring the decrease of comet tail fluorescence over time.
Comet tails disassemble very slowly when the assembly mixture is replaced with buffer alone or buffer containing latrunculin to sequester free G actin (Fig. 1, A and C). Thus, the intrinsic disassembly rate of comet tails is slow (comet tail t 1/2 is >15 min). In contrast, comet tails rapidly depolymerized when the assembly mixture was replaced with a high speed supernatant of thymus extract diluted into buffer (comet tail t 1/2 of ∼30 s in 500 μg/ml of total protein), implying that thymus contains high concentrations of depolymerization factors (Fig. 1, B and C). By Western blotting, undiluted thymus extract contains ∼20 μM cofilin (unpublished data). Pure Escherichia coli–expressed cofilin was also able to depolymerize Listeria actin comet tails under these conditions, but the rates of disassembly appeared to be slower compared with those detected with dilute thymus cytosol. To quantitatively compare thymus extract to pure cofilin, we normalized activity to cofilin concentration (Fig. 1, D and E). In these and subsequent experiments, we measured the t 1/2 of disassembly, and we plot the data as an apparent disassembly rate (apparent kdis) versus concentration, where apparent kdis = ln2/t 1/2. From this analysis, thymus extract is 5–10 times more potent than pure cofilin for disassembling Listeria actin comet tails, implying that it contains factors in addition to cofilin that accelerate actin disassembly.
Figure 1.

Fast disassembly of Listeria actin comet tails requires cellular factors. (A) Frames from a time-lapse sequence showing the slow decay of comet tails diluted into latrunculin. (B) Time-lapse images of a Listeria comet tail in thymus extract. Frames in A and B are 30 s apart. (C) Actin intensity decay profiles of comet tails in latrunculin or thymus extract. Each point is the normalized mean of at least 30 comet tails from two experiments. (D and E) Apparent disassembly rates of comet tails in thymus extract (D) or pure cofilin alone (E). Mean ± SD (error bars); n = 3 experiments/point. Bar, 10 μm.
To identify these factors, thymus cytosol was separated on an anion exchange column (DE52), and the depolymerizing activity of the flow through fraction was compared with that of the bound material (Fig. 2 A). Although the flow through fraction was sufficient to depolymerize comet tails, it was less potent than the starting material. The DE52-bound fraction contained no detectable depolymerizing activity; however, it potentiated the activity of the flow through. By Western blotting, all of the endogenous cofilin was present in the DE52 flow through (unpublished data). Normalizing the flow through fraction to cofilin concentration revealed that thymus DE52 flow through is only twice as potent as pure E. coli–expressed cofilin alone. To test whether pure cofilin could substitute for DE52 flow through, we titrated pure cofilin down to a concentration that is insufficient to disassemble comet tails on its own. Potent depolymerizing activity could be regenerated by combining this subthreshold amount of pure cofilin with the DE52-bound cofilin-free fraction (Fig. 2 B). These results suggest that besides cofilin, thymus cytosol contains at least one additional factor involved in actin depolymerization that can be separated from cofilin by fractionation on DE52.
Figure 2.

Thymus extract contains multiple factors that contribute to comet tail disassembly. (A) Thymus extract contains at least two factors involved in comet tail disassembly. One unit of activity is the amount of protein required to fully disassemble all comet tails in <5 min. (B) Cofilin substitutes for the thymus DE52 flow through fraction, whereas the DE52-bound fraction potentiates cofilin activity. (C) The DE52-bound cofilin-potentiating fraction consists of at least two factors. Specific activity is as described in A.
The ability of the DE52-bound material to induce a subthreshold amount of cofilin to disassemble Listeria actin comet tails provided a simple visual assay to fractionate for the potentiating factors. However, substantial amounts of activity were lost upon further fractionation of the DE52-bound cofilin-potentiating fraction. Therefore, we screened several columns and conditions to test the possibility that the DE52-bound material in fact contains multiple factors that must act together to facilitate cofilin-mediated depolymerization. Separation of the DE52-bound fraction on cation exchange resulted in the loss of all activity (Fig. 2 C). However, combining limiting cofilin with the S flow resulted in the appearance of a new peak of activity bound to the S column. Therefore, the cofilin-potentiating activity in thymus cytosol consists of at least two factors. The S flow through fraction was designated fraction X, and the S-bound material was designated fraction Y.
Factor X was purified from fraction X through a series of chromatographic separations. The activity of factor X was monitored using the Listeria comet tail depolymerization assay in the presence of limiting cofilin and fraction Y. With this assay, factor X fractionated as a single biochemical activity (Table I), resulting in the isolation of a single polypeptide (Fig. 3 A) that was identified by mass spectrometry as Aip1 (55.4% amino acid and 54.9% mass coverage; Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200603149/DC1).
Table I.
Purification of Aip1
| Step | Protein | Activity | Specific activity | Percent yield |
|---|---|---|---|---|
| mg | U/mg | |||
| Extract | 9,000 | NA | NA | NA |
| DE52 | 770 | 200,000 | 260 | NA |
| SFT | 323 | 66,667 | 206 | 33 |
| Phenyl | 30 | 47,000 | 1,567 | 23.5 |
| MonoQ | 10.3 | 33,000 | 3,204 | 16.5 |
| HAP | 2.2 | 17,000 | 7,727 | 8.5 |
| Gel filter | 0.53 | 6,333 | 11,949 | 3.7 |
SFT, S flow through; HAP, hydroxyapatite; NA, not applicable.
Figure 3.

Purification of cofilin-potentiating factors from thymus extract. (A) Coomassie-stained gel showing the purification of factor X (Aip1). (B) Coomassie-stained gel showing the purification of factor Y (coronin-1A). Numbers left of the gels correspond to molecular mass standards in kilodaltons.
A limiting amount of cofilin was used in combination with Aip1 to isolate factor Y through a series of chromatographic separations. With this assay, factor Y fractionated as a single biochemical activity (Table II), resulting in the isolation of two closely migrating polypeptides on SDS-PAGE (Fig. 3 B) that were both identified by mass spectrometry as coronin-1A (43.4% amino acid and 43.2% mass coverage; Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200603149/DC1).
Table II.
Purification of coronin
| Step | Protein | Activity | Specific activity | Percent yield |
|---|---|---|---|---|
| mg | U/mg | |||
| Extract | 9,000 | NA | NA | NA |
| DE52 | 770 | 200,000 | 260 | NA |
| S bound | 43 | 34,000 | 790 | 17 |
| Heparin | 10 | 25,000 | 2,500 | 12.5 |
| HAP | 4.2 | 22,000 | 5,200 | 11 |
| MonoQ | 3.2 | 18,000 | 5,600 | 9 |
HAP, hydroxyapatite; NA, not applicable.
