

Abstract
Differential modifications of proliferating cell nuclear antigen (PCNA) determine DNA repair pathways at stalled replication forks. In yeast, PCNA monoubiquitination by the ubiquitin ligase (E3) yRad18 promotes translesion synthesis (TLS), whereas the lysine-63–linked polyubiquitination of PCNA by yRad5 (E3) promotes the error-free mode of bypass. The yRad5-dependent pathway is important to prevent genomic instability during replication, although its exact molecular mechanism is poorly understood. This mechanism has remained totally elusive in mammals because of the lack of apparent RAD5 homologues. We report that a putative tumor suppressor gene, SHPRH, is a human orthologue of yeast RAD5. SHPRH associates with PCNA, RAD18, and the ubiquitin-conjugating enzyme UBC13 (E2) and promotes methyl methanesulfonate (MMS)–induced PCNA polyubiquitination. The reduction of SHPRH by stable short hairpin RNA increases sensitivity to MMS and enhances genomic instability. Therefore, the yRad5/SHPRH-dependent pathway is a conserved and fundamental DNA repair mechanism that protects the genome from genotoxic stress.
Introduction
DNA damage blocks the progression of replicative DNA polymerases and causes stalled replication forks at S phase. Persistent stalled replication forks collapse and cause genomic instability or cell death (Kolodner et al., 2002). In Saccharomyces cerevisiae, stalled replication forks are resolved either by bypassing DNA damage with translesion synthesis (TLS) polymerases or by template switching to the nascent strand of sister chromatid (Smirnova and Klein, 2003; Ulrich, 2005). The selection of these pathways is regulated through the modification of proliferating cell nuclear antigen (PCNA), a homotrimeric complex that encircles DNA strands and functions as a loading dock for DNA polymerases as well as various DNA repair machinery.
PCNA is monoubiquitinated at the lysine 164 (K164) by the ubiquitin ligase (E3) yRad18 with the ubiquitin-conjugating enzyme (E2) yRad6/Ubc2 after the cells are treated with a low concentration (0.02%) of the alkylating agent methyl methanesulfonate (MMS; Hoege et al., 2002). Monoubiquitinated PCNA switches the replicative DNA polymerase δ to nonessential polymerases specialized for TLS (Fig. 1 A; Friedberg et al., 2005). After the treatment of yeast cells with MMS, the same monoubiquitinated lysine residue of PCNA is further modified with a noncanonical lysine 63 (K63)–linked polyubiquitin chain by yRad5 (E3) along with the yUbc13–yMms2 (E2 and E2 variant) heterodimer complex (Hoege et al., 2002). This modification of PCNA presumably promotes the error-free mode of bypass, which is thought to use a template-switch type of recombination through reversal of stalled forks (Lawrence, 1994; Ulrich, 2005). However, nothing is known about molecular mechanisms downstream from PCNA polyubiquitination.
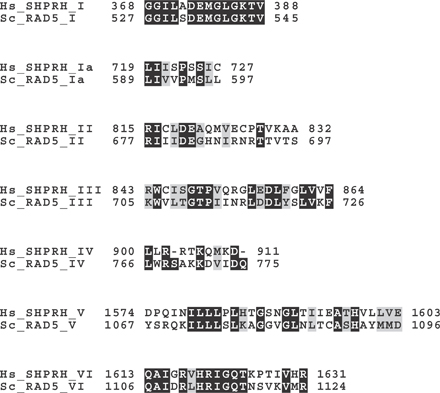
Figure 1.

Human SHPRH and yeast Rad5 share the domain structure. (A) Differential modifications of PCNA determine DNA repair pathways. R18, Rad18; R5, Rad5; R6, Rad6; 13, Ubc13; 2, Mms2. (B) Schematic representation of human (Hs) SHPRH and yeast (Sc) Rad5. SWI2/SNF2 (subdomains I, Ia, II, III, IV, V, and VI) and RING finger domains are indicated. Multiple alignments of RING finger domains of human (Hs), rat (Rn), mouse (Mm), zebrafish (Dr) SHPRH homologues, fission yeast (Sp) rad8 (a Rad5 homologue), and budding yeast (Sc) Rad5. Dots indicate the conserved cysteines and histidine. The cysteine mutated in this study (C1432) is indicated with an asterisk.
