

Abstract
Approximately 2% of mammalian genes encode proteases. Comparative genomics reveals that those involved in immunity and reproduction show the most interspecies diversity and evidence of positive selection during evolution. This is particularly true of granzymes, the cytotoxic proteases of natural killer cells and CD8+ T cells. There are 5 granzyme genes in humans and 10 in mice, and it is suggested that granzymes evolve to meet species-specific immune challenge through gene duplication and more subtle alterations to substrate specificity. We show that mouse and human granzyme B have distinct structural and functional characteristics. Specifically, mouse granzyme B is 30 times less cytotoxic than human granzyme B and does not require Bid for killing but regains cytotoxicity on engineering of its active site cleft. We also show that mouse granzyme A is considerably more cytotoxic than human granzyme A. These results demonstrate that even “orthologous” granzymes have species-specific functions, having evolved in distinct environments that pose different challenges.
Introduction
Granzymes are cytotoxic serine proteases used by cytotoxic lymphocytes (CLs) to destroy virus-infected and malignant cells. They are delivered into the cytoplasm of the target cell by the pore-forming protein perforin. Once inside the target cell, granzymes cleave specific proteins and trigger apoptosis (for review see Russell and Ley, 2002).
Humans have 5 granzymes (A, B, H, K, and M), whereas mice have 10 granzymes, having duplicated the ancestral granzyme B/H gene. In the absence of reported human granzyme deficiency, understanding of granzymes has been built by experimental approaches involving knockout mice or the use of purified granzymes on cells or cell extracts. For example, loss of granzyme A (GrA) or granzyme B (GrB) in mice is not catastrophic, but loss of both results in a severe immunological defect. Numerous studies using purified human and rodent granzymes have identified substrates, regulators, and trafficking pathways, whereas granzyme- deficient mice are widely used to probe the contribution and importance of granzymes to immunity (Russell and Ley, 2002).
GrA and GrB are the major and most-studied granzymes. GrA cleaves substrates after basic residues and induces caspase-independent apoptosis (for review see Lieberman and Fan, 2003). GrB cleaves after acidic residues, primarily Asp, and induces apoptosis by directly or indirectly activating caspases, and by cleaving other proapoptotic proteins such as Bid or ICAD/DFF45 (for review see Trapani and Sutton, 2003). Studies using CL from knockout mice confirm that GrA and GrB trigger distinct apoptotic pathways and are thus complementary (Pardo et al., 2004).
The high sequence homology and conserved primary cleavage specificity of human and mouse granzymes has led to widespread and interchangeable use of human and mouse enzymes in experimental systems, usually without side-by-side comparisons being made. Because GrA is a unique gene in both species, it is reasonable to assume an orthologous relationship between mouse GrA (mGrA) and human GrA (hGrA), but recent structural data show that they have distinct extended substrate specificities (Bell et al., 2003), raising the possibility that they possess distinct functions. The situation with GrB is even more complicated. hGrB has multiple paralogues in the mouse and rat, suggesting that the roles fulfilled by a single enzyme in humans may be split among several in rodents (Pham et al., 1996; Revell et al., 2005).
Without detailed comparative knowledge of the characteristics of human and mouse granzymes, data from mice cannot be extrapolated with confidence to humans. Here, we show that the cytotoxicity, substrate preferences, and inhibitor interactions of hGrB and mGrB are substantially different. We also extend the study of Bell et al. (2003) to show that hGrA and mGrA have markedly different cytotoxic potential. Such differences probably reflect the two species' response to differing immune evolutionary pressure.
Results
hGrB is more cytotoxic than mGrB
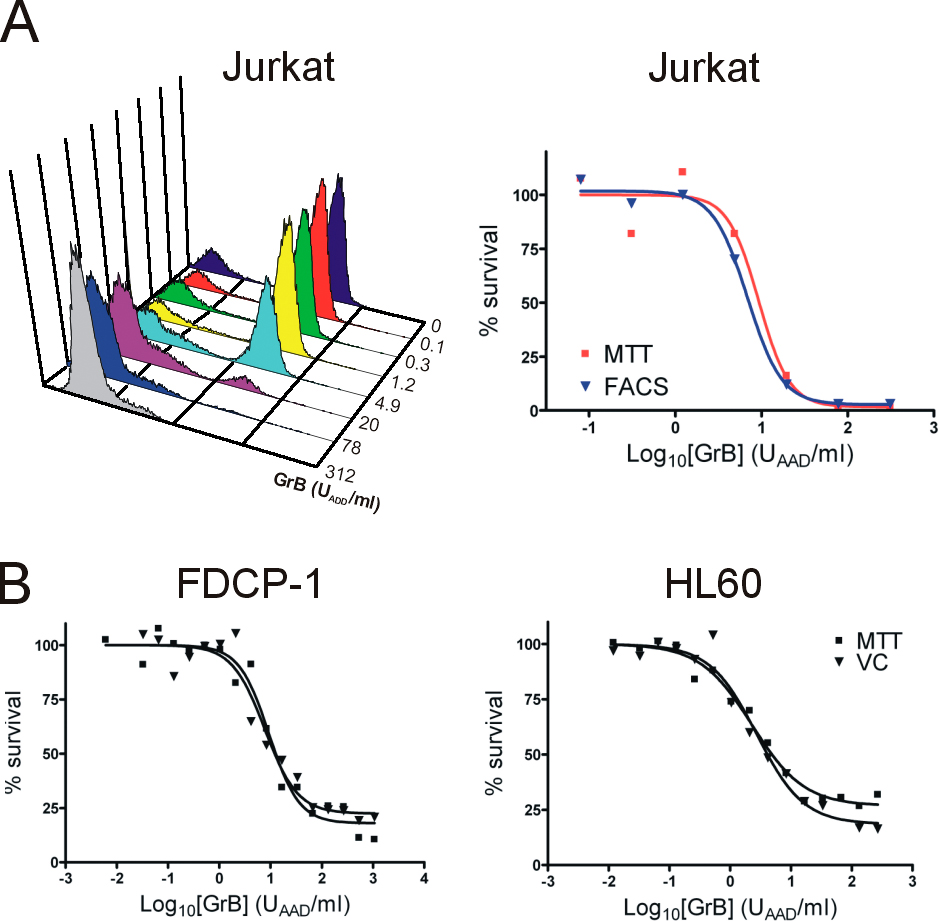
Granzyme cytotoxicity can be studied in vitro by adding purified perforin and granzyme to cultured cells and monitoring cell survival (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200606073/DC1). Keeping the concentration of perforin constant, granzyme cytotoxicity is quantitated using a dose–response approach, in which the amount of granzyme required to kill 50% of the target cells (EC50) is measured (Sun et al., 2004). Initial experiments using this system with either human or mouse cells showed that hGrB is substantially more cytotoxic than mGrB.
This apparent difference in cytotoxicity between mGrB and hGrB was confirmed using several different targets, as cell type–dependent differences in susceptibility to granzymes have been reported (Pardo et al., 2002). In one human cell line and three mouse cell lines examined in detail, hGrB was consistently more cytotoxic than mGrB (Fig. 1). With the exception of YAC-1, cells were 10–60-fold more sensitive to hGrB (Fig. 1). Killing of mouse YAC-1 cells required similarly high levels of hGrB or mGrB, suggesting that a key substrate is low or absent in these cells. mGrB was also 30-fold less effective in killing mouse lymphoma cells (Fig. 2 C).
Figure 1.

hGrB is more cytotoxic than mGrB in perforin-mediated cell death. (A) Cytotoxicity profiles of mGrB and hGrB on mouse and human cells. Cells were incubated for 1 h with hGrB (solid line) or mGrB (dashed line) in the presence of perforin. Cell survival was assessed after 24 h by MTT assay. A representative experiment is shown for each cell line, and the amount of enzyme required to kill 50% of the cells (EC50) is indicated on the graph. (B) Summary of data from replicate experiments. (C) Comparison of GrB cytotoxicity in the presence of SLO. P815 cells were exposed to mGrB or hGrB as described in A, except SLO was used instead of perforin.
Figure 2.

mGrB and hGrB have distinct substrate profiles and cytotoxic mechanisms. (A) mGrB inefficiently cleaves an optimal hGrB peptide substrate. Michaelis plots of peptide substrate hydrolysis by human (h) and mouse (m) GrB. Calculated specificity constants for each enzyme–substrate pair are tabulated below the plots. (B) mGrB inefficiently cleaves Bid. 35S-labeled mouse procaspase 3 or mouse Bid were incubated with hGrB or mGrB at 37°C for 30 min. 100 μM z-VAD was included in parallel reactions to prevent the second step in caspase 3 activation. Products were separated by 15% SDS-PAGE and visualized by fluorography. (C) mGrB does not require Bid to kill cells. Bid+/+ (solid line) or Bid−/− (dashed line) mouse B cell lymphomas (Waterhouse et al., 2005) were incubated for 1 h with hGrB or mGrB in the presence of perforin. Cell survival was assessed after 24 h by MTT assay. EC50 values from replicate experiments are indicated below.
