

Abstract
Cancer cells exhibit increased glycolysis for ATP production due, in part, to respiration injury (the Warburg effect). Because ATP generation through glycolysis is less efficient than through mitochondrial respiration, how cancer cells with this metabolic disadvantage can survive the competition with other cells and eventually develop drug resistance is a long-standing paradox. We report that mitochondrial respiration defects lead to activation of the Akt survival pathway through a novel mechanism mediated by NADH. Respiration-deficient cells (ρ-) harboring mitochondrial DNA deletion exhibit dependency on glycolysis, increased NADH, and activation of Akt, leading to drug resistance and survival advantage in hypoxia. Similarly, chemical inhibition of mitochondrial respiration and hypoxia also activates Akt. The increase in NADH caused by respiratory deficiency inactivates PTEN through a redox modification mechanism, leading to Akt activation. These findings provide a novel mechanistic insight into the Warburg effect and explain how metabolic alteration in cancer cells may gain a survival advantage and withstand therapeutic agents.
Introduction
Over 70 yr ago, Warburg discovered that cancer cells are more dependent on glycolysis for generation of ATP, even when abundant oxygen is present in the cellular environment (Warburg, 1930). During the past several decades, this metabolic alteration has been observed in many cancer types, including solid tumors and leukemia. It is now recognized that the Warburg effect represents a prominent metabolic characteristic of malignant cells. Although the exact mechanisms responsible for this metabolic alteration remain to be elucidated, malfunction of mitochondrial respiration or “respiration injury” due, in part, to mitochondrial DNA (mtDNA) mutations/deletions is thought to be an important contributing factor (Warburg, 1956; Wallace, 1999; Simonnet et al., 2002; Xu et al., 2005). Recent studies revealed that cancer cells of various tissue origins exhibit frequent mutations in their mtDNA (Carew et al., 2003; Copeland et al., 2002; Nomoto et al., 2002). Because mtDNA encodes for 13 protein components of the mitochondrial respiratory chain, it is likely that certain mtDNA mutations may cause malfunction of the respiratory chain, forcing the cells to increase glycolysis to maintain their ATP supply. The active metabolism in cancer cells requires a constant supply of sufficient ATP. Paradoxically, generation of ATP through glycolysis (two ATPs per glucose) is far less efficient than ATP production through mitochondrial oxidative phosphorylation (36 ATPs per glucose). It is unclear how cancer cells, with this apparent disadvantage in energy metabolism, can survive the competition with other cells in vivo and develop as a malignant cell population with drug-resistant potential.
Environmental factors, especially hypoxic conditions in the tumor tissue microenvironment, may also force cancer cells to use glycolytic pathways to generate ATP to meet their energy supply. Hypoxia-induced metabolic adaptations cause the hypoxic cells to exhibit certain biochemical characteristics similar to those of mitochondrial respiratory-defective cells. Under hypoxic conditions, cancer cells have to use the “energy-inefficient” glycolytic pathway to generate ATP, leading to accumulation of a high level of NADH, which is normally channeled to the electron transport chain as the energy fuel in respiration-competent cells.
Cancer cells use multiple pathways to enhance their survival and prevent apoptosis under various conditions. Increased expression of Bcl-2 and -XL antiapoptotic factors and activation of NFκB and phosphatidylinositol 3-kinase (PI3K)–Akt pathways are among the well-characterized mechanisms by which cancer cells promote their survival capacity. Overexpression of Bcl-2 and/or -XL counteracts the proapoptotic effects of Bax and Bak and inhibits the mitochondria-mediated cell death pathway (Chan and Yu, 2004). NFκB can be activated by various stimuli, such as cytokines, DNA-damaging agents, and reactive oxygen species (ROS). Its antiapoptotic effect is mediated mainly by promoting expression of a variety of cell survival factors through a transcriptional activation mechanism (Aggarwal, 2004). The PI3K–Akt pathway promotes cell survival and proliferation through a series of downstream events, including enhancing nutrient uptake and energy metabolism through activation of mTOR (Edinger and Thompson, 2002), stimulating aerobic glycolysis (Elstrom et al., 2004), and suppressing apoptosis by phosphorylation of the Bad protein (Downward, 2004). The Akt survival pathway is positively and negatively regulated by PI3K and PTEN, respectively, through their opposing effects on phosphatidylinositol-3′-phosphate (PIP3) generation (Stanbolic et al., 1998). Despite the fact that the regulatory mechanisms of these pathways have been characterized in great detail, their potential roles in promoting the survival of cancer cells with mitochondrial respiration defects remain largely unknown.
Because mitochondrial respiratory defects are frequently observed in human cancers, owing to genetic alterations of mtDNA and/or hypoxic conditions in tumor tissue environment, elucidation of the molecular and biochemical mechanisms contributing to the survival of cancer cells with such metabolic defects is obviously important in understanding the biology of the Warburg effect and in developing new strategies to overcome drug resistance. This study used experimental model systems to investigate the possible survival mechanisms in cells with mitochondrial genetic defects and in mitochondrial respiration-competent cells under conditions where the respiratory activity is compromised by specific pharmacological inhibitors or by hypoxia.
Results
Cancer cells with mitochondrial respiratory defects exhibit survival advantage
To investigate the molecular events contributing to the survival mechanisms in cancer cells with mitochondrial respiration defects, we first derived multiple clones of respiration-deficient cells (ρ-) with altered mtDNA from two different human cancer cell lines, HL-60 (leukemia), and Raji (lymphoma), and then examined potential molecular alterations that promote cell survival. mtDNA was preferentially damaged