Mammalian PCNA also undergoes monoubiquitination after a low dose of MMS (0.02%) and UV irradiation, and monoubiquitinated PCNA preferentially binds to TLS polymerases (Kannouche et al., 2004; Friedberg et al., 2005). So far, no evident PCNA polyubiquitination has been observed in mammals. Furthermore, even though homologues of yRad18 (RAD18), yRad6 (HHR6A and HHR6B), yUbc13 (UBC13), yMms2 (MMS2/ UEV2), and downstream TLS polymerases have been identified (Koken et al., 1991; Hofmann and Pickart, 1999; Johnson et al., 1999; Masutani et al., 1999; Tateishi et al., 2000), no apparent RAD5 homologues have been discovered. Therefore, it has been controversial whether the mammalian error-free mode of bypass exists, and PCNA regulation through differential ubiquitinations is a conserved and fundamental mechanism in mammals.
SHPRH is a putative tumor suppressor gene encoding a large protein of 1,683 amino acids with various predicted functional domains, including SWI2/SNF2 and RING domains (Fig. 1 B; Sood et al., 2003). The human SHPRH gene was mapped to chromosome 6q24, which has been suggested to contain tumor suppressor genes. Four point mutations in the SHPRH gene were identified in melanoma and ovarian cancer–derived cell lines, although roles of SHPRH in cancer development have been largely unexplored (Sood et al., 2003).
In an effort to investigate whether mammals have a yeast Rad5–like pathway that prevents genomic instability, we identified SHPRH as a human RAD5 orthologue. We demonstrate that SHPRH has conserved biochemical properties with yeast Rad5 and suppresses genomic instability by promoting K63-linked polyubiquitination of PCNA.
Results and discussion
Human SHPRH promotes noncanonical K63-linked polyubiquitination of PCNA
Yeast Rad5 is a member of the SWI2/SNF2 family of helicases with the E3 activity (Johnson et al., 1992). A unique structural feature of yRad5 is that the RING domain for its E3 activity is embedded between the conserved motifs IV and V of the SWI2/SNF2 domain (Fig. 1 B). To identify human proteins with this unique domain structure, we performed a SMART search (http://smart.embl-heidelberg.de/) and identified a putative tumor suppressor gene, SHPRH. SHPRH and yRad5 show 45.5 and 36.4% identities and 62.1 and 47.3% similarities in the SWI2/SNF2 and RING domains, respectively, but have little homology in other sequences (Fig. 1 B and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200606145/DC1). SHPRH also contains predicted linker-histone and PHD-finger domains, which are not found in yRad5. Just like mammalian RAD18, which shares even higher sequence similarity with yRAD18 (Tateishi et al., 2000), SHPRH expression could not rescue the UV sensitivity of the rad5 strain (Fig. S2).
To test whether SHPRH is a functional orthologue of yRad5, we examined whether SHPRH could polyubiquitinate PCNA. The overexpression of HA-tagged ubiquitin (HA-Ub), FLAG-tagged PCNA (FLAG-PCNA), and myc-His–tagged SHPRH (SHPRH-myc-His) in human embryonic kidney (HEK) 293T cells induced mono- and polyubiquitinations of PCNA (Fig. 2 A, lane 5). PCNA polyubiquitination by SHPRH was further enhanced by UBC13–MMS2 (Fig. 2 A, lane 7) but not by RAD6 (lane 6) or UBC13 with the C87A mutation, which inactivates the E2 activity of UBC13 (lane 8). In contrast, RAD18 exclusively induced PCNA monoubiquitination with RAD6 (Fig. S3 A, lanes 6 and 7, available at http://www.jcb.org/cgi/content/full/jcb.200606145/DC1). Enhancement of PCNA monoubiquitination by SHPRH may be caused by competition between SHPRH and the deubiquitin enzyme USP1, which removes monoubiquitin from PCNA (Huang et al., 2006). To clarify the role of the RING domain in SHPRH, we created two proteins: one with a mutation at the conserved cysteine 1432 in the RING domain (C1432A) and the other with a deletion of the entire RING domain at the C terminus (ΔRING). Both mutants showed reduced levels of PCNA polyubiquitinations (Fig. 2 B, lane 4; and Fig. S3 B), suggesting a role of the RING domain of SHPRH. The remaining levels of PCNA ubiquitinations observed in these mutants imply that SHPRH mutants may be able to “recruit” or “nucleate” endogenous SHPRH or other E3 ligases. Supporting the specificity of overexpressed SHPRH to PCNA, SHPRH expression did not affect the basal polyubiquitination level of His6-c-JUN, a nuclear protein modified with a lysine 48–linked polyubiquitin chain by several ubiquitin ligases, such as SCFFbw7 (Nateri et al., 2004; Fig. S3 C).