To test whether poorer uptake of mGrB explains its inefficient killing, we compared accumulation of fluoresceinated mGrB and hGrB in Jurkat cells (Bird et al., 2005). No impairment in the rate or extent of accumulation of mGrB versus hGrB was noted in these cells (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200606073/DC1) or in the mouse P815 and lymphoma cell lines used in this study. The differences in cytotoxicity between mGrB and hGrB also remained when the bacterial porin, streptolysin O (SLO) was used instead of perforin (Fig. 1 C). SLO admits proteins directly to the cytoplasm via transient plasma membrane pores (Walev et al., 2001) and provides a perforin-independent mechanism for granzyme uptake, as shown by its ability to deliver mutant granzymes that cannot be delivered by perforin (Bird et al., 2005). The fact that mGrB killing is as inefficient in SLO as it is in perforin suggests that there is no restriction in perforin-mediated delivery of mGrB. Thus, differences in uptake or delivery of hGrB and mGrB do not explain their markedly different cytotoxic potential.
mGrB-mediated cytotoxicity does not require Bid
To investigate the basis for the differences in cytotoxicity between mGrB and hGrB, we compared their activities on peptide substrates and on the natural substrates procaspase 3 and Bid (Metkar et al., 2003; Waterhouse et al., 2005). Combinatorial peptide substrate analysis has identified the peptide IleGluXxxAsp↓ as an optimal recognition and cleavage sequence for hGrB and rat GrB (Thornberry et al., 1997; Harris et al., 1998). This appears in human and mouse Bid, and both are cleaved at this site by hGrB (Pinkoski et al., 2001). Although mGrB cleaves Bid (Thomas et al., 2001), the cleavage site has not been determined. A similar P4-P1 cleavage sequence is present on procaspase 3, but the downstream (P′) sequence differs from Bid. The nomenclature of cleavage site positions of a protease substrate follows Schechter and Berger (1967). The protease cleaves the peptide bond between P1 and P1′. Adjacent residues in the N-terminal direction are numbered P2, P3, P4, etc. On the carboxyl side of the cleavage site, residues are numbered P2′, P3′, P4′, etc. The P1 residue is accommodated in the S1 pocket of the catalytic cleft, the P2 residue is accommodated in the S2 pocket, and so forth.
mGrB and hGrB both cleaved the small peptide substrate Boc-AlaAlaAsp-thio benzyl ester (AAD-sbzl) with similar efficiency (Fig. 2 A, right), consistent with their known Aspase activities, indicating that their apparent functional distinctions are not due to differences in ability to hydrolyse a P1(Asp)-P1′ bond. As expected, hGrB also efficiently cleaved IleGluThrAsp-pNA (IETD-pNA; Fig. 2 A, left). In contrast, mGrB hydrolyzed IETD-pNA 30-fold less efficiently than hGrB (Fig. 2 A, left), suggesting that it cannot comfortably accommodate this peptide in its substrate binding pocket.
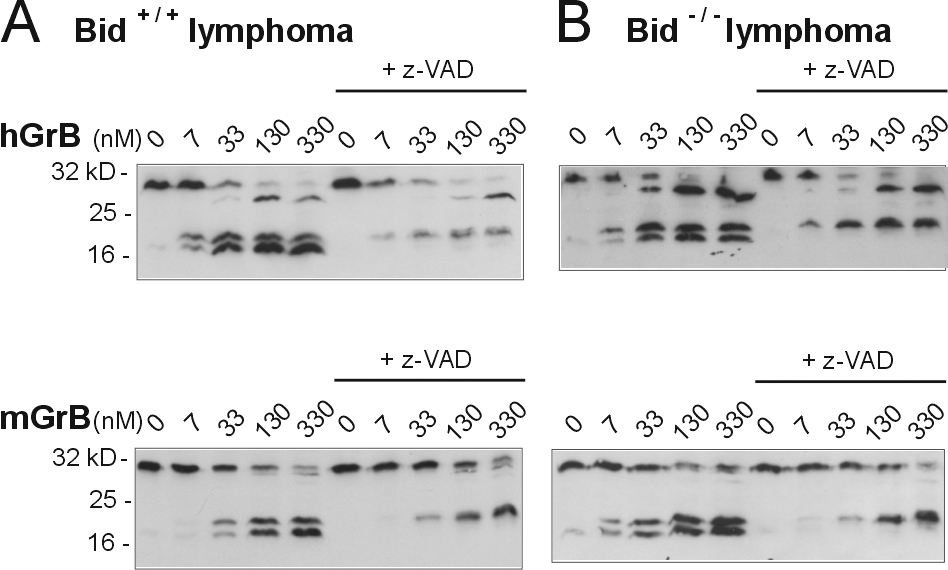
The markedly inferior ability of mGrB to cleave IETD-pNA also suggested that it would not cleave full-length Bid or procaspase 3 efficiently. To test this, we produced 35S-labeled mouse Bid and mouse procaspase 3 and incubated the protein with hGrB or mGrB. Under these conditions, complete cleavage of Bid was achieved in 25 nM hGrB, whereas 2 μM mGrB failed to completely cleave Bid (Fig. 2 B, right). Thus, mGrB cleaves Bid 80–100-fold less efficiently than hGrB. In contrast, the two granzymes cleaved procaspase 3 with similar efficiency (Fig. 2 B, left), suggesting that other factors such as P′ residues also influence cleavage (Table I and Fig. 3).
Table I.
Cleavage of Bid and phage display consensus peptides
| Substratea | mGrB
|
hGrB
|
||||
|---|---|---|---|---|---|---|
| k cat | K M | k cat/K M | k cat | K M | k cat/K M | |
| s−1 | μM | M−1s−1 | s−1 | μM | M−1s−1 | |
| Abz-IEPD↓SESQK(dnp)S (wt Bid cleavage site) |
0.049 ± 0.004 | 30.88 ± 2.48 | 1,575 ± 172 | 1.32 ± 0.09 | 18.96 ± 2.45 | 69,397 ± 10,161 |
| Abz-IEPD↓SGSQK(dnp)S (Bid site with P2′Gly) |
0.65 ± 0.18 | 59.67 ± 2.92 | 10,915 ± 3,101 | 5.11 ± 0.27 | 22.06 ± 2.67 | 231,449 ± 30,585 |
| Abz-LEYD↓LGALK(dnp)S (Phage display sequence) |
2.51 ± 0.16 | 23.48 ± 3.68 | 106,948 ± 18,094 | 0.008 ± 0.0004 | 122.9 ± 6.8 | 61.3 ± 4.5 |
Values presented are the mean ± SD (n = 3).
Peptide substrates include aminobenzoyl (Abz), dinitrophenyl (dnp), and C-terminal serine to enhance peptide solubility. Arrow indicates P1-P1′ bond and cleavage site.
Figure 3.

Comparison of hGrB and mGrB cleavage specificities by substrate phage display. (A) Statistical distribution of amino acids in sequences enriched in a P1 (Asp) substrate phage library cleaved by hGrB or mGrB (n = 70 and 102 peptides, respectively). Δσ is a measure of the difference between observed and expected frequencies in terms of standard deviation for each amino acid at each subsite (Matthews et al., 1994). A value of Δσ ≥ 5 (black bars) is considered to represent a significant deviation from the expected frequency. (B) Consensus cleavage sequences for mGrB and hGrB derived from phage display.
Previous studies using hGrB have indicated that Bid cleavage is a key event in GrB-mediated apoptosis (Goping et al., 2003; Sutton et al., 2003; Waterhouse et al., 2005). GrB initiates procaspase 3 activation by cleaving it once, but full activation requires a second cleavage, autocatalytically or via another caspase. This is illustrated in Fig. 2 B and Fig. S3 (available at http://www.jcb.org/cgi/content/full/jcb.200606073/DC1) by the inclusion of the caspase inhibitor z-VAD in the reactions. In vivo, this second cleavage step is prevented by inhibitor of apoptosis protein (IAP) bound to caspase 3 and will not occur unless IAP is displaced by Smac/Diablo released from mitochondria by Bid (Goping et al., 2003; Sutton et al., 2003). Thus, in the absence of Bid, hGrB cannot drive full caspase 3 activation.
To examine the importance of Bid in mGrB-mediated apoptosis, we compared the ability of hGrB and mGrB to kill mouse cells lacking Bid. As reported previously (Waterhouse et al., 2005), hGrB efficiently killed mouse lymphoma cells containing Bid (EC50 2.5 U/ml), but was >1,000-fold less cytotoxic toward lymphoma cells lacking Bid (Fig. 2 C, left). In contrast, mGrB killed the Bid+/+ and Bid−/− cells with similar efficiency, indicating that Bid cleavage is not essential for mGrB cytotoxicity (Fig. 2 C, right). These observations cannot be explained by differences in procaspase 3 levels between these lines or by differences in the ability of mGrB or hGrB to cleave procaspase 3 either in vitro or in cell extracts (compare Fig. 2 B and Fig. S3). It is also unlikely that mGrB can circumvent Bid by directly and fully activating procaspase 3, as z-VAD blocks the second cleavage event when procaspase 3 is exposed to mGrB (Fig. 2 B and Fig. S3). Collectively, the aforementioned results indicate that mGrB and hGrB have similar but distinct substrate specificities and that they trigger death differently.