To simplify the assay during the purification procedure, depolymerization activity was assessed by determining the amount of Aip1, coronin, or cofilin required to depolymerize all of the comet tails to completion in a set amount of time. Although this assay facilitated purification, it is presumably nonlinear and, thus, is not sufficiently accurate to determine the amount of each factor required to disassemble comet tails. To obtain a better measurement of the effective concentrations of each component, we varied the concentration of one of the three factors while holding the other two constant and measured the apparent rate of actin comet tail disassembly by quantifying the total fluorescence within the comet tails as a function of time using time-lapse fluorescence microscopy.
Fig. 4 A displays the apparent rate of comet tail disassembly as a function of cofilin concentration using cofilin alone or in the presence of the other depolymerizing factors. The coronin and Aip1 concentrations were set at saturating levels for this experiment (1 and 0.2 μM, respectively). Cofilin alone was sufficient to disassemble the comet tails with an IC50 of ∼3–4 μM. The addition of coronin alone or Aip1 alone to cofilin had no substantial effect on the amount of cofilin required to disassemble the comet tails. However, the addition of both coronin and Aip1 accelerated cofilin-mediated depolymerization and lowered the IC50 of cofilin 10 times to ∼0.3 μM. To determine the IC50s of Aip1 and coronin, we used a constant limiting amount of cofilin (1 μM) and a constant saturating amount of either Aip1 (0.2 μM) or coronin (2 μM) while varying the concentration of the other potentiating factor. By this method, we estimate an IC50 for Aip1 of 30 nM (Fig. 4 B) and 150 nM for coronin (Fig. 4 C). These potent effects argue for biochemical specificity of the reaction.
Figure 4.

Dose-response analysis of cofilin, Aip1, and coronin in Listeria actin comet tail disassembly. (A) Amount of cofilin required to disassemble comet tails in the presence or absence of Aip1 and coronin. (B) Titration of the amount of Aip1 required to enhance the depolymerization of Listeria comet tails in the presence of 1 μM of limiting cofilin and 1 μM of saturating coronin. (C) Titration of the amount of coronin required to enhance the depolymerization of Listeria comet tails in the presence of 1 μM of limiting cofilin and 0.2 μM of saturating Aip1. Each plot shows the mean ± SD (error bars); n = 3 experiments/point.
All of the aforementioned experiments test the ability of the three depolymerizers to rapidly disassemble actin comet tails into dilute buffer. In cells, actin disassembly must occur in the presence of high concentrations of polymerizable G actin. To examine actin comet tail disassembly under polymerizing conditions, we added a source of polymerizable G actin to the depolymerizing mixture. G actin was provided in the form of a profilin G–ATP actin complex that is currently thought to represent the physiological state of polymerizable actin in cells (Pollard and Borisy, 2003). Profilin suppresses spontaneous nucleation but has little effect on the ability of actin to add to the barbed ends of existing actin filaments. Because profilin also promotes disassembly and ATP exchange on cofilin–actin–ADP complexes, the presence of profilin should prevent the accumulation of these complexes in our reaction.
Cofilin in the presence or absence of either coronin or Aip1 alone was tested to see whether any of these combinations were sufficient to disassemble actin comet tails in the presence of profilin–actin (Fig. 5 A). A high concentration of cofilin (10 μM) was used for these experiments to obtain reasonably fast baseline disassembly rates. The addition of increasing amounts of profilin–actin in the disassembly reaction blocked comet tail disassembly by cofilin alone. Disassembly by a combination of 10 μM cofilin and coronin was also highly sensitive to the competing profilin–actin complex. In contrast, the combination of Aip1 and a high concentration of cofilin permitted comet tail disassembly in the presence of the competing polymerizable profilin–actin complex. All three factors together also disassembled comet tails in the presence of profilin–actin, but the rate of disassembly was markedly enhanced (Fig. 5 B). In the presence of all three factors, as little as 1 μM cofilin was sufficient to disassemble comet tails in the presence of 8 μM profilin–actin at a rate three times faster than a combination of 10 μM cofilin and Aip1 alone. These experiments demonstrate that although cofilin alone is sufficient to disassemble comet tails into buffer, it is not sufficient to disassemble comet tails under more physiological conditions in which a source of polymerizable actin is present. Aip1 appears to be the most important factor conferring resistance to actin monomer. However, comet tail disassembly by cofilin and Aip1 alone requires high concentrations of cofilin. The addition of coronin lowers the cofilin requirement 10-fold even when the reaction is challenged with high concentrations of polymerizable actin. Therefore, coronin appears to either promote cofilin binding to comet tails or alters filament structure, thereby making the filaments more sensitive to cofilin action.
Figure 5.

Disassembly of Listeria actin comet tails in the presence of profilin–actin. (A) Comparison of comet tail disassembly rates as a function of profilin–actin concentration in the presence of a high concentration (10 μM) of cofilin alone versus 10 μM cofilin + 0.1 μM Aip1 or 10 μM cofilin + 1 μM coronin. (B) Comparison of comet tail disassembly rates as a function of profilin–actin concentration in the presence of all three depolymerizing factors. In this experiment, the cofilin was lowered to 1 μM while Aip1 was maintained at 0.1 μM and coronin was maintained at 1 μM. Each plot shows the mean ± SD (error bars); n = 3 experiments/point.
To examine these possibilities, we first tested whether any individual or combination of two out of the three disassembly factors could act in a preincubation step or whether all three factors had to be present at once to disassemble comet tails. To do this, comet tails were assembled in perfusion chambers, and the assembly mixture was replaced with a solution containing only one or two of three disassembly factors. After an incubation period, the preincubation solution was replaced with a solution containing the remaining factors, and comet tail disassembly was monitored by fluorescence imaging. Comet tail disassembly under these conditions was compared with a positive control in which comet tails were preincubated in buffer alone and were chased with all three factors at once. Actin comet tails preincubated in coronin and chased with cofilin and Aip1 disassembled as rapidly as washing into all three factors at once. However, comet tails were stable under all other conditions (Fig. 6 A). Therefore, coronin binding to the actin comet tail alters the substrate either by promoting cofilin binding or altering actin filament structure, thereby making the filament more sensitive to cofilin action.
Figure 6.