Figure 2.

SHPRH is a functional orthologue of yeast Rad5. (A) SHPRH promotes PCNA polyubiquitination. HEK 293T cells were transfected with plasmids expressing 0.5 μg HA-ubiquitin (HA-Ub), 0.5 μg FLAG-PCNA, 2.0 μg SHPRH-myc-His, 100 ng RAD6-HA, 50 ng UBC13-HA, 50 ng UBC13(C87A)-HA, and 50 ng MMS2-HA in the combinations indicated. Lysates were immunoprecipitated with an anti-FLAG antibody, and ubiquitinated PCNAs were detected by an anti-HA antibody. Total PCNAs were probed with an anti-FLAG antibody. PCNA-Ub1 and PCNA-UbN indicate mono- and polyubiquitinated PCNAs, respectively. Mono- and diubiquitinated PCNAs are marked with single and double asterisks, respectively. Expression of SHPRH and E2s was analyzed by blotting lysates with specific anti-epitope tag antibodies. (B) E3 ligase activity of SHPRH for PCNA polyubiquitination is perturbed by a mutation in the conserved cysteine of the RING domain (C1432A). (C) HEK 293T cells were transfected as in A, except using ubiquitin mutants (left), PCNA-K164R mutant (middle), or lentivirus-infected 293T cells (right). The dot represents the IgG heavy chain. Lanes 4 and 5 of the top panel were exposed longer than lanes 6 and 7. (D) RAD18 and SHPRH cooperate to promote PCNA polyubiquitination. Cells were transfected as indicated with the same amounts of plasmids as in A, and endogenous PCNA was immunoprecipitated with an anti-PCNA antibody.
Conserved roles of SHPRH as a human Rad5 orthologue
To determine whether SHPRH polyubiquitinates the conserved K164 of PCNA, which is also targeted by yRad5, PCNA (either wild type or K164R mutant) was immunoprecipitated and the level of mono- and polyubiquitinations was analyzed. PCNA with the K164R mutation was defective in mono- and polyubiquitinations (Fig. 2 C). Furthermore, the SHPRH-promoted polyubiquitination of PCNA was reduced by the overexpression of the ubiquitin(K63R) mutant but was enhanced by the ubiquitin(K48R) mutant (Fig. 2 C). We therefore concluded that SHPRH functions with the UBC13–MMS2 complex to modify the K164 of PCNA with a noncanonical K63-linked polyubiquitin chain in vivo.
Previous yeast studies suggested that the monoubiquitination of PCNA by yRad18 precedes polyubiquitination by yRad5 (Hoege et al., 2002). It is consistent with genetic observations that yRAD18 is epistatic to yRAD5 after exposure to various DNA-damaging agents (Johnson et al., 1992; Ulrich and Jentsch, 2000; Motegi et al., 2006). Notably, stable knockdown of RAD18 by short hairpin RNA (shRNA) substantially reduced SHPRH-mediated PCNA polyubiquitination (Fig. 2 C). Furthermore, coexpression of RAD18 and RAD6 with SHPRH and UBC13–MMS2 synergistically promoted the polyubiquitination of endogenous PCNA (Fig. 2 D). These results clearly indicate that the PCNA monoubiquitination by RAD18–RAD6 and polyubiquitination by SHPRH–UBC13–MMS2 are sequential, rather than competitive, events.