Substrate phage display confirms that hGrB and mGrB have distinct specificities
To compare the substrate specificities of hGrB and mGrB, we used substrate phage display. Phage carry a variable (randomized) nonameric peptide linked to a His tag, fused to a coat protein Phage adsorbed to nickel-agarose are treated with protease, and cleaved phage are analyzed after several rounds of capture and release.
The granzymes were used to probe a library in which the fourth residue of the nonamer had been fixed as Asp. As shown in Fig. 3, phage enriched by hGrB treatment displayed a P4-P3-P2-P1-P1′-P2′-P3′ consensus sequence, [I/V][G/E]ADVLV. This closely matches the P4-P1 sequence previously identified by combinatorial peptide analysis (Thornberry et al., 1997; Harris et al., 1998) and validates the phage display approach. In contrast, treatment with mGrB yielded the consensus sequence [I/L]X[F/Y]DXGV. Specifically, the preferences at the P4, P2, and P2′ positions are different between mGrB and hGrB. mGrB shows an equal preference for Ile or Leu at P4 and an aromatic residue at P2, whereas hGrB strongly prefers Ile at P4 and has a relaxed requirement at P2. mGrB also has a very strong preference for Gly at P2′, whereas hGrB has no obvious preference at this position.
To confirm that mGrB can cleave a substrate with the aforementioned characteristics, we synthesized a quenched-fluorescence substrate peptide based on one of the phage sequences that met the consensus [L/I]X[F/Y]DXGX. This peptide abz-LEYDLGALK(dnp)S was cleaved by both mGrB and hGrB, but kinetic analysis showed that it is a 370-fold better substrate for mGrB (Table I, compare specificity constants). These results show that the consensus substrate sequences for hGrB and mGrB differ at the key P4, P2, and P2′ positions and indicate structural differences in the corresponding pockets in the catalytic clefts of the two enzymes.
Efficient cleavage of Bid by mGrB requires a P2′ Gly
As shown in Fig. 2, mGrB cleaves Bid poorly but procaspase 3 relatively well. The substrate phage display results suggest that mGrB requires Gly at the P2′ to efficiently cleave substrates, but both mouse and human Bid have Glu at this position. To determine whether P2′ Gly enhances cleavage by mGrB, we synthesized a peptide substrate based on the GrB cleavage site in mouse Bid (IEPD↓SESQ) and a derivative with P2′ Gly (IEPD↓SGSQ). Both hGrB and mGrB cleaved the wild-type (wt) Bid substrate, but hGrB cleaved it >40-fold more efficiently (Table I). This is consistent with the 30-fold difference in the ability of the two proteases to cleave IETD-pNA (Fig. 2 A). In contrast, mGrB cleaved the P2′ Gly substrate sevenfold better than the wt Bid substrate, whereas hGrB showed a threefold improvement over cleavage of the wt sequence, consistent with the slight overrepresentation of Gly at P2′ in the phage display data (Fig. 3). Thus, mGrB prefers P2′ Gly in natural substrates, and its poor cleavage of Bid can be explained partly by lack of Gly at this position. Given the conservation of the P4-P1 sequence in Bid and procaspase 3 (IEXD), these results also explain why procaspase 3 is apparently a better substrate of mGrB than Bid (Fig. 2 B): mouse procaspase 3 has a P2′ Gly at its GrB cleavage site.
mGrB can be “humanized” by alteration of its S4/S3 subsite
The inability of mGrB to cleave after IETD suggested differences between mGrB and hGrB in the substrate binding cleft, presumably in the pockets that interact with the side chains of the P4, P3, or P2 residues of a substrate. Inspection of the structure of hGrB bound to the tetrapeptide aldehyde inhibitor IEPD-CHO (Rotonda et al., 2001) revealed that Asn218 in the S3 pocket forms hydrogen bonds with the P3 Glu of the substrate, whereas Tyr174 in the S4 pocket undergoes hydrophobic interactions with P4 Ile of the substrate (Fig. 4 A). These two residues are conserved in rat GrB and have been shown to influence substrate specificity (Ruggles et al., 2004). In contrast, they are not conserved in mGrB (Fig. 4 B, Arg174 and Lys218). Thus, we predicted that mutating mGrB toward hGrB by replacing Arg174 with Tyr and Lys218 with Asn would enable it to more efficiently cleave IETD and Bid.
Figure 4.

Structural differences in the S4 and S3 subsites of human and mouse GrB. (A) Interaction of the IEPD peptide with the active site cleft of hGrB. Shown is a section of the structure of hGrB (green ribbon) complexed to the inhibitor IEPD-CHO (red stick; available from Protein Data Bank under accession no. 1IAU; Rotonda et al., 2001). Highlighted are the GrB residues Y174 and N218 predicted to interact with the P4 and P3 residues of the peptide. The active site serine (S195) is also indicated. (B) Alignment of the human, mouse, and rat GrB sequences. This illustrates that Y174 and N218 are not conserved in the mouse. R226 (shown in purple), which is at the bottom of the S1 pocket and interacts with P1 Asp, is conserved. Catalytic triad residues are indicated in yellow.
As shown in Fig. 5 A, mutated mGrB retained the ability to cleave AAD-sbzl and showed substantially enhanced activity on IETD-pNA, with k cat/K m values on both substrates similar to those of hGrB (compare Fig. 5 A with Fig. 2 A). In addition, the mutant mGrB showed enhanced ability to cleave Bid, together with a slight increase in the ability to cleave procaspase 3 (Fig. 5 B). This translated into an increased ability to kill the Bid+/+ lymphomas (Fig. 5 C). Interestingly, the mutated mGrB also showed an increased ability to kill the Bid−/− lymphomas (Fig. 5 C), suggesting that rather than being simply converted to hGrB selectivity, it has gained the ability to cleave hGrB-specific targets (e.g., Bid) while retaining the ability to cleave mGrB-specific targets. Finally, mutant mGrB showed 100-fold enhanced ability to kill P815 cells, with similar efficiency to hGrB (Fig. 5 D, left).
Figure 5.

Humanized mGrB shows improved substrate cleavage and cytotoxic potential. (A) Michaelis plots of peptide substrate cleavage by wt and humanized (mut) mGrB. Calculated specificity constants for each enzyme–substrate pair are tabulated below the plots. (B) Cleavage of mouse Bid and mouse procaspase 3 by wt and humanized mGrB. 35S-labeled mouse procaspase 3 or Bid were incubated with wt mGrB or humanized mGrB at 37°C for 30 min. 100 μM z-VAD was included in parallel reactions to prevent the second step in caspase 3 activation. Products were separated by 15% SDS-PAGE and visualized by fluorography. (C) Increased cytotoxicity of humanized mGrB on Bid+/+ and Bid−/− mouse lymphoma cells. Cells were incubated for 1 h with hGrB (closed squares; solid line), wt mGrB (closed triangles; solid line), or humanized mGrB (open circles; dashed line) in the presence of perforin. Cell survival was assessed after 24 h by MTT assay. Collated EC50 values from these experiments and those described in Figs. 1 and 2 C are indicated below representative curves. (D) Increased cytotoxicity and lack of regulation of humanized mGrB by SPI6 in P815 cells. Symbols and lines are described in C.
At present, the reason for the difference in mutant mGrB killing of the lymphoma cells (two- to fourfold increase) compared with P815 cells (100-fold increase) is unclear, but it is possible that the altered mGrB cleaves a target in P815 cells that is absent from the lymphoma cells. Nevertheless, collectively, the aforementioned results confirm that mGrB has a substrate binding cleft that is structurally distinct from hGrB, whichexplains its different substrate specificity profile and killing mechanism.
Serpins that regulate hGrB and mGrB cytotoxicity are not conserved
hGrB is regulated by the intracellular protease inhibitor, PI-9, a member of the serpin superfamily (Bird et al., 1998; Hirst et al., 2003). The serpin inhibitory mechanism requires recognition of an exposed reactive center loop (RCL) as a potential substrate by a cognate protease, followed by cleavage between the P1 and P1′ residues. This triggers conformational change in the serpin, distorting the active site of the protease and trapping an intermediate consisting of the protease bound to the serpin (for review see Gettins, 2002). hGrB cleaves the PI-9 RCL after Glu, consistent with its preference for acidic residues, and is very rapidly trapped (Sun et al., 2001).
Mice have seven PI-9 homologues (Kaiserman et al., 2002), but the counterpart of PI-9 is not immediately obvious. The closest homologue, SPI6, has not been tested with mGrB but inhibits hGrB 20-fold slower than PI-9 (Sun et al., 1997). When overexpressed, SPI6 protects dendritic cells (Medema et al., 2001) and T cells (Phillips et al., 2004) from CL attack and increases the survival of CD8+ memory cells (Phillips et al., 2004). Loss of SPI6 results in diminished survival of CTL (Zhang et al., 2006). However, none of these studies has demonstrated that SPI6 directly or efficiently inhibits mGrB. To determine whether SPI6 is a mouse GrB inhibitor, we identified the cleavage site for mGrB in the SPI6 RCL and measured the kinetics of the interaction, assessed the ability of SPI6 and mGrB to interact in CL extracts, and tested whether SPI6 protects cells against death mediated by purified mGrB and perforin.