Coronin promotes cofilin binding to actin comet tails. (A) Apparent comet tail disassembly rates after the preincubation of comet tails in either buffer (φ), 1 μM coronin (Cor), 3 μM cofilin (Cfn), 0.3 μM Aip1, or a combination of any two factors. After preincubation, these solutions were replaced with the remaining factors at the same listed concentrations, and comet tail disassembly was measured by time-lapse imaging. Each experiment plots the mean ± SD (error bars); n = 4. (B) Paired fluorescent images of labeled cofilin bound to actin comet tails in the presence or absence of a coronin preincubation. (a and b) No preincubation. (c and d) Comet tails preincubated with 1 μM coronin. Both samples were then incubated in 4 μM of labeled cofilin before glutaraldehyde fixation and imaging. (a and c) Actin; (b and d) cofilin. (C) Ratio of cofilin fluorescence to actin fluorescence in the presence or absence of a coronin preincubation. n = 3 experiments for each point. (D) Images from a time-lapse sequence showing the dissociation of labeled cofilin from actin comet tails after a preincubation in buffer alone or cofilin from comet tails preincubated in 1 μM coronin. Numbers refer to time in seconds. (E) Comparison of cofilin dissociation rates from comet tails preincubated in buffer or 1 μM coronin. (C and D) Each point is the mean ± SD; n = 4 experiments. au, arbitrary units. Bars, 10 μm.
To test whether coronin promotes cofilin binding, a point mutation was generated in cofilin by changing Ser108 to Cys to allow labeling with fluorescent dyes. This reagent was then used to measure cofilin binding to actin comet tails with or without a coronin preincubation using fluorescence imaging to quantify the intensity of bound cofilin normalized to actin fluorescence in the comet tail. AlexaFluor488 actin was used to label the comet tails, and AlexaFluor568 cofilin was used to quantify cofilin. Comet tails disassembled using this combination of fluorophores in the presence of AlexaFluor568 cofilin, coronin, and Aip1 with kinetics similar to those detected with wild-type cofilin (unpublished data).
Using 4 μM of labeled cofilin, comet tails bound little cofilin in the absence of a coronin preincubation. However, comet tails bound readily detectable amounts of cofilin after a coronin preincubation (Fig. 6 B). To quantify this effect, we compared the amount of cofilin bound to comet tails normalized to the amount of actin (Fig. 6 C). In the absence of coronin preincubation, the amount of cofilin bound to actin comet tails rose steadily with increasing amounts of cofilin and appeared to approach saturation near 10 μM cofilin. Unfortunately, it was not possible to extend the analysis to higher concentrations of cofilin because the comet tails washed out of the perfusion chamber upon fixation. Comet tails bound approximately four to five times more cofilin after a coronin preincubation and appeared to approach saturation near 8 μM cofilin. Despite the high amount of cofilin bound under these conditions, relative to in the absence of coronin, the comet tails were stable, which is consistent with our previous experiment demonstrating that coronin and cofilin together do not disassemble comet tails much faster than cofilin alone (Fig. 4 A). These results demonstrate that coronin alters the comet tail substrate by promoting cofilin binding.
We repeated the binding experiments using fluorescently labeled actophorin (Ser88 to Cys mutation) from Acanthamoeba castellanii and obtained similar results. Therefore, coronin-dependent increased cofilin binding to comet tails is not peculiar to the point mutation used for labeling, and, in fact, the coronin effect probably extends to all members of the ADF/cofilin family across phyla. The four- to fivefold coronin-dependent increase in cofilin binding is close to the 10-fold reduction in the amount of cofilin required to disassemble comet tails in the presence of coronin and Aip1 relative to cofilin alone. Therefore, coronin's effect on actin comet tail disassembly can largely be attributed to its ability to promote cofilin binding. We also tried to measure cofilin binding in the presence of coronin and Aip1, but comet tails disassembled too rapidly to make such measurements, even at acidic pH.
Coronin could promote cofilin binding either by creating new binding sites or increasing the affinity of cofilin's interaction to existing binding sites. To distinguish between these possibilities, the mutant fluorescently labeled cofilin was used to monitor its dissociation from the comet tail after a preincubation in buffer alone or 1 μM coronin. After this step, the buffer-treated tails were incubated in 6 μM of labeled cofilin, and the coronin-treated comet tails were incubated in 3 μM of labeled cofilin. The chambers were then washed with three chamber volumes of buffer while acquiring a fluorescence time-lapse sequence. Cofilin appeared to dissociate from comet tails with similar kinetics irrespective of a coronin preincubation step. Fig. 6 D shows representative frames from these experiments after preincubation in buffer alone or coronin. Note that these images are not showing comet tail disassembly; the actin comet tails are, in fact, stable. Rather, they show the loss of cofilin signal from a constant density of actin. The dissociation rates were quantified by measuring the decay of cofilin fluorescence over time (Fig. 6 E). The results confirm that cofilin dissociates from comet tails in the presence or absence of exogenous coronin with similar, perhaps identical, kinetics. Therefore, coronin appears to create new cofilin-binding sites of similar, if not identical, affinity to those that occur in the absence of additional coronin.
All of the previous assays used Listeria actin comet tails as a physiologically relevant actin gel for disassembly. Thus, the generality of the activity of the three factors was tested on pure actin filaments in solution using a sedimentation assay (Fig. 7 A). 2.5 μM actin was polymerized in vitro, mixed with combinations of the disassembly factors, and the resulting products were separated by ultracentrifugation. This assay separates long filaments (>20 subunits) from fragments and actin monomer. Under starting conditions, nearly all of the actin in a pure actin solution alone pellets, which is consistent with actin's submicromolar critical concentration. Neither 1 μM coronin alone, 0.1 μM Aip1 alone, nor the combination of coronin and Aip1 altered the distribution of actin between supernatant and pellet. Thus, with this assay, Aip1 and coronin in the absence of cofilin do not appear to promote actin depolymerization, which is consistent with our observations of Listeria comet tails. 2.5 μM cofilin alone resulted in a slight increase in the amount of actin in the supernatant, which is consistent with reports that stoichiometric cofilin scores only as a weak depolymerizer (Yonezawa et al., 1988; Okada et al., 2002; Yeoh et al., 2002; Chen et al., 2004). This distribution was not further altered by the addition of either coronin alone or Aip1 alone. In contrast, in the presence of all three factors together (2.5 μM cofilin + 1 μM coronin + 0.1 μM Aip1), more than half of the actin was found in the supernatant. Therefore, the three disassembly factors together are more potent at depolymerizing actin than any factor alone or the combination of any two. These data are consistent with the results from the fractionation scheme using Listeria comet tails.
Figure 7.

Sedimentation analysis of actin disassembly. (A) Coomassie gel of the distribution of combinations of actin, cofilin, coronin, and Aip1 between the supernatant (S) and pellet (P). (B) Coomassie gel of cofilin binding to F actin in the presence or absence of coronin. M, molecular mass markers. (C) Densitometry of gel shown in B. Numbers on the left of A and B are the molecular masses of standards in kilodaltons.