SHPRH interacts with PCNA, UBC13, and RAD18 and self-multimerizes
Yeast Rad5 interacts with PCNA, yUbc13, and yRad18 and self-multimerizes (Ulrich and Jentsch, 2000). We observed that SHPRH physically interacted with GST-fused PCNA (wild type or K164R mutant) and GST-UBC13, but not with GST-MMS2, GST-RAD6, or GST alone (Fig. 3, A and B). The interactions between the PCNA(K164R) mutant and SHPRH or RAD18 suggest that the ubiquitination of PCNA is not essential for these interactions. SHPRH interacted with UBC13 and weakly with UBC13(C87A) in vivo (Fig. 3, C and D). Although structural studies predicted that the interaction site on UBC13 with RING domains is distinct from the cysteine C87 (VanDemark et al., 2001), C87 may affect this interaction in vivo. In addition, we observed an increased level of SHPRH protein in cells coexpressing wild-type UBC13 and, to a lesser extent, UBC13(C87A), suggesting the stabilization of SHRPH through the complex formation with UBC13. We also identified the self-association of SHPRH and the interaction between SHPRH and RAD18 by coimmunoprecipitation experiments (Fig. 3, E and F). These observations demonstrate that SHPRH has interaction features similar to yRad5.
Figure 3.

SHPRH physically interacts with PCNA, UBC13, and RAD18 and multimerizes. (A and B) SHPRH associates with PCNA and UBC13 in vitro. SHPRH-FLAG was pulled down with GST-fused PCNA (wild type or K164R mutant; A) or with GST-fused E2s (B) and analyzed by blotting with an anti-FLAG antibody. RAD18-FLAG was also pulled down with GST-PCNA (A). (C and D) SHPRH associates with UBC13 in vivo. SHPRH-myc-His (C) or SHPRH-FLAG (D) was coexpressed with wild-type UBC13-HA, UBC13(CA)-HA, or MMS2-HA in the combinations indicated. Anti-myc (C) or anti-HA (D) immunoprecipitates were blotted with an anti-HA (C) or anti-FLAG (D) antibody. Note that the level of SHPRH is enhanced in the presence of wild-type UBC13 and, to a lesser extent, UBC13(CA). SHPRH showed a weak but reproducible interaction with UBC13(CA). Blots with anti–α-tubulin or anti-GFP serve as loading or transfection control, respectively. (E and F) SHPRH interacts with RAD18 and self-multimerizes in vivo. In F, cell lysates expressing SHPRH-FLAG and SHPRH-myc-His were immunoprecipitated with anti-myc antibody and blotted with an anti-FLAG antibody. Normal IgG antibody was used for control.
SHPRH suppresses genomic instability through PCNA polyubiquitination
To investigate the biological significance of PCNA polyubiquitination in mammalian cells, we transfected HEK 293T cells with low amounts of plasmids that express HA-ubiquitin and FLAG-PCNA, with or without SHPRH-myc-His, and treated with various DNA-damaging agents. No apparent changes in PCNA polyubiquitination were detected after treating cells with 30 J/m2 UV or 0.3 mM of the DNA cross-linking agent mitomycin C (MMC; Fig. 4 A). In contrast, PCNA polyubiquitination was induced after treating cells with 0.01% MMS (Fig. 4 A, lane 7). MMS-induced PCNA polyubiquitination was strongly enhanced by the additional expression of SHPRH. We also noticed that the signals of SHPRH in the cell extracts were reduced only when cells were treated with MMS (Fig. 4 A, lane 8), implying that SHPRH was redistributed to the insoluble (chromatin bound) fraction. MMS treatment of HCT116 human colon carcinoma cells (without any transfections) also showed endogenous PCNA polyubiquitination in a dose-dependent manner (Fig. 4 B). To examine the cell cycle specificity of PCNA polyubiquitination, we treated cells arrested in G1 phase or 4 h after release from the G1 arrest with MMS. We observed a somewhat substantial level of PCNA polyubiquitination in untreated cells in G1 phase, but not in S phase (Fig. 4 C, lanes 1 and 3). Importantly, PCNA polyubiquitination was most efficiently induced in mid–S phase (Fig. 4 C, lane 4).