The hGrB–PI-9 interaction requires cleavage of the PI-9 RCL at Glu340 (Sun et al., 2001; Fig. 6 A), and mGrB also cleaves PI-9 at this point (unpublished data). N-terminal sequencing of the 6-kD fragment generated by treatment of SPI6 with mGrB or hGrB showed that cleavage occurs at Cys339, adjacent to the predicted P1 Cys340 (Fig. 6 A). This is a previously unrecognized cleavage specificity of GrB and broadens the range of potential substrates.
Figure 6.

hGrB and mGrB are regulated by distinct, species-specific serpins. (A) Comparison of PI-9 and SPI6 RCLs showing GrB cleavage sites identified by N-terminal sequencing. The predicted P1 residue is indicated in large, bold font. (B) Serpins, granzymes, and complexes in CL extracts were visualized by sequential immunoblotting. Lysis of cells in LSB traps preformed complex. Cell disruption in NP-40 and incubation for 10 min at 37°C allows postlysis formation of complexes. (C) Species-specific protection by serpins from GrB-mediated death. Human SKW6.4 cells (closed squares; solid line) or SKW6.4 cells expressing PI-9 (open squares; dashed line) were incubated for 1 h with hGrB or mGrB in the presence of perforin. Cell survival was assessed after 24 h by MTT assay. Similar experiments were performed using mouse P815 cells (closed triangles; solid line) or P815 cells expressing SPI6 (open triangles; dashed line). Collated EC50 values from these experiments and those described in Fig. 1 are indicated below representative curves.
Although cleavage at Cys339 might allow capture of some mGrB by SPI6, the kinetics of the interaction will indicate whether mGrB is effectively regulated by SPI6 in vivo. Accordingly, we measured the rate constant (k ass) and stoichiometry of inhibition of SPI6 and mGrB complex formation (Table II). The results strongly suggest that SPI6 is a poor inhibitor of mGrB. The stoichiometry of inhibition indicates that fewer than one out of five contacts results in mGrB trapping, and the low k ass indicates that complex formation is very slow. SPI6 is even less effective in inhibiting hGrB (Table II). To put these results into context, PI-9 inhibits hGrB with equimolar stoichiometry and a rate of 1.2 × 106 M−1s−1 (Sun et al., 2001). Furthermore, the inhibition of mGrB by SPI6 is slower than the inhibition of hGrB by the viral serpin CrmA (Quan et al., 1995), which does not block GrB-mediated apoptosis in vivo.
Table II.
Inhibition of GrB by SPI6
| Parameter | n | mGrB | hGrB |
|---|---|---|---|
| Stoichiometry of inhibition | 3 | 5.8 ± 0.5 | >10 |
| Rate constant (k ass M−1s−1) | 2 | 5.6 ± 1.4 × 104 | 4.0 ± 0.3 × 103 |
Values presented are the mean ± SD.
Another signature of a serpin–protease interaction is the formation of SDS-stable complexes. For example, when human CLs are lysed, GrB is released from granules and rapidly binds cytosolic PI-9, and because GrB is in excess, essentially all of the PI-9 moves into complex (Fig. 6 B, left; Hirst et al., 2003). Similarly, in extracts of mouse CLs probed with anti-mGrB or anti-SPI6 antibodies, a complex containing mGrB and SPI6 was evident (Fig. 6 B, right). However, less than half of the SPI6 moved into complex, consistent with a less efficient interaction between this serpin and GrB, and indicating that the slow in vitro kinetics are not due to lack of a SPI6 cofactor present in CLs. To confirm that complex formation is GrB dependent, extracts were prepared from CLs taken from mice lacking GrB. No complex was evident in these cells (unpublished data).
If mouse and human GrB are orthologous, PI-9 and SPI6 should provide cross-species protection from GrB-mediated cell death. To test this, we challenged mouse P815 cells expressing SPI6 with hGrB and human SKW6.4 cells expressing PI-9 with mGrB (Fig. 6 C). As expected, PI-9 protected SKW6.4 cells against hGrB (Fig. 6 C, top left), increasing the EC50 ∼20-fold. SPI6 protected P815 cells against mGrB (Fig. 6 C, bottom right), increasing the EC50 >14-fold. Furthermore, the inactive SPI6T327R hinge mutant did not prevent mGrB-mediated death, indicating that the classic serpin inhibitory mechanism is involved in SPI6 cytoprotection (unpublished data). However, PI-9 did not protect cells against mGrB (Fig. 6 C, top right), nor did SPI6 effectively protect cells against hGrB (Fig. 6 C, bottom left).
Finally, the humanized mutant mGrB almost completely lost its ability to be regulated by SPI6, exhibiting a similar cytotoxicity profile to hGrB on SPI6-expressing cells (Fig. 5 D, right). This is consistent with the alterations to its active site cleft and substrate interactions and emphasizes the high specificity of a cognate serpin–protease interaction. From the aforementioned experiments, we concluded that mGrB is indirectly inhibited by SPI6 and that PI-9 and SPI6 are not functionally interchangeable. These results confirm that mGrB and hGrB have distinct substrate specificities and indicate that they are regulated differently in vivo.
Mouse and human GrA have significantly different cytotoxic potential
The functional differences that we identified between mGrB and hGrB raised the possibility that such interspecies differences would also be evident in other granzymes. Although mouse and human GrA cleave the peptide substrate benzyloxy-carbonyl-Lys-thiobenzyl ester with similar efficiency (Fig. 7 A; Odake et al., 1991), structural and substrate preference analysis of GrA has identified differences between the mouse and human enzymes in the active site cleft (Bell et al., 2003). This study identified an optimal P4-P1 substrate for hGrA as VANR↓, whereas the optimal mGrA substrate was identified as GYFR↓. Mutation of the hGrA S4 and S3 pockets converted it to a mouse-like GrA (Bell et al., 2003).
Figure 7.

Mouse but not human GrA induces perforin-dependent cell death. (A) Progress curves of mGrA (closed boxes) and hGrA (open boxes) cleaving benzyloxy-carbonyl-Lys-thiobenzyl ester, where 1 U of enzyme increases absorbance by 1 milli-OD/min. mGrA contains 144 ± 5 U/μg, whereas hGrA contains 18 ± 2 U/μg. This is consistent with the difference in kcat/KM previously reported (Odake et al., 1991), confirming that the enzymes are equally active. (B) 300 ng of recombinant hGrA was added to LSB with or without DTT and then boiled for 5 min before 10% gel SDS-PAGE and immunoblotting for hGrA. The image shows separated lanes originating from the same gel. (C) Human Jurkat cells or mouse lymphoma cells were incubated for 1 h with recombinant mGrA or recombinant hGrA in the presence or absence of perforin (Pf). Cell survival was assessed 24 h later by MTT assay. (D) HeLa cells were incubated for 4 h with recombinant hGrB, recombinant mGrA, or native hGrA in perforin. Cell survival was assessed by 51Cr release assay. Error bars indicate standard deviation (n = 8).
To determine whether these structural differences translate into differences in cytotoxic potential, we compared the ability of mGrA and hGrA to kill human or mouse cells in the presence of perforin. We used two different death assays: our standard MTT (3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) test, which measures loss of mitochondrial respiration (Fig. 7 C), and 51Cr release, which measures loss of membrane integrity (Fig. 7 D). As shown in Fig. 7 (C and D), mGrA efficiently killed mouse and human cells, but hGrA did not. mGrA-induced death was perforin dependent, as cells exposed to mGrA alone survived (Fig. 7 C). The inability of hGrA to kill cells was not related to the source of the protease, as both recombinant and native forms were used. All of the hGrA used was in the physiological 60-kD dimeric form, which could be reduced to the 30-kD monomeric form (Fig. 7 B). Monomeric hGrA also failed to kill cells (unpublished data).
Discussion
GrA and GrB are believed to be the primary cytotoxins in human and mouse CLs. The evidence presented here clearly demonstrates substantial interspecies distinctions that suggest caution when using mouse models to elucidate the molecular and pathophysiological functions of these proteases in humans. For example, loss of GrB may be much more devastating to humans than the mild phenotype of GrB-deficient mice would suggest, given that hGrB is more efficient than mGrB and that hGrA is apparently nonlethal and could not compensate for GrB absence.
Such interspecies differences between granzymes are illustrated by the GrB–Bid interaction, which indicates that distinct death pathways are initiated by hGrB and mGrB (Fig. 8 A). Our results show that hGrB is a much more effective killer than mGrB because it can cleave and activate Bid, whereas mGrB cleaves Bid poorly and does not require it for initiating apoptosis. The requirement for Bid in hGrB-mediated killing is not an artifact arising from its use on mouse target cells, as Bcl-2 overexpression in human cells blocks hGrB cytotoxicity but can be relieved by Bid coexpression (Waterhouse et al., 2006). The failure of hGrB to kill in the absence of Bid indicates that it lacks the capacity to independently and fully activate effector caspases, as suggested previously (Goping et al., 2003; Sutton et al., 2003). Its ability to cleave other substrates must also be insufficient to cause death. In contrast, mGrB probably directly activates another effector caspase, such as procaspase 7, or it relieves IAP inhibition of caspase 3 independently of Bid. It may also cause caspase-independent death by cleaving substrates such as ICAD or as-yet-undefined mitochondrial targets (Thomas et al., 2001).