Although coronin in combination with cofilin did not alter the distribution of actin, coronin increased the amount of cofilin bound to filamentous actin (F actin) in the pellet (Fig. 7 A), which is consistent with results using Listeria comet tails. To compare coronin-dependent increased binding on pure actin filaments with that seen on comet tails, the amount of cofilin bound to F actin as a function of increasing cofilin concentration in the presence or absence of coronin was measured in a sedimentation assay. A representative gel from such an experiment is shown in Fig. 7 B, and a plot from the densitometry of this gel is shown in Fig. 7 C. Although coronin promotes cofilin binding to pure actin filaments, the effect is not nearly as pronounced as it was on Listeria comet tails. With pure actin filaments, coronin increased the amount of cofilin bound to F actin by 1.5 times compared with the four- to fivefold increase detected on comet tails. The quantitative discrepancy could be the result of several factors. One possibility is that the sedimentation assay is simply not very sensitive or quantitative because sedimentation alters the equilibrium. Alternatively, other factors within the comet tail in addition to coronin might influence cofilin binding. Nevertheless, results from the sedimentation experiments qualitatively support the conclusion from comet tails that coronin promotes cofilin binding to F actin.
Discussion
Cofilin is thought to be the key factor promoting fast depolymerization of actin filaments in cells, but other cellular factors clearly contribute to this reaction. Although cofilin alone is sufficient to depolymerize Listeria actin comet tails, it is unable to rapidly disassemble comet tails under more physiological conditions in which depolymerization is challenged by a physiological concentration of G actin presented as a profilin–actin–ATP complex. Similar results were obtained using actin–ATP alone (unpublished data). We identified Aip1 and coronin as two factors in thymus extracts that facilitate cofilin-mediated actin disassembly. Together, Aip1 and coronin accelerate the cofilin-mediated disassembly of Listeria comet tails, reduce the amount of cofilin required for this reaction, and permit efficient disassembly of the comet tail in the presence of a source of polymerizable G actin. The combination of these factors provides a means whereby Listeria actin comet tail filaments can efficiently depolymerize while suppressing assembly.
Aip1 has already been implicated as a cofactor in cofilin-mediated depolymerization by both biochemical and genetic evidence (Iida and Yahara, 1999; Okada et al., 1999; Rodal et al., 1999), although the relative importance of Aip1 appears to vary between organisms and cell types. In Drosophila melanogaster S2 cells (Rogers et al., 2003) and in Caenorhabditis elegans embryos (Ono, 2001), the inhibition of Aip1 function leads to severe defects in actin organization and accumulation of excess actin polymer. Deletion of Aip1 in yeast (Rodal et al., 1999) and Dictyostelium discoideum (Konzok et al., 1999) also perturbs actin dynamics and function, but the phenotypes are comparatively less severe. Our biochemical data implicate Aip1 in the disassembly of the highly branched Listeria comet tail that is assembled primarily by frequent actin nucleation by the Arp2/3 complex (Brieher et al., 2004). Cells can also assemble more highly bundled actin arrays through formin-mediated polymerization (Higashida et al., 2004), but the mechanisms that disassemble these arrays are not known. Variations in the severity of Aip1 (as well as coronin and cofilin) deletion between organisms and cell types might reflect differences in the relative importance of Arp2/3 versus formin-mediated actin arrays in different cells and alternative disassembly mechanisms.
In contrast to Aip1, coronin has not previously been implicated in actin depolymerization. However, genetic data and its intracellular localization are consistent with a role in actin turnover. Coronin shows synthetic genetic interactions with nonlethal point mutations in cofilin as well as with mutations in actin that slow ATP hydrolysis and, therefore, stabilize actin filaments (Goode et al., 1999). Coronin preferentially localizes to dynamic actin structures such as yeast cortical actin patches (Goode et al., 1999) as well as lamellipodia and phagocytic cups in higher eukaryotes (de Hostos et al., 1991, 1993; Mishima and Nishida, 1999). It is also abundant in Listeria comet tails (David et al., 1998). Our biochemical data suggest that one of the functions of coronin within these structures is to facilitate cofilin recruitment to these structures and, thus, fast turnover.
The combination of Aip1 and coronin lowers the concentration of cofilin required to disassemble actin comet tails at rates comparable with those seen in cells. Cellular cofilin concentrations have only been measured in a few cases, and the reported concentrations vary widely between organisms and cell types. For example, Xenopus laevis egg extracts contain 3 μM cofilin compared with 30 μM in platelets (Pollard et al., 2000). However, total cofilin measured in cells cannot be compared directly with the concentrations we used in Figs. 1, 4, and 5 because cofilin in cells is probably present partly in the form of cofilin–actin–ADP complexes, which are presumably inactive in depolymerization, as well as in the form of phosphocofilin, which is also relatively inactive (Abe et al., 1996; Moriyama et al., 1996). In the future, it will be important to measure the concentrations of free actin, cofilin, coronin, and Aip1 that are available for depolymerization. Our dose-response analysis suggests that low micromolar concentrations of recombinant cofilin are sufficient for fast disassembly in the presence of competing G actin as long as coronin and Aip1 are present in the 10–100 nM range. In yeast, total Aip1 (Rodal et al., 1999) and coronin (Goode et al., 1999) are approximately equimolar with cofilin. Coronin and Aip1 concentrations in other cells have not been measured. From our purification tables, we roughly estimate that our starting thymus cytosol contains at least 0.5 μM Aip1 and 1.4 μM coronin.
The response of disassembly to polymerizable G actin suggests the factor that prevents polymerization of barbed ends in Listeria tails is primarily Aip1 acting in cooperation with coronin and cofilin. Aip1 is known to suppress polymerization from barbed ends of cofilin-decorated filaments (Okada et al., 2002; Balcer et al., 2003; Ono et al., 2004). Others have cast capping protein in this role (Marchand et al., 1995), and the role of Aip1 in cofilin-mediated disassembly might simply be to cap barbed ends, thereby suppressing further growth and preventing the reannealing of severed filaments. Although Aip1 suppresses filament elongation, it is not as effective of a capping agent as cytochalasin D or the gelsolin–actin complex. However, it is more effective than either of these agents in facilitating cofilin-mediated actin disassembly, suggesting that Aip1 actively contributes to the disassembly reaction (Ono et al., 2004). Our purification strategy did not reveal a role for capping protein, and, in preliminary tests, we have not observed the potentiation of cofilin-mediated depolymerization by recombinant capping protein. Therefore, like Ono et al. (2004), we favor a more active role for Aip1 in actin disassembly. Our data suggest that Aip1 in conjunction with coronin and cofilin contributes to a rate-limiting transition that converts stable, growing filaments into highly unstable filaments that rapidly disassemble (Fig. 8).