Figure 4.

PCNA polyubiquitination is induced by MMS. (A) HEK 293T cells transfected with plasmids expressing 0.2 μg HA-ubiquitin (Ub) and 0.2 μg FLAG-PCNA with or without 0.6 μg SHPRH-myc-His were treated with 30 J/m2 UV, 0.3 mM MMC, or 0.01% MMS for 2 h. Anti-FLAG immunoprecipitated PCNA was analyzed with an anti-HA antibody. (B) MMS treatment induces polyubiquitination of endogenous PCNA. HCT116 cells were treated with the indicated doses of MMS for 2 h. Immunoprecipitated PCNA was analyzed with an anti-ubiquitin (P4D1) antibody. (C) HCT116 cells in G1 (lanes 1 and 2) or S phase (lanes 3 and 4) were treated with 0.01% MMS for 2 h. A polyubiquitinated species of PCNA was detected with anti-polyubiquitin (FK2) antibody.
Mutations in genes in the yRad5 pathway cause genomic instability and increased cell sensitivity to various DNA-damaging agents (Smirnova and Klein, 2003; Stelter and Ulrich, 2003; Smith et al., 2004; Motegi et al., 2006). To determine whether reduced expression of SHPRH could cause similar cellular phenotypes, we transduced HCT116 cells with lentiviral vectors that express two different SHPRH-interfering shRNAs (constructs B or C; Fig. 5 A). SHPRH-silenced cells (by construct C) showed a substantial reduction in MMS-induced PCNA polyubiquitination compared with control (Fig. 5 B, lane 4). All three clones with reduced SHPRH expression showed higher sensitivity to MMS than wild type or the two control clones infected with an empty lentivirus (Fig. 5 C). In contrast, SHPRH-silenced cells showed no substantial sensitivity to UV mimetic 4-nitroquinoline 1-oxide (4-NQO), MMC, or γ-irradiation (unpublished data). Moreover, SHPRH-silenced clones (B2 and C4) showed a greater number of chromosome breaks than did wild type after 0.01% MMS treatment (Fig. 5, D and E). Notably, the levels of reduction of SHPRH expression (Fig. 5 A) were well correlated with their levels of sensitivity to MMS and the frequencies of chromosome breaks (Fig. 5, A, D, and E). These observations suggest that SHPRH is involved in MMS-induced DNA-damage responses.
Figure 5.

SHPRH suppresses MMS-induced genomic instability. (A) Expression level of SHPRH in individual clones carrying an integrated SHPRH-shRNA vector was analyzed by RT-PCR (top) and Western blot with anti-SHPRH antibody (bottom). (B) SHPRH-silenced cells show a reduced level of MMS-induced PCNA polyubiquitination. HCT116 cells with control (vector) or SHPRH-silencing (shRNA) lentivirus were treated with 0.01% MMS for 2 h. (C) The reduced expression of SHPRH sensitizes cells to MMS. Three independent shRNA clones were treated with increasing concentrations of MMS, and serially diluted cells were plated. Surviving colonies were counted and quantified. Doses of MMS for 10% survival are 0.0145% (wild type [wt]), 0.0110% (B2), 0.0090% (C4), and 0.0085% (B11). (D and E) Reduction of SHPRH enhances chromosome breaks by MMS. Metaphase chromosomes were analyzed 24 h after a 1-h treatment of different clones with 0.01% MMS. (D) One example from each clone is shown. (E) Noticeable chromosome breakages per cell were quantified from 100 metaphases per each clone.
Accumulating evidence suggests that DNA damage bypass followed by PCNA modifications is important for suppressing genomic instability and cancers. For example, the overexpression of HHR6B was implicated in the chromosomal instability phenotypes of human breast cancer cells (Shekhar et al., 2002). The targeted disruptions of the RAD18 or REV3L gene in either mouse embryonic stem cells or mouse embryonic fibroblasts, respectively, increased genomic instability, including sister chromatid exchange, homologous recombination, and illegitimate recombination (Tateishi et al., 2003; Wittschieben et al., 2006). Mutations of XPV, which encodes a TLS polymerase η, were found in cancer-prone xeroderma pigmentosum variant syndrome (Johnson et al., 1999; Masutani et al., 1999). Our observations prove that the regulatory mechanisms of PCNA through differential modifications by RAD18 and yRad5/ SHPRH are fully conserved and constitute a fundamental mechanism to prevent genomic instability throughout evolution.