Figure 8.
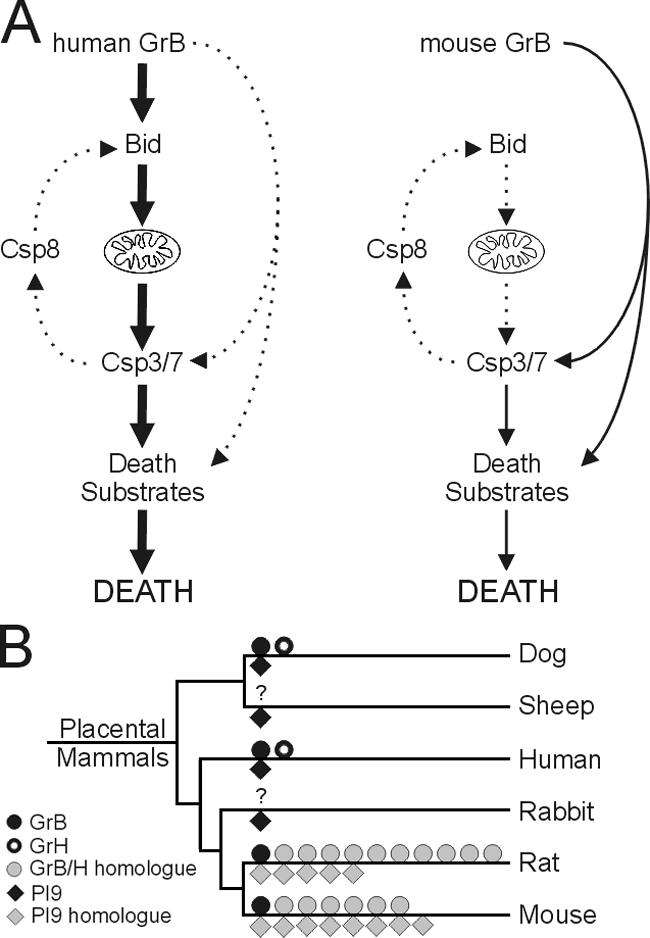
Functional divergence of human and mouse GrB. (A) hGrB and mGrB differ in cytotoxicity because hGrB has the capacity to efficiently activate Bid and initiate the mitochondrial death pathway, whereas mGrB lacks this capacity and must proceed through less efficient alternate pathways leading to activation of effector caspases or degradation of other death substrates. Activation of effector caspases 3 and 7 may also lead to a caspase amplification loop via caspase 8–mediated Bid cleavage, although the contribution of this loop to cell death is unclear. (B) Phylogram of selected mammalian species indicating expansion and divergence of GrB, GrH, and PI-9 homologues in rodents. GrB, GrH, and their homologues are indicated above the relevant branch, and PI-9 and its homologues are below. Dog, sheep, and rabbit have PI-9 genes with a conserved P1 Glu, and are thus considered orthologous to human. The question marks indicate genomes in which the GrB/H locus has not been sequenced or is too fragmented for analysis.
The differences between hGrB and mGrB are also emphasized by our ability to humanize mGrB and improve its capacity to cleave Bid. However, humanized mGrB remains relatively inefficient compared with hGrB, indicating that additional changes to the mGrB substrate binding pocket are required to fully accommodate Bid. Supporting evidence for differences between mGrB and hGrB can also be found in previous studies. For example, synthetic catalytic inhibitors designed to hGrB do not inhibit mGrB (Willoughby et al., 2002); the human adenoviral protein L4-100K inhibits hGrB but not mGrB (Andrade et al., 2003); mGrB does not process human Bid (Adrain et al., 2005); and mGrB does not require Bax or Bak for cytotoxicity, whereas hGrB does (Thomas et al., 2001; Cartron et al., 2003).
Similarly, hGrA and mGrA do not appear to be functionally equivalent, as we have shown that mGrA is cytotoxic and that hGrA is not. This is supported by several other lines of evidence. Modeling of mGrA on the structure of hGrA has identified differences in their active site clefts, substantiated by differences in peptide substrate preference and the conversion of hGrA to an mGrA-like protease by mutation of the S4/S3 pockets (Bell et al., 2003). Early work showed that mGrA is cytotoxic when produced with perforin in rat basophilic leukemia cells (Shiver et al., 1992), and many studies on the mechanisms of GrA-mediated death have used CTL and target cells from mice (Pardo et al., 2002, 2004). In contrast, recent studies using specific inhibitors show that hGrA is a minor contributor to CL cytotoxicity (Mahrus and Craik, 2005). Such findings do not necessarily overturn the idea that hGrA engages apoptotic pathways by cleaving specific subcellular proteins and generating reactive oxygen species (Pardo et al., 2004; Martinvalet et al., 2005), but they suggest that it does so much less efficiently than mGrA and that it is not sufficient to induce cell death.
The differences between hGrB and mGrB also extend to their regulation, as indicated by differences between PI-9 and SPI6. PI-9 is an efficient hGrB inhibitor that protects CLs and bystanders from misdirected GrB (Sun et al., 1996; Bird et al., 1998; Hirst et al., 2003), and SPI6 is a putative counterpart of PI-9 (Sun et al., 1997; Medema et al., 2001; Phillips et al., 2004). Mice lacking SPI6 have fewer CTL, and remaining CTL have fewer granules and less evidence of cytoplasmic mGrB (Zhang et al., 2006). Crossing SPI6-deficient mice with mGrB-deficient mice rescues the CTL defect, but because the latter mice also lack several mGrB paralogues (Pham et al., 1996), it cannot be assumed that SPI6 directly controls mGrB. Although the accumulating evidence is consistent with a cytoprotective role for both PI-9 and SPI6 in CLs, our present work strongly suggests that the question of the mechanism of cytoprotection by SPI6 remains open, as it is not an effective mGrB inhibitor. Given that other mGrB paralogues are cytotoxic, four possibilities exist: the primary target of SPI6 is a cytotoxic mGrB paralogue; SPI6 simultaneously controls multiple mGrB paralogues through a one-size-fits-all RCL; mGrB or paralogues activate a downstream apoptotic protease, which is efficiently inhibited by SPI6 (and not activatable by hGrB); or SPI6 protects granule integrity independently of granzyme inhibition (in which case, PI-9 would be predicted to control GrB and independently protect granule integrity). We consider the last two possibilities more likely because they explain why the rat, which has a more human-like GrB (Harris et al., 1998), has an orthologue of SPI6 rather than PI-9.
Among mammals, the multiple granzyme and serpin genes found in rodents appear to be the exception rather than the rule (Fig. 8 B), and it is evident that GrB and PI-9 paralogues emerged after divergence of Lagomorpha and Rodentia. A priori, the disappearance of the PI-9 RCL sequence from rodents suggests a fundamental change to the structure of mGrB, which we have confirmed by demonstrating that mGrB is catalytically distinct from hGrB and is not controlled by PI-9. Conversely, we have shown that hGrB is not controlled by SPI6. Although mGrB has changed, the GrB cleavage site in mouse Bid is conserved and is accessible to hGrB. Thus, it can be surmised that mGrB has lost cytotoxicity rather than hGrB gaining it. Conservation of the mouse Bid cleavage site suggests that another protease attacks it or that it serves another function in the molecule.
Why have these changes occurred in rodents? Given its role in immunity, the simplest explanation is that mGrB has evolved to target a virulence factor from a particular pathogen or to avoid inhibition by a specific pathogen protein. Specific targeting of virulence factors has been demonstrated for another leukocyte protease, neutrophil elastase (Weinrauch et al., 2002). Duplication of mGrB to generate paralogues may have allowed compensating cytotoxic mechanisms to evolve and/or multiple virulence factors to be targeted. The former possibility is supported by the reported cytotoxic functions of GrC and GrF (Revell et al., 2005). The appearance of cytotoxic mGrB paralogues may have then driven the evolution of cognate PI-9 paralogues.
Although functioning primarily as a cytotoxin, GrB probably has other roles. For example, in humans it is found in testis and has been implicated in reproduction (Hirst et al., 2001). It may also be involved in extracellular matrix remodeling (Buzza et al., 2005). With multiple substrates and roles, it is easy to envisage that particular functions of GrB may have devolved to separate mGrB paralogues in rodents. For example, mouse granyzme N appears to be restricted to the testis and may have assumed the testicular role of hGrB (Takano et al., 2004).