Figure 8.
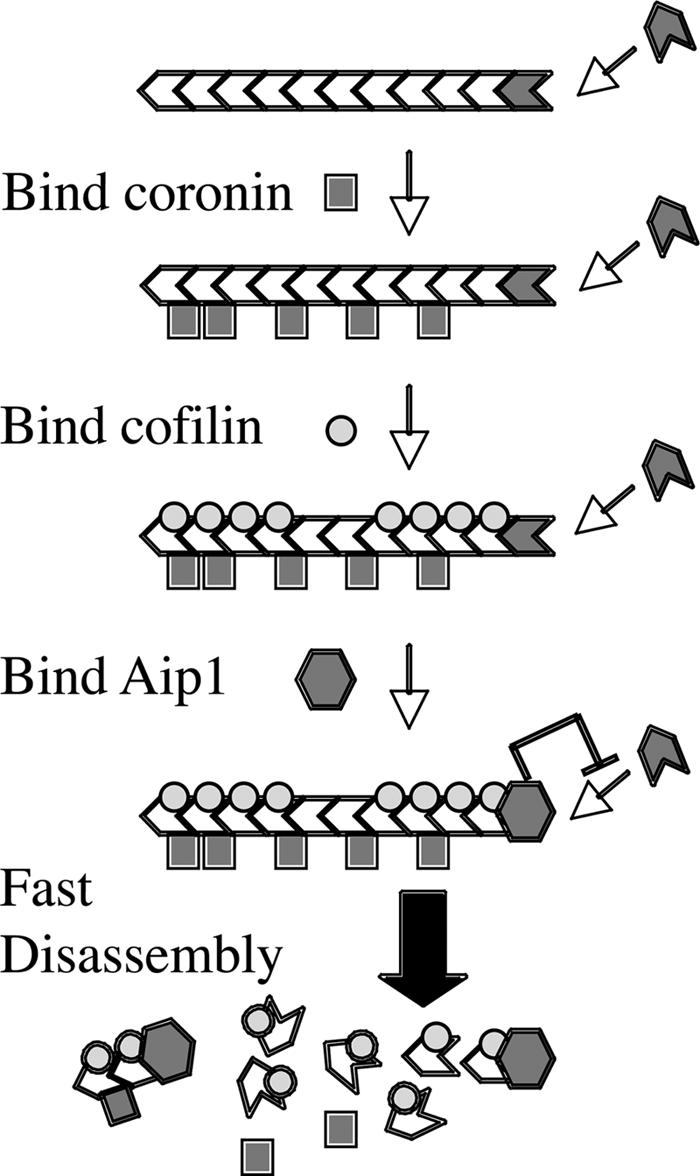
Model of actin filament disassembly by cofilin, coronin, and Aip1. Binding of coronin to a growing filament promotes cofilin binding, which, in turn, promotes binding of Aip1 to the barbed end. Aip1 inhibits elongation of the filament and triggers its fast disassembly. The binding of coronin, cofilin, and Aip1 contributes to a rate-limiting transition that converts a stable filament that can grow to an unstable filament, which rapidly shrinks.
Coronin promotes cofilin binding to actin comet tails as well as pure F actin filaments, and the increased binding might account for most, if not all, of coronin function in actin disassembly. Actin subunits within filaments can deviate from their canonical helical organization (Holmes et al., 1990) to one in which the subunits are rotated to a lesser extent, resulting in a change in the twist of the filament. Cofilin binds to these imperfections along the filament and, thus, destabilizes it (Galkin et al., 2003). Coronin could promote cofilin binding either by increasing the affinity of cofilin for these untwisted sites or by generating additional untwisted sites. We favor the latter because the rate of cofilin dissociation from comet tails was similar in both the presence and absence of exogenous coronin. Coronin had a greater effect on cofilin binding to Listeria comet tails than to pure actin filaments, suggesting that filaments within comet tails might be less disordered than pure filaments and therefore resist cofilin binding. If this is true, coronin binding to the comet tail somehow makes it easier for filaments within the tail to alter their twist and bind more cofilin.
Cells contain structurally distinct actin arrays that disassemble at different rates and possibly disassemble through different mechanisms. For example, the highly branched actin arrays that characterize both lamellipodia (Theriot and Mitchison, 1991) and Listeria actin comet tails (Theriot et al., 1992) turnover rapidly with a t 1/2 near 30 s, whereas the parallel bundles found in filopodia disassemble with a t 1/2 as long as 25 min (Mallavarapu and Mitchison, 1999). The biochemical mechanisms underlying these differences are not known. In the future, it will be important to determine the relative contributions of cofilin, coronin, Aip1, and other disassembly factors such as gelsolin to the disassembly of different actin arrays. Understanding the biochemical mechanisms controlling actin disassembly is essential for understanding the spatial organization of the actin cytoskeleton.
Materials and methods
Proteins
Rabbit skeletal muscle actin, bovine Arp2/3 (Brieher et al., 2004), and bovine profilin (Rozycki et al., 1991) were purified as described previously. Recombinant human cofilin (a gift from A. Weeds [Medical Research Council, Cambridge, United Kingdom] and A. McGough [Purdue University, West Lafayette, IN]) was purified as described previously (Maciver et al., 1998). Purified, recombinant, rhodamine-labeled actophorin-S88C from A. castellanii was provided by D. Mullins (University of California, San Francisco, San Francisco, CA). Actin was labeled with tetramethylrhodamine or AlexaFluor488 N-hydroxysuccinimidyl esters as described previously (Brieher et al., 2004). A point mutant of human cofilin (Ser108 to Cys) was generated by standard techniques expressed in bacteria and purified in the same way as wild-type cofilin. The mutant cofilin was labeled on the introduced cysteine with AlexaFluor568 maleimide.
HeLa cell extracts
Frozen cell pellets from 10 liters of culture were obtained from the National Cell Culture Center. Frozen pellets were thawed at 37°C and lysed with a 2:1 buffer (cell pellet volume of 20 mM Hepes, pH 7.4, 100 mM KCl, 2 mM MgCl2, 2 mM EGTA, 2% Triton X-100, 14.3 mM β-mercaptoethanol, and 0.2 mM PMSF). The lysate was centrifuged at 100,000 g for 1 h at 4°C, and the supernatant was dialyzed overnight against the lysis buffer lacking Triton X-100. The dialyzed sample was centrifuged at 100,000 g for 1 h at 4°C, and the supernatant was frozen in liquid nitrogen and stored at −80°C.