Materials and methods
Cell culture, reagents, and antibodies
HEK 293T and HCT116 cells were cultured in DME and McCoy's media supplemented with 10% fetal bovine serum, respectively. Commercially available anti-PCNA (PC10), anti-ubiquitin (P4D1), anti-polyubiquitin (FK2), anti-myc (9E10), anti-HA (12CA5), anti-FLAG (M2), anti-V5 antibodies, and anti-RAD18 (K-15) were used. Polyclonal anti-SHPRH antibody was previously described (Sood et al., 2003). MMS, MMC, and mimosine were purchased from Sigma-Aldrich.
Construction of various expression plasmids
Full-length cDNA of human SHPRH was obtained by PCR with IMAGE clones (available from GenBank/EMBL/DDBJ under accession nos. AI888407 and AA255592) and RACE-PCR products as templates. cDNA encoding human RAD18, PCNA, MMS2 (UEV2), and RAD6 (HHR6B) were obtained from Mammalian Genome Collection (RAD18, MHS1010-52750; PCNA, MHS1011-58526; MMS2, MHS1011-62471; HHR6B, MHS1011-62750). Expression plasmids were constructed by subcloning each cDNA into pcDNA3.1-myc-His, p3XFLAG-CMV, or pGEX-6P-1. UBC13(wild type or C87A)-HA–expressing plasmids were gifts from D. Bohmann (European Molecular Biology Laboratory, Heidelberg, Germany) and J. Kehrl (National Institute of Allergy and Infectious Diseases, Bethesda, MD), respectively. Point mutations in the RING finger domain of SHPRH(C1432A) and in PCNA(K164R) were introduced by using an in vitro mutagenesis method (QuikChange; Stratagene).
Coimmunoprecipitation and GST pull-down assays
For coimmunoprecipitation assay, HEK 293T cells were transfected with various combinations of expression plasmids using FuGENE 6 transfection reagent (Roche) and lysed in the TNE buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.5% NP-40, 1 mM EDTA, 8% glycerol, 0.5 mM DTT, 50 mM NaF, 1 mM PMSF, 1 μg/ml aprotinin, and 1 μg/ml leupeptin). For the GST pull-down assay, various GST fusion proteins were expressed in Escherichia coli strain BL21 (DE3)-RIL (Stratagene) and purified using glutathione–Sepharose beads (GE Healthcare). 20 μg of GST fusion proteins were used for pulling down SHPRH-FLAG or RAD18-FLAG expressed in HEK 293T cells.
Generation of lentivirus-mediated stable knockdown cells
SHPRH-silencing vectors were constructed by cloning the target sequences of 5′-TTCAATGCCCTCCTACAC-3′ (construct B) and 5′-AGTGTCCATCCTTTCCAT-3′ (construct C) into the lentivirus-based expression vector pLL3.7 (a gift from V. Parijs, Massachusetts Institute of Technology, Cambridge, MA). RAD18-silencing lentivirus vector was purchased from Open Biosystems. Lentivirus packaging plasmids were gifts from F. Candotti (National Human Genome Research Institute, Bethesda, MD). Lentivirus-infected cells were selected by the expression of GFP (SHPRH) or by puromycin (RAD18). The expression level of SHPRH was examined by Western blot or by RT-PCR with primers 5′-GAGCAACTCTGATCATCTCTCCAAG-3′ and 5′-GATAGAGAAGTCGAACCCACCAGTG-3′. Primers used for amplifying ACTIN as a control were 5′-GCTCGTCGTCGACAACGGCTC-3′ and 5′-CAAACATGATCTGGGTCATCTTCTC-3′. SHPRH-silenced clones B2, B11 (construct B), and C4 (construct C) were used in this study.