In closing, we have shown that mGrB is structurally and enzymatically distinct from hGrB and has a different killing mechanism. We have also shown that human GrA is not cytotoxic, whereas mGrA is substantially cytotoxic. Such species differences may also extend to granzymes M and K, but appropriate comparisons have yet to be made. It may be that the overall killing efficiency of human and mouse CLs is comparable and that the loss of GrB cytotoxicity in the mouse has been offset by a gain in GrA cytotoxicity or the evolution of GrB paralogues. Nevertheless, use of a granzyme from one species on cells or extracts from another species may produce misleading results because key substrates are missing or are inefficiently cleaved because recognition sites are not conserved. Use of mice to test inhibitors of human granzymes will fail because differences in active site topology of the mouse enzymes will prevent inhibitor binding. Likewise, use of mice to study viral infection may be compromised, as conserved virus inhibitors (e.g., SPI2/CrmA, Bcl-2 homologues, and AdL4-100K homologues) will interact differently with or fail to control mouse granzymes. Finally, use of mouse models to study the impact of granzymes on tumorigenesis is also problematic because the species-specific death pathways and substrates outlined here imply that tumors will subvert the immune system differently in mice and humans by up- or down-regulating different proteins.
Materials and methods
Granzymes and serpins
Recombinant serpins were produced in Pichia pastoris as described previously (Sun et al., 1995, 1997). Recombinant granzymes were also produced from P. pastoris (Sun et al., 1999, 2004). To improve expression levels, the wt mGrB cDNA was modified by QuikChange site-directed mutagenesis (Stratagene) to remove a cryptic polyadenylation site, using the oligonucleotide 5′-CACTGTGAAGGAAGTATAATCAATGTCACTTTGGGGGC-3′ and its complement. The S4/S3 subsites in mGrB were mutated using the oligos 5′-GAGTCCTACTTTAAAAATTATTACAACAAAACCAATCAG-3′ and its complement (R174Y) and 5′-TCCTATGGATATAATGATGGTTCACCTCC-3′ and its complement (K218N). Activity of recombinant GrB was assessed by cleavage of the peptide substrate benzyloxycarbonyl- AlaAlaAsp-thiobenzyl ester (Sun et al., 1999, 2004). The specific activity of hGrB was 90 U/μg, and the specific activity of mGrB was 120 U/μg. The proportion of active enzyme in each batch of purified protein was estimated by the ability to complex with PI-9 or SPI-6. Routinely, >90% of granzyme in a batch could complex with the appropriate serpin. Native human GrA was from lymphokine-activated killer cells. Activity of GrA was assessed by cleavage of the substrate benzyloxycarbonyl- Lys-thiobenzyl ester.
Kinetic analysis of substrate cleavage by granzymes and inhibition by serpins
Quenched fluorescence substrates were synthesized and used as described previously (Sun et al., 2001). Activity of granzymes on the peptide substrates Boc-AAD-thiobenzyl ester (BIOMOL Research Laboratories, Inc.) and IETD-pNA (Calbiochem) was assessed as described previously (Sun et al., 2001). Inhibition of granzymes by serpins was measured using standard approaches (Hopkins and Stone, 1995; Sun et al., 2001). Granzyme cleavage sites in SPI6 and PI-9 were identified by N-terminal sequencing (Sun et al., 2001).
Production of granzyme substrates via in vitro transcription and translation
35S-labeled mouse procaspase 3 or mouse Bid was produced from cDNAs in the expression vector pSVTf via in vitro transcription and translation (Sun et al., 2004). Cleavage by granzymes was assessed by adding a specified amount of protease directly to the translation mix and incubating for 30 min at 37°C. The caspase inhibitor z-VAD was obtained from Calbiochem. Products were separated by SDS-PAGE and visualized by fluorography.
Analysis of granzyme substrate specificity by phage display
Construction of substrate libraries based on bacteriophage T7 used the T7Select1-1b phage display system (Novagen). Each library was made by synthesizing a degenerate oligonucleotide, annealing it to complementary half-site oligonucleotides, ligating the resulting heteroduplex to vector arms, and adding it to a T7 phage packaging extract. The half-site oligonucleotides were 5′-GCCGCCTGGAGTGAGAG-3′ and 5′-AGCTTAGTGATGGTGATGGTGATG-3′. One library was constructed using the degenerate oligonucleotide 5′-AATTCTCTCACTCCAGGCGGC(NNK)9CATCACCATCACCATCACA-3′ (where N represents any nucleotide and K represents T/C). This added a randomized nonameric peptide and a hexahistidine tag to the C terminus of the 10B coat protein. The complexity of this “random” library was 7 × 106 plaque-forming units. Another library was constructed using 5′-AATTCTCTCACTCCAGGCGGC(NNK)3GAC(NNK)5CATCACCATCACCATCACA-3′. This encoded an aspartate near the middle of the randomized peptide. The complexity of this “fixed” library was 4 × 106 plaque-forming units. Each library was amplified to >1010 plaque-forming units. About 109 plaque-forming units of amplified phage were bound to nickel-chelated Sepharose beads at 4°C. Unbound phage was removed by washing the beads in phosphate-buffered saline containing 850 mM NaCl and 0.1% (vol/vol) Tween 20. After two further washes in phosphate-buffered saline containing 1 mM MgSO4, the suspension was split into two parts (treatment and control; control treatments were performed to assess the extent of phage release from beads in the absence of protease). 200 nM granzyme was added to the treatment tube, and both tubes were incubated overnight at 37°C. Plaque-forming units in the supernatants representing cleaved or released phage were then counted and amplified to form sublibraries for the next round of selection. Phage remaining bound to the beads was eluted with 0.5 M imidazole, and plaque-forming units were counted to assess cleavage efficiency. After several selection rounds, individual plaques were chosen at random for sequence analysis. Phage DNA was amplified by PCR using dedicated primers (T7Select cloning kit; Novagen). Sequencing of PCR products using the same primers was performed using the Big Dye 3.1 kit (GE Healthcare).
The sequencing results were analyzed to determine the statistical distribution of each amino acid at each position of the nonamer (Matthews et al., 1994), allowing for the redundancy of the code, the fact that only 32 out of 64 codons are represented by NNK, and the exclusion from the analysis of any sequences encoding a stop codon in the nonamer. In a binomial distribution of amino acids, Δσ yields the difference of the observed frequency from the expected frequency in terms of standard deviations:
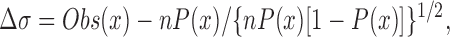 |
where Obs(x) is the number of times amino acid x occurs in the selected sequences, P(x) is the theoretical probability of amino acid x occurring, and n is the total number of sequences analyzed.
Cells, cell culture, and transfections
Human YT and Jurkat cells were maintained as described previously (Bird et al., 1998). Eμ-myc/bid−/− and Eμ-myc/bid+/+ mouse B cell lymphomas (Waterhouse et al., 2005) were obtained from R. Johnstone (Peter MacCallum Cancer Institute, Melbourne, Australia). Human SKW6.4 and mouse EL4 and YAC-1 cells were maintained in Dulbecco's modified Eagle medium containing 10% heat-inactivated fetal calf serum, 2 mM glutamine, and 55 μM β-mercaptoethanol. P815 cells were maintained in RPMI 1640 medium containing 10% heat-inactivated fetal calf serum, 2 mM glutamine, and 55 μM β-mercaptoethanol. Isolation and culture of mouse splenocytes followed standard procedures.
To generate SKW6.4 cells stably expressing PI-9, a PI-9 expression vector (Bird et al., 1998) was cotransfected by electroporation with a plasmid expressing neomycin resistance, and selection was performed in 1 mg/ml G418 (Sigma-Aldrich). Clones were established by limiting dilution and screened for PI-9 expression by indirect immunofluorescence and immunoblotting. FLAG-tagged SPI6 and SPI6T327R (containing an inactivating mutation in the proximal hinge) cDNAs were generated by PCR from the appropriate templates using the primer pair 5′-CGGGATCCATGGACTACAAAGACGATGACGATAAAGGGAATACTCTGTCTGAAG-3′ and 5′-GCTCTAGATTATGGAGATGAGAACCTGCCACA-3′. Products were cloned into the BamHI and XbaI sites of the expression vector pEF-PGKpuropA (Huang et al., 1997). 1.25 × 107 P815 cells were electroporated at 330 μF and 350 V with either pEFpuro/FlagSPI6 or pEFpuro/FlagSPI6T327R vectors linearized with SalI. Transfected pools were grown for 3 d in 1.1 μg/ml puromycin, and subsequently 45 SPI6 and 28 SPI6T327R lines were cloned by limiting dilution in puromycin selection. Clones were scored for relative SPI6 expression by indirect immunofluorescence using the α-FLAG M2 antibody. Expression was confirmed by immunoblotting using rabbit antiserum (R25) raised to recombinant SPI6. This antiserum does not recognize the highly related mouse serpins (Serpinb9b, Serpinb9e, Serpinb6, Serpinb6b, and Serpinb1) and can be considered specific for SPI6.
Cytotoxicity assays
Cell death mediated by granzymes and recombinant perforin was assessed as previously described (Sun et al., 2004; Bird et al., 2005). Recombinant perforin was produced using a baculovirus expression system (Liu et al., 1994). Recombinant SLO was prepared and used according to Walev et al. (2001).