Listeria comet tail assembly
Chemically inactivated Listeria were prepared and introduced into perfusion chambers as described previously (Brieher et al., 2004). Comet tails were assembled by mixing 20 μl of assay buffer (100 mM Hepes, pH 7.8, 50 mM KCl, 2 mM MgCl2, 2 mM ATP, and 14.3 mM β-mercaptoethanol) with 5 μl HeLa extract, 0.1 μM Arp2/3, 4 μM actin, and 1 μM tetramethylrhodamine-actin. Comet tail assembly was allowed to proceed for 8–10 min at room temperature before proceeding with the disassembly reaction.
Listeria comet tail disassembly
After comet tails were assembled, the contents of the chamber were washed once with assay buffer containing 100 mM DTT, 4.5 mg/ml glucose, 250 μg/ml glucose oxidase, and 30 μg/ml catalase to scavenge oxygen and free radicals. The wash buffer was then replaced with either purified depolymerization factors or biochemical fractions from bovine thymus to induce disassembly. Depending on conditions, fluorescent images were taken every 5–20 s through a 20× NA 0.75 objective (Nikon) on a stand (90i; Nikon) using a cooled CCD camera (Orca-ER; Hamamatsu) and MetaMorph imaging software (Molecular Devices) using 2 × 2 binning.
To examine actin comet tail disassembly in the presence of the profilin–actin complex, equimolar amounts of profilin and G actin were combined to make a stock solution of 50 μM profilin and 50 μM actin in G buffer (2 mM Tris, pH 8, 0.2 mM CaCl2, 0.2 mM ATP, and 1 mM DTT) and incubated on ice for at least 1 h before mixing it with the appropriate depolymerizers just before assaying the mixture. Comet tail disassembly was quantified by first thresholding the images from a time-lapse stack two SDs above background fluorescence for each experiment. Background fluorescence was determined from the mean fluorescence intensity of regions within the frame that did not contain any actin comet tails or clouds. The thresholded image stack was then used to generate a binary image for each image from the time-lapse sequence. The binary image was multiplied by the corresponding raw image from the original time lapse using the Logical AND command in MetaMorph to generate a stack of images corrected for background fluorescence. Decay of the integrated intensity from these corrected images was used to determine comet tail t 1/2. The data were converted to an apparent disassembly rate by plotting ln2/t 1/2. Comet tail disassembly in vitro under our conditions does not necessarily show exponential kinetics, as is implied through this treatment of the data. However, it does serve as a convenient means to display the data. Therefore, we refer to these values as an apparent off rate.
Purification of depolymerizing factors from bovine thymus
200 g of frozen bovine calf thymus was thawed overnight at 4°C. The tissue was cut into small pieces and homogenized in buffer A (20 mM Tris, pH 7.4, 25 mM NaCl, 1 mM EGTA, 2 mM MgCl2, and 14 mM β-mercaptoethanol) in a Waring-type blender. The homogenate was centrifuged for 30 min at 8,000 g, and insoluble material was discarded. Polyethyleneimine was added to the supernatant to a final concentration of 0.05% and stirred for 30 min at 4°C. The slurry was centrifuged at 8,000 g for 30 min, and the pellet was discarded. The supernatant was centrifuged at 150,000 g at a k factor of 133 for 90 min. The supernatant was mixed with 200 ml DE52 (Whatman), stirred for 60 min, and allowed to settle. The liquid was decanted off the beads, and the beads were resuspended in 100 ml of buffer A. The slurry was poured into a column, and the flow through was combined with the decanted solution to generate the DE52 flow through fraction. The DE52 column was washed with an additional 400 ml of buffer A and eluted with an 800-ml gradient to 400 mM NaCl in buffer A to generate the DE52-bound fraction. DE52-bound factors that facilitated cofilin-mediated depolymerization eluted between 80–140 mM NaCl.
The DE52-bound fraction was dialyzed into 20 mM Pipes, pH 6.8, 25 mM NaCl, and 14 mM β-mercaptoethanol (buffer B), centrifuged at 100,000 g with a k factor of 220 for 1 h, and applied to a 70-ml S Sepharose column equilibrated in buffer B. The column was washed with two column volumes of buffer B and eluted with a 1-liter gradient to 400 mM NaCl in buffer B. The S flow through was designated fraction X and contains Aip1. Factor Y (coronin) elutes from the column near 270 mM NaCl.
Factor X (Aip1) was purified from the S flow through fraction. Solid (NH4)2SO4 was added slowly to the S flow through fraction to a final concentration of 2 M. The sample was centrifuged at 15,000 g for 30 min, and the pellet was discarded. The supernatant was applied to a 45-ml phenyl Sepharose HP column equilibrated in buffer B containing 2 M (NH4)2SO4. The column was washed with 1 vol of the same buffer and eluted with a 600-ml gradient to buffer B. Factor X eluted from the column near 500 mM (NH4)2SO4. Active fractions were dialyzed into 20 mM Tris, pH 8, 25 mM NaCl, and 14 mM β-mercaptoethanol (buffer C) and were applied to a 6-ml MonoQ column. The column was eluted with a 150-ml gradient to 500 mM NaCl in buffer C. Factor X eluted at 150 mM NaCl. Active fractions were dialyzed against buffer B and applied to a 5-ml hydroxyapatite column equilibrated in buffer B. The column was eluted with a 100-ml gradient to 500 mM sodium phosphate, pH 6.8. Factor X eluted at 175 mM sodium phosphate. Active fractions were concentrated using a centrifugal ultrafiltration device with a nominal cutoff of 30 kD. The retentate was loaded on a 1 × 30-cm Superose 12 gel filtration column. Activity eluted near 65 kD. At this point, factor X was pure and was identified by mass spectrometry.
Factor Y was purified from the S-bound material. Active S-bound fractions were dialyzed into buffer D (20 mM Hepes, pH 7.4, 25 mM NaCl, and 14 mM β-mercaptoethanol). The dialyzed sample was applied to a 10-ml heparin column equilibrated in buffer D. The column was eluted with a gradient to 1 M NaCl in buffer D. Factor Y eluted near 350 mM NaCl. Active fractions were dialyzed into buffer B and applied to a 5-ml hydroxyapatite column equilibrated in buffer B. The column was eluted with a 75-ml gradient to 500 mM sodium phosphate. Factor Y eluted near 80 mM sodium phosphate. Active fractions were dialyzed into buffer C and applied to a 1-ml MonoQ column equilibrated in buffer C. The column was eluted with a 20-ml gradient to 400 mM NaCl in buffer C. Factor Y eluted from the column near 150 mM NaCl. At this point, factor Y was pure and was identified by mass spectrometry.