Detection of MMS-induced PCNA polyubiquitination, sensitivity assay, and chromosome breakage analysis
HCT116 cells were treated with MMS for 2 h, washed with PBS, and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.1% Triton X-100, 0.5 mM DTT, 50 mM NaF, 1 mM PMSF, 1 μg/ml aprotinin, and 1 μg/ml leupeptin). In some experiments, cells were treated with 0.5 mM mimosine for 20 h. For measuring MMS sensitivity, SHPRH-knockdown cells were treated with increasing concentrations of MMS for 1 h, washed with PBS three times, replated onto 6-well plates at defined cell densities, and cultured for 10 d. Colonies grown from the surviving cells were counted and expressed as a survival fraction (%) compared with untreated cells as 100%. For analyzing MMS-induced chromosomal breaks, SHPRH-knockdown cells were treated with 0.01% MMS for 1 h, washed with PBS three times, and cultured for 24 h. At least 100 metaphase spreads from each clone were analyzed.
Online supplemental material
Fig. S1 shows moderate homology between the SWI2/SNF2 domains in SHPRH and yeast Rad5. Fig. S2 shows that SHPRH expression in the yeast rad5 strain could not complement UV sensitivity. Fig. S3 shows that RAD18 and RAD6B induced exclusive monoubiquitination of PCNA, the reduced activity of SHPRH(ΔRING) for PCNA polyubiquitination, and that SHPRH did not promote polyubiquitination of c-JUN. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200606145/DC1.
Supplementary Material
Acknowledgments
We thank A. Dutra (National Human Genome Research Institute [NHGRI]) and E. Pak (NHGRI) for metaphase chromosome analysis; J. Kehrl, Z.J. Chen (University of Texas), D. Bohmann, L. van Parijs, and F. Candotti for expression plasmids; B. Berman (NHGRI), S. Burgess (NHGRI), P. Meltzer (NHGRI), A. Nussenzweig (National Cancer Institute [NCI]), R. Nussbaum (NHGRI), J. Puck (NHGRI), and members of the Myung laboratory for comments on the manuscript; D. Bodine (NHGRI), M. Lichten (NCI), and Y. Shiloh (Tel Aviv University) for helpful discussion. K. Myung especially thanks E. Cho.
This work was supported by funds from the Japan Society for the Promotion of Science (to A. Motegi) and the intramural research program of the National Human Genome Research Institute, National Institutes of Health (to P.P. Liu and K. Myung).
Abbreviations used in this paper: HEK, human embryonic kidney; MMC, mitomycin C; MMS, methyl methanesulfonate; PCNA, proliferating cell nuclear antigen; shRNA, short hairpin RNA; TLS, translesion synthesis.
References
- Friedberg, E.C., A.R. Lehmann, and R.P. Fuchs. 2005. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol. Cell. 18:499–505. [DOI] [PubMed] [Google Scholar]
- Hoege, C., B. Pfander, G.L. Moldovan, G. Pyrowolakis, and S. Jentsch. 2002. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 419:135–141. [DOI] [PubMed] [Google Scholar]
- Hofmann, R.M., and C.M. Pickart. 1999. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell. 96:645–653. [DOI] [PubMed] [Google Scholar]
- Huang, T.T., S.M. Nijman, K.D. Mirchandani, P.J. Galardy, M.A. Cohn, W. Haas, S.P. Gygi, H.L. Ploegh, R. Bernards, and A.D. D'Andrea. 2006. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 8:339–347. [DOI] [PubMed] [Google Scholar]
- Johnson, R.E., S.T. Henderson, T.D. Petes, S. Prakash, M. Bankmann, and L. Prakash. 1992. Saccharomyces cerevisiae RAD5-encoded DNA repair protein contains DNA helicase and zinc-binding sequence motifs and affects the stability of simple repetitive sequences in the genome. Mol. Cell. Biol. 12:3807–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, R.E., C.M. Kondratick, S. Prakash, and L. Prakash. 1999. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 285:263–265. [DOI] [PubMed] [Google Scholar]
- Kannouche, P.L., J. Wing, and A.R. Lehmann. 2004. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell. 14:491–500. [DOI] [PubMed] [Google Scholar]