Preparation of cell extracts, antibodies, and immunoblotting
For analysis of serpin complexes, cells were lysed in either 1 volume of Laemmli sample buffer (LSB) or 1/2 volume of NP-40 lysis buffer (50 mM Tris HCl, pH 8.0, 10 mM EDTA, and 1% [vol/vol] Nonidet P40), followed by incubation at 37°C for 10 min, after which 1/2 volume of 2× LSB was added. Viscosity was reduced by mechanically shearing the DNA using a needle and syringe. For analysis of caspase 3 cleavage, cells were lysed in NP-40 lysis buffer containing 1 μg/ml pepstatin, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 10 μg/ml PMSF, and a postnuclear supernatant was prepared by centrifugation at 16,000 g. Antibodies to PI-9 (R15) and hGrB (2C5 hybridoma supernatant) were used for immunoblotting at 1:2,000 and 1:100, respectively. A rat monoclonal antibody against mGrB was obtained from eBioscience (clone 16G6) and was used at 1:1,000. Goat anti-actin sera (Santa Cruz Biotechnology, Inc.) was used at 1:1,000. Rabbit antisera to SPI6 (R25) and hGrA (R045) was raised against recombinant protein purified from P. pastoris following standard procedures and was used at 1:1,000. Rabbit antisera to caspase 3 (Cell Signaling Technology) was used at 1:100. HRP-conjugated secondary antibodies was used at 1:5,000 and detected using enhanced chemiluminescence.
Image acquisition and manipulation
Images of blots were initially captured on x-ray films, which were subsequently scanned into Photopaint (Corel). Adjustments to brightness or contrast of digital images were applied to the whole image. No nonlinear adjustments were made.
Online supplemental material
Fig. S1 shows a comparison of methods used to assess cell survival after GrB treatment. Fig. S2 shows a comparison of uptake of mGrB and hGrB into target cells. Fig. S3 shows GrB-mediated procaspase 3 cleavage in Bid+/+ or Bid−/− mouse B cell lymphoma extracts. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200606073/DC1.
Supplementary Material
Acknowledgments
We thank K. Sedelies for assistance with 51Cr assays, Q. Zhou for help with construction of mutant mGrB, Dr. R. Johnstone for providing the Eμ-myc cells, and Dr. N. Waterhouse (Peter McCallum Cancer Institute) for advice and discussions.
This work was supported by program grants to P.I. Bird, J.C. Whisstock (284233), and J.A. Trapani from the National Health and Medical Research Council (Australia).
D. Kaiserman and C.H. Bird contributed equally to this paper.
Abbreviations used in this paper: CL, cytotoxic lymphocyte; IAP, inhibitor of apoptosis protein; MTT, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; RCL, reactive center loop; SLO, streptolysin O; wt, wild-type.
References
- Adrain, C., B.M. Murphy, and S.J. Martin. 2005. Molecular ordering of the caspase activation cascade initiated by the cytotoxic T lymphocyte/natural killer (CTL/NK) protease granzyme B. J. Biol. Chem. 280:4663–4667. [DOI] [PubMed] [Google Scholar]
- Andrade, F., L. Casciola-Rosen, and A. Rosen. 2003. A novel domain in adenovirus L4-100K is required for stable binding and efficient inhibition of human granzyme B: possible interaction with a species-specific exosite. Mol. Cell. Biol. 23:6315–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, J.K., D.H. Goetz, S. Mahrus, J.L. Harris, R.J. Fletterick, and C.S. Craik. 2003. The oligomeric structure of human granzyme A is a determinant of its extended substrate specificity. Nat. Struct. Biol. 10:527–534. [DOI] [PubMed] [Google Scholar]
- Bird, C.H., V.R. Sutton, J. Sun, C.E. Hirst, A. Novak, J.A. Trapani, and P.I. Bird. 1998. Selective regulation of apoptosis: the cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol. Cell. Biol. 18:6387–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird, C.H., J. Sun, K. Ung, D. Karambalis, J. Whisstock, J.A. Trapani, and P.I. Bird. 2005. Cationic sites on granzyme B contribute to cytotoxicity by promoting its uptake into target cells. Mol. Cell. Biol. 25:7854–7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzza, M.S., L. Zamurs, J. Sun, C.H. Bird, A.I. Smith, J.A. Trapani, C.J. Froelich, E.C. Nice, and P.I. Bird. 2005. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J. Biol. Chem. 280:23549–23558. [DOI] [PubMed] [Google Scholar]
- Cartron, P.F., P. Juin, L. Oliver, S. Martin, K. Meflah, and F.M. Vallette. 2003. Nonredundant role of Bax and Bak in Bid-mediated apoptosis. Mol. Cell. Biol. 23:4701–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gettins, P.G. 2002. Serpin structure, mechanism, and function. Chem. Rev. 102:4751–4804. [DOI] [PubMed] [Google Scholar]
- Goping, I.S., M. Barry, P. Liston, T. Sawchuk, G. Constantinescu, K.M. Michalak, I. Shostak, D.L. Robert, A.M. Hunter, R. Korneluk, and R.C. Bleackley. 2003. Granzyme B-induced apoptosis requires both direct caspase activation and relief of caspase inhibition. Immunity. 18:355–365. [DOI] [PubMed] [Google Scholar]
- Harris, J.L., E.P. Peterson, D. Hudig, N.A. Thornberry, and C.S. Craik. 1998. Definition and redesign of the extended substrate specificity of granzyme B. J. Biol. Chem. 273:27364–27373. [DOI] [PubMed] [Google Scholar]
- Hirst, C.E., M.S. Buzza, V.R. Sutton, J.A. Trapani, K.L. Loveland, and P.I. Bird. 2001. Perforin-independent expression of granzyme B and proteinase inhibitor 9 in human testis and placenta suggests a role for granzyme B-mediated proteolysis in reproduction. Mol. Hum. Reprod. 7:1133–1142. [DOI] [PubMed] [Google Scholar]
- Hirst, C.E., M.S. Buzza, C.H. Bird, H.S. Warren, P.U. Cameron, M. Zhang, P.G. Ashton-Rickhardt, and P.I. Bird. 2003. The intracellular granzyme B inhibitor, proteinase inhibitor 9, is up-regulated during accessory cell maturation and effector cell degranulation, and its overexpression enhances CTL potency. J. Immunol. 170:805–815. [DOI] [PubMed] [Google Scholar]
- Hopkins, P.C., and S.R. Stone. 1995. The contribution of the conserved hinge region residues of α1-antitrypsin to its reaction with elastase. Biochemistry. 34:15872–15879. [DOI] [PubMed] [Google Scholar]
- Huang, D.C., S. Cory, and A. Strasser. 1997. Bcl-2, Bcl-XL and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene. 14:405–414. [DOI] [PubMed] [Google Scholar]
- Kaiserman, D., S. Knaggs, K.L. Scarff, A. Gillard, G. Mirza, M. Cadman, R. McKeone, P. Denny, J. Cooley, C. Benarafa, et al. 2002. Comparison of human chromosome 6p25 with murine chromosome 13 reveals a greatly expanded ov-serpin gene repertoire in the mouse. Genomics. 79:349–362. [DOI] [PubMed] [Google Scholar]
- Lieberman, J., and Z. Fan. 2003. Nuclear war: the granzyme A-bomb. Curr. Opin. Immunol. 15:553–559. [DOI] [PubMed] [Google Scholar]
- Liu, C.C., P.M. Persechini, and J.D. Young. 1994. Characterization of recombinant mouse perforin expressed in insect cells using the baculovirus system. Biochem. Biophys. Res. Commun. 201:318–325. [DOI] [PubMed] [Google Scholar]
- Mahrus, S., and C.S. Craik. 2005. Selective chemical functional probes of granzymes A and B reveal granzyme B is a major effector of natural killer cell-mediated lysis of target cells. Chem. Biol. 12:567–577. [DOI] [PubMed] [Google Scholar]
- Martinvalet, D., P. Zhu, and J. Lieberman. 2005. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity. 22:355–370. [DOI] [PubMed] [Google Scholar]
- Matthews, D.J., L.J. Goodman, C.M. Gorman, and J.A. Wells. 1994. A survey of furin substrate specificity using substrate phage display. Protein Sci. 3:1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema, J.P., D.H. Schuurhuis, D. Rea, J. van Tongeren, J. de Jong, S.A. Bres, S. Laban, R.E. Toes, M. Toebes, T.N. Schumacher, et al. 2001. Expression of the serpin serine protease inhibitor 6 protects dendritic cells from cytotoxic T lymphocyte-induced apoptosis: differential modulation by T helper type 1 and type 2 cells. J. Exp. Med. 194:657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metkar, S.S., B. Wang, M.L. Ebbs, J.H. Kim, Y.J. Lee, S.M. Raja, and C.J. Froelich. 2003. Granzyme B activates procaspase-3 which signals a mitochondrial amplification loop for maximal apoptosis. J. Cell Biol. 160:875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odake, S., C.M. Kam, L. Narasimhan, M. Poe, J.T. Blake, O. Krahenbuhl, J. Tschopp, and J.C. Powers. 1991. Human and murine cytotoxic T lymphocyte serine proteases: subsite mapping with peptide thioester substrates and inhibition of enzyme activity and cytolysis by isocoumarins. Biochemistry. 30:2217–2227. [DOI] [PubMed] [Google Scholar]