Mass spectrometry
Gel bands were excised, diced, and subjected to in-gel digestion with 12.5 ng/μl of sequencing grade trypsin (Promega) in 50 mM ammonium bicarbonate overnight at 37°C. Peptides were extracted with 50% acetonitrile (MeCN) and 2.5% formic acid (FA) and were then dried. Peptides were resuspended in 2.5% MeCN and 2.5% FA and were loaded using an autosampler onto a microcapillary column packed with 5 μm of reverse-phase MagicC18 material (200 Å; Michrom Bioresources, Inc.). Elution was performed with a 5–35% MeCN (0.1% FA) gradient over 60 min after a 15-min isocratic loading at 2.5% MeCN and 0.5% FA. Mass spectra were acquired in a linear ion trap mass spectrometer (Finnigan LTQ; Termo Electron) over the entire run using 10 mass spectrometry/mass spectrometry scans after each survey scan. Raw data were searched for fully tryptic peptides against the National Center for Biotechnology Information nonredundant indexed database using Sequest software (Thermo Finnigan) and a mass tolerance of 2 D. After the identification of AIP1 and coronin-1A in the respective bands, a BLASTP search was performed to identify related sequences from Bos taurus. Two such sequences were identified for AIP1 (gi 76620344 and gi 86821067), and four were identified for coronin-1A (gi 27806251, gi 73587271, gi 76639125, and gi 76676005). Mass spectrometry data were then researched simultaneously against either the set of two sequences for AIP1 or the set of four sequences for coronin-1A with trypsin off and a mass tolerance of 2 D.
Cofilin binding to Listeria actin comet tails
Cofilin binding to actin comet tails was measured using the mutant cofilin (S108C) labeled with AlexaFluor568 on the introduced cysteine. Comet tails were assembled in perfusion chambers in the presence of AlexaFluor488-labeled actin to mark the comet tails. The chambers were then washed twice in assay buffer and incubated either in assay buffer or 1 μM coronin in assay buffer for 5 min. The chambers were washed twice with assay buffer, incubated with varying concentrations of labeled cofilin for 2 min, and fixed with 1% glutaraldehyde in the same buffer. Fluorescent images were collected as described above for Listeria comet tail disassembly except a 40× NA 0.95 objective (Nikon) was used instead of a 20× objective. Fluorescence intensity of the actin and cofilin signals was corrected for background and integrated as described for Listeria comet tail disassembly, and the corrected cofilin signal was normalized to the corrected actin signal.
Dissociation of S108C cofilin from comet tails was measured by washing the perfusion chamber with three chamber volumes of assay buffer and acquiring a time-lapse sequence. The amount of cofilin remaining bound to the comet tails over time was normalized to the amount bound immediately after the washing step (t = 0) using the same processing methods described above.
Actin sedimentation assays
5 μM actin was polymerized in assay buffer for 1 h at room temperature and mixed with combinations of coronin, cofilin, and Aip1 at the concentrations indicated in additional assay buffer to give a final concentration of actin of 2.5 μM. After 15 min at room temperature, the samples were centrifuged at 486,000 g for 30 min at 20°C. Equal portions of the supernatant and pellet were separated by SDS-PAGE and stained with Coomassie. Densitometry of gel bands was performed using ImageJ software (National Institutes of Health).
Online supplemental material
Figures show amino acid sequences of Aip1 (Fig. S1), coronin (Fig. S2), and tryptic peptides identified by mass spectrometry with Xcorr values of >1.8 for doubly charged ions and 2 for triply charged ions. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200603149/DC1.
Supplementary Material
Acknowledgments
We thank members of the Mitchison laboratory for many insightful comments.
This work was supported by National Institutes of Health grant GM 48027. H.Y. Kueh is a Howard Hughes Medical Institute Predoctoral Fellow.
Abbreviations used in this paper: FA, formic acid; F actin, filamentous actin.
References
- Abe, H., T. Obinata, L.S. Minamide, and J.R. Bamburg. 1996. Xenopus laevis actin-depolymerizing factor/cofilin: a phosphorylation-regulated protein essential for development. J. Cell Biol. 132:871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcer, H.I., A.L. Goodman, A.A. Rodal, E. Smith, J. Kugler, J.E. Heuser, and B.L. Goode. 2003. Coordinated regulation of actin filament turnover by a high-molecular-weight Srv2/CAP complex, cofilin, profilin, and Aip1. Curr. Biol. 13:2159–2169. [DOI] [PubMed] [Google Scholar]
- Bamburg, J.R. 1999. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 15:185–230. [DOI] [PubMed] [Google Scholar]
- Brieher, W.M., M. Coughlin, and T.J. Mitchison. 2004. Fascin-mediated propulsion of Listeria monocytogenes independent of frequent nucleation by the Arp2/3 complex. J. Cell Biol. 165:233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron, L.A., T.M. Svitkina, D. Vignjevic, J.A. Theriot, and G.G. Borisy. 2001. Dendritic organization of actin comet tails. Curr. Biol. 11:130–135. [DOI] [PubMed] [Google Scholar]
- Chen, H., B.W. Bernstein, J.M. Sneider, J.A. Boyle, L.S. Minamide, and J.R. Bamburg. 2004. In vitro activity differences between proteins of the ADF/cofilin family define two distinct subgroups. Biochemistry. 43:7127–7142. [DOI] [PubMed] [Google Scholar]
- David, V., E. Gouin, M.V. Troys, A. Grogan, A.W. Segal, C. Ampe, and P. Cossart. 1998. Identification of cofilin, coronin, Rac and capZ in actin tails using a Listeria affinity approach. J. Cell Sci. 111:2877–2884. [DOI] [PubMed] [Google Scholar]
- de Hostos, E.L., B. Bradtke, F. Lottspeich, R. Guggenheim, and G. Gerisch. 1991. Coronin, an actin binding protein of Dictyostelium discoideum localized to cell surface projections, has sequence similarities to G protein subunits. EMBO J. 10:4097–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hostos, E.L., C. Rehfuess, B. Bradtke, D.R. Waddell, R. Albrecht, J. Murphy, and G. Gerisch. 1993. Dictyostelium mutants lacking the cytoskeletal protein coronin are defective in cytokinesis and cell motility. J. Cell Biol. 120:164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin, V.E., A. Orlova, M.S. VanLoock, A. Shvetsov, E. Reisler, and E.H. Egelman. 2003. ADF/cofilin use an intrinsic mode of F-actin instability to disrupt actin filaments. J. Cell Biol. 163:1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, M., X. Song, G. Mouneimne, M. Sidani, D.S. Lawrence, and J.S. Condeelis. 2004. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 304:743–746. [DOI] [PubMed] [Google Scholar]