- Koken, M.H., P. Reynolds, I. Jaspers-Dekker, L. Prakash, S. Prakash, D. Bootsma, and J.H. Hoeijmakers. 1991. Structural and functional conservation of two human homologs of the yeast DNA repair gene RAD6. Proc. Natl. Acad. Sci. USA. 88:8865–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodner, R.D., C.D. Putnam, and K. Myung. 2002. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 297:552–557. [DOI] [PubMed] [Google Scholar]
- Lawrence, C. 1994. The RAD6 DNA repair pathway in Saccharomyces cerevisiae: what does it do, and how does it do it? Bioessays. 16:253–258. [DOI] [PubMed] [Google Scholar]
- Masutani, C., R. Kusumoto, A. Yamada, N. Dohmae, M. Yokoi, M. Yuasa, M. Araki, S. Iwai, K. Takio, and F. Hanaoka. 1999. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 399:700–704. [DOI] [PubMed] [Google Scholar]
- Motegi, A., K. Kuntz, A. Majeed, S. Smith, and K. Myung. 2006. Regulation of gross chromosomal rearrangements by ubiquitin and SUMO ligases in Saccharomyces cerevisiae. Mol. Cell. Biol. 26:1424–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nateri, A.S., L. Riera-Sans, C. Da Costa, and A. Behrens. 2004. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 303:1374–1378. [DOI] [PubMed] [Google Scholar]
- Shekhar, M.P., A. Lyakhovich, D.W. Visscher, H. Heng, and N. Kondrat. 2002. Rad6 overexpression induces multinucleation, centrosome amplification, abnormal mitosis, aneuploidy, and transformation. Cancer Res. 62:2115–2124. [PubMed] [Google Scholar]
- Smirnova, M., and H.L. Klein. 2003. Role of the error-free damage bypass postreplication repair pathway in the maintenance of genomic stability. Mutat. Res. 532:117–135. [DOI] [PubMed] [Google Scholar]
- Smith, S., J.-Y. Hwang, S. Banerjee, A. Majeed, A. Gupta, and K. Myung. 2004. Mutator genes for suppression of gross chromosomal rearrangements identified by a genome-wide screening in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 101:9039–9044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood, R., I. Makalowska, M. Galdzicki, P. Hu, E. Eddings, C.M. Robbins, T. Moses, J. Namkoong, S. Chen, and J.M. Trent. 2003. Cloning and characterization of a novel gene, SHPRH, encoding a conserved putative protein with SNF2/helicase and PHD-finger domains from the 6q24 region. Genomics. 82:153–161. [DOI] [PubMed] [Google Scholar]
- Stelter, P., and H.D. Ulrich. 2003. Control of spontaneous and damage- induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 425:188–191. [DOI] [PubMed] [Google Scholar]
- Tateishi, S., Y. Sakuraba, S. Masuyama, H. Inoue, and M. Yamaizumi. 2000. Dysfunction of human Rad18 results in defective postreplication repair and hypersensitivity to multiple mutagens. Proc. Natl. Acad. Sci. USA. 97:7927–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi, S., H. Niwa, J. Miyazaki, S. Fujimoto, H. Inoue, and M. Yamaizumi. 2003. Enhanced genomic instability and defective postreplication repair in RAD18 knockout mouse embryonic stem cells. Mol. Cell. Biol. 23:474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich, H.D. 2005. The RAD6 pathway: control of DNA damage bypass and mutagenesis by ubiquitin and SUMO. Chembiochem. 6:1735–1743. [DOI] [PubMed] [Google Scholar]
- Ulrich, H.D., and S. Jentsch. 2000. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 19:3388–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDemark, A.P., R.M. Hofmann, C. Tsui, C.M. Pickart, and C. Wolberger. 2001. Molecular insights into polyubiquitin chain assembly: crystal structure of the Mms2/Ubc13 heterodimer. Cell. 105:711–720. [DOI] [PubMed] [Google Scholar]
- Wittschieben, J.P., S.C. Reshmi, S.M. Gollin, and R.D. Wood. 2006. Loss of DNA polymerase ζ causes chromosomal instability in mammalian cells. Cancer Res. 66:134–142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}