- Pardo, J., S. Balkow, A. Anel, and M.M. Simon. 2002. The differential contribution of granzyme A and granzyme B in cytotoxic T lymphocyte-mediated apoptosis is determined by the quality of target cells. Eur. J. Immunol. 32:1980–1985. [DOI] [PubMed] [Google Scholar]
- Pardo, J., A. Bosque, R. Brehm, R. Wallich, J. Naval, A. Mullbacher, A. Anel, and M.M. Simon. 2004. Apoptotic pathways are selectively activated by granzyme A and/or granzyme B in CTL-mediated target cell lysis. J. Cell Biol. 167:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham, C.T.N., D.M. MacIvor, B.A. Hug, J.W. Heusel, and T.J. Ley. 1996. Long-range disruption of gene expression by a selectable marker cassette. Proc. Natl. Acad. Sci. USA. 93:13090–13095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, T., J.T. Opferman, R. Shah, N. Liu, C.J. Froelich, and P.G. Ashton-Rickhardt. 2004. A role for the granzyme B inhibitor serine protease inhibitor 6 in CD8+ memory cell homeostasis. J. Immunol. 173:3801–3809. [DOI] [PubMed] [Google Scholar]
- Pinkoski, M.J., N.J. Waterhouse, J.A. Heibein, B.B. Wolf, T. Kuwana, J.C. Goldstein, D.D. Newmeyer, R.C. Bleackley, and D.R. Green. 2001. Granzyme B-mediated apoptosis proceeds predominantly through a Bcl-2-inhibitable mitochondrial pathway. J. Biol. Chem. 276:12060–12067. [DOI] [PubMed] [Google Scholar]
- Quan, L.T., A. Caputo, R.C. Bleackley, D.J. Pickup, and G.S. Salvesen. 1995. Granzyme B is inhibited by the cowpox virus serpin cytokine response modifier A. J. Biol. Chem. 270:10377–10379. [DOI] [PubMed] [Google Scholar]
- Revell, P.A., W.J. Grossman, D.A. Thomas, X. Cao, R. Behl, J.A. Ratner, Z.H. Lu, and T.J. Ley. 2005. Granzyme B and the downstream granzymes C and/or F are important for cytotoxic lymphocyte functions. J. Immunol. 174:2124–2131. [DOI] [PubMed] [Google Scholar]
- Rotonda, J., M. Garcia-Calvo, H.G. Bull, W.M. Geissler, B.M. McKeever, C.A. Willoughby, N.A. Thornberry, and J.W. Becker. 2001. The three-dimensional structure of human granzyme B compared to caspase-3, key mediators of cell death with cleavage specificity for aspartic acid in P1. Chem. Biol. 8:357–368. [DOI] [PubMed] [Google Scholar]
- Ruggles, S.W., R.J. Fletterick, and C.S. Craik. 2004. Characterization of structural determinants of granzyme B reveals potent mediators of extended substrate specificity. J. Biol. Chem. 279:30751–30759. [DOI] [PubMed] [Google Scholar]
- Russell, J.H., and T.J. Ley. 2002. Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 20:323–370. [DOI] [PubMed] [Google Scholar]
- Schechter, I., and A. Berger. 1967. On the size of the active site in proteases. Biochem. Biophys. Res. Commun. 27:157–162. [DOI] [PubMed] [Google Scholar]
- Shiver, J.W., L. Su, and P.A. Henkart. 1992. Cytotoxicity with target DNA breakdown by rat basophilic leukemia cells expressing both cytolysin and granzyme A. Cell. 71:315–322. [DOI] [PubMed] [Google Scholar]
- Sun, J., P. Coughlin, H. Salem, and P. Bird. 1995. Production and characterization of recombinant human proteinase inhibitor 6 expressed in Pichia pastoris. Biochim. Biophys. Acta. 1252:28–34. [DOI] [PubMed] [Google Scholar]
- Sun, J., C.H. Bird, V. Sutton, L. McDonald, P.B. Coughlin, T.A. De Jong, J.A. Trapani, and P.I. Bird. 1996. A cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier A is present in cytotoxic lymphocytes. J. Biol. Chem. 271:27802–27809. [DOI] [PubMed] [Google Scholar]
- Sun, J., L. Ooms, C.H. Bird, V.R. Sutton, J.A. Trapani, and P.I. Bird. 1997. A new family of 10 murine ovalbumin serpins includes two homologs of proteinase inhibitor 8 and two homologs of the granzyme B inhibitor (proteinase inhibitor 9). J. Biol. Chem. 272:15434–15441. [DOI] [PubMed] [Google Scholar]
- Sun, J., C.H. Bird, M.S. Buzza, K.E. McKee, J.C. Whisstock, and P.I. Bird. 1999. Expression and purification of recombinant human granzyme B from Pichia pastoris. Biochem. Biophys. Res. Commun. 261:251–255. [DOI] [PubMed] [Google Scholar]
- Sun, J., J.C. Whisstock, P. Harriott, B. Walker, A. Novak, P.E. Thompson, A.I. Smith, and P.I. Bird. 2001. Importance of the P4' residue in human granzyme B inhibitors and substrates revealed by scanning mutagenesis of the proteinase inhibitor 9 reactive center loop. J. Biol. Chem. 276:15177–15184. [DOI] [PubMed] [Google Scholar]
- Sun, J., C.H. Bird, K.Y. Thia, A.Y. Matthews, J.A. Trapani, and P.I. Bird. 2004. Granzyme B encoded by the commonly occurring human RAH allele retains pro-apoptotic activity. J. Biol. Chem. 279:16907–16911. [DOI] [PubMed] [Google Scholar]
- Sutton, V.R., M.E. Wowk, M. Cancilla, and J.A. Trapani. 2003. Caspase activation by granzyme B is indirect, and caspase autoprocessing requires the release of proapoptotic mitochondrial factors. Immunity. 18:319–329. [DOI] [PubMed] [Google Scholar]
- Takano, N., H. Matsui, and T. Takahashi. 2004. Granzyme N, a novel granzyme, is expressed in spermatocytes and spermatids of the mouse testis. Biol. Reprod. 71:1785–1795. [DOI] [PubMed] [Google Scholar]
- Thomas, D.A., L. Scorrano, G.V. Putcha, S.J. Korsmeyer, and T.J. Ley. 2001. Granzyme B can cause mitochondrial depolarization and cell death in the absence of BID, BAX, and BAK. Proc. Natl. Acad. Sci. USA. 98:14985–14990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry, N., T.A. Rano, E.P. Peterson, D.M. Rasper, T. Timkey, M. Garcia-Calvo, V.M. Houtzager, P.A. Nordstrom, S. Roy, J.P. Vaillancourt, et al. 1997. A combinatorial approach defines specificities of members of the caspase family and granzyme B. J. Biol. Chem. 272:17907–17911. [DOI] [PubMed] [Google Scholar]
- Trapani, J.A., and V.R. Sutton. 2003. Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr. Opin. Immunol. 15:533–543. [DOI] [PubMed] [Google Scholar]
- Walev, I., S.C. Bhakdi, F. Hofmann, N. Djonder, A. Valeva, K. Aktories, and S. Bhakdi. 2001. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc. Natl. Acad. Sci. USA. 98:3185–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse, N.J., K.A. Sedelies, K.A. Browne, M.E. Wowk, A. Newbold, V.R. Sutton, C.J.P. Clarke, J. Oliaro, R.K. Lindemann, P.I. Bird, et al. 2005. A central role for Bid in granzyme B-induced apoptosis. J. Biol. Chem. 280:4476–4482. [DOI] [PubMed] [Google Scholar]
- Waterhouse, N.J., K.A. Sedelies, V.R. Sutton, M.J. Pinkoski, K.Y. Thia, R.W. Johnstone, P.I. Bird, D.R. Green, and J.A. Trapani. 2006. Functional dissociation of DeltaPsim and cytochrome c release defines the contribution of mitochondria upstream of caspase activation during granzyme B-induced apoptosis. Cell Death Differ. 13:607–618. [DOI] [PubMed] [Google Scholar]
- Weinrauch, Y., D. Drujan, S.D. Shapiro, J. Weiss, and A. Zychlinsky. 2002. Neutrophil elastase targets virulence factors of enterobacteria. Nature. 417:91–94. [DOI] [PubMed] [Google Scholar]
- Willoughby, C.A., H.G. Bull, M. Garcia-Calvo, J. Jiang, K.T. Chapman, and N.A. Thornberry. 2002. Discovery of potent, selective human granzyme B inhibitors that inhibit CTL mediated apoptosis. Bioorg. Med. Chem. Lett. 12:2197–2200. [DOI] [PubMed] [Google Scholar]
- Zhang, M., S.M. Park, Y. Wang, R. Shah, N. Liu, A.E. Murmann, C.R. Wang, M.E. Peter, and P.G. Ashton-Rickhardt. 2006. Serine protease inhibitor 6 protects cytotoxic T cells from self-inflicted injury by ensuring the integrity of cytotoxic granules. Immunity. 24:451–461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}