- Goode, B.L., J.J. Wong, A.C. Butty, M. Peter, A.L. McCormack, J.R. Yates, D.G. Drubin, and G. Barnes. 1999. Coronin promotes the rapid assembly and cross-linking of actin filaments and may link the actin and microtubule cytoskeletons in yeast. J. Cell Biol. 144:83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashida, C., T. Miyoshi, A. Fujita, F. Oceguera-Yanez, J. Monypenny, Y. Andou, S. Narumiya, and N. Watanabe. 2004. Actin polymerization-driven molecular movement of mDia1 in living cells. Science. 303:2007–2010. [DOI] [PubMed] [Google Scholar]
- Holmes, K.C., D. Popp, W. Gebhard, and W. Kabsch. 1990. Atomic model of the actin filament. Nature. 347:44–49. [DOI] [PubMed] [Google Scholar]
- Ichetovkin, I., W. Grant, and J. Condeelis. 2002. Cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the Arp2/3 complex. Curr. Biol. 12:79–84. [DOI] [PubMed] [Google Scholar]
- Iida, K., and I. Yahara. 1999. Cooperation of two actin-binding proteins, cofilin and Aip1, in Saccharomyces cerevisiae. Genes Cells. 4:21–32. [DOI] [PubMed] [Google Scholar]
- Konzok, A., I. Weber, E. Simmeth, U. Hacker, M. Maniak, and A. Muller-Taubenberger. 1999. DAip1, a Dictyostelium homologue of the yeast actin-interacting protein 1, is involved in endocytosis, cytokinesis, and motility. J. Cell Biol. 146:453–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciver, S.K., B.J. Pope, S. Whytock, and A.G. Weeds. 1998. The effect of two actin depolymerizing factors (ADF/cofilins) on actin filament turnover: pH sensitivity of F-actin binding by human ADF, but not of Acanthamoeba actophorin. Eur. J. Biochem. 256:388–397. [DOI] [PubMed] [Google Scholar]
- Mallavarapu, A., and T. Mitchison. 1999. Regulated actin cytoskeleton assembly at filopodium tips controls their extension and retraction. J. Cell Biol. 146:1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand, J.B., P. Moreau, A. Paoletti, P. Cossart, M.F. Carlier, and D. Pantaloni. 1995. Actin-based movement of Listeria monocytogenes: actin assembly results from the local maintenance of uncapped filament barbed ends at the bacterium surface. J. Cell Biol. 130:331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima, M., and E. Nishida. 1999. Coronin localizes to leading edges and is involved in cell spreading and lamellipodium extension in vertebrate cells. J. Cell Sci. 112:2833–2842. [DOI] [PubMed] [Google Scholar]
- Mitchison, T.J. 1992. Compare and contrast actin filaments and microtubules. Mol. Biol. Cell. 3:1309–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama, K., K. Iida, and I. Yahara. 1996. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells. 1:73–86. [DOI] [PubMed] [Google Scholar]
- Okada, K., T. Obinata, and H. Abe. 1999. XAIP1: a Xenopus homologue of yeast actin interacting protein 1 (AIP1), which induces disassembly of actin filaments cooperatively with ADF/cofilin family proteins. J. Cell Sci. 112:1553–1565. [DOI] [PubMed] [Google Scholar]
- Okada, K., L. Blanchoin, H. Abe, H. Chen, T.D. Pollard, and J.R. Bamburg. 2002. Xenopus actin-interacting protein 1 (XAip1) enhances cofilin fragmentation of filaments by capping filament ends. J. Biol. Chem. 277:43011–43016. [DOI] [PubMed] [Google Scholar]
- Ono, S. 2001. The Caenorhabditis elegans unc-78 gene encodes a homologue of actin-interacting protein 1 required for organized assembly of muscle actin filaments. J. Cell Biol. 152:1313–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, S., K. Mohri, and K. Ono. 2004. Microscopic evidence that actin-interacting protein 1 actively disassembles actin-depolymerizing factor/Cofilin-bound actin filaments. J. Biol. Chem. 279:14207–14212. [DOI] [PubMed] [Google Scholar]
- Pollard, T.D., and G.G. Borisy. 2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 112:453–465. [DOI] [PubMed] [Google Scholar]
- Pollard, T.D., L. Blanchoin, and R.D. Mullins. 2000. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 29:545–576. [DOI] [PubMed] [Google Scholar]
- Ponti, A., P. Vallotton, W.C. Salmon, C.M. Waterman-Storer, and G. Danuser. 2003. Computational analysis of F-actin turnover in cortical actin meshworks using fluorescent speckle microscopy. Biophys. J. 84:3336–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodal, A.A., J.W. Tetreault, P. Lappalainen, D.G. Drubin, and D.C. Amberg. 1999. Aip1p interacts with cofilin to disassemble actin filaments. J. Cell Biol. 145:1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, S.L., U. Wiedemann, N. Stuurman, and R.D. Vale. 2003. Molecular requirements for actin-based lamella formation in Drosophila S2 cells. J. Cell Biol. 162:1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt, J., B.J. Agnew, H. Abe, J.R. Bamburg, and T.J. Mitchison. 1997. Xenopus actin depolymerizing factor/cofilin (xac) is responsible for the turnover of actin filaments in Listeria monocytogenes tails. J. Cell Biol. 136:1323–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozycki, M., C.E. Schutt, and U. Lindberg. 1991. Affinity chromatography-based purification of profilin:actin. Methods Enzymol. 196:100–118. [DOI] [PubMed] [Google Scholar]
- Theriot, J.A., and T.J. Mitchison. 1991. Actin microfilament dynamics in locomoting cells. Nature. 352:126–131. [DOI] [PubMed] [Google Scholar]
- Theriot, J.A., T.J. Mitchison, L.G. Tilney, and D.A. Portnoy. 1992. The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature. 357:257–260. [DOI] [PubMed] [Google Scholar]
- Welch, M.D., A. Iwamatsu, and T.J. Mitchison. 1997. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature. 385:265–269. [DOI] [PubMed] [Google Scholar]
- Yeoh, S., B. Pope, H.G. Mannherz, and A. Weeds. 2002. Determining the differences in actin binding by human ADF and cofilin. J. Mol. Biol. 315:911–925. [DOI] [PubMed] [Google Scholar]
- Yonezawa, N., E. Nishida, S. Maekawa, and H. Sakai. 1988. Studies on the interaction between actin and cofilin purified by a new method. Biochem. J. 251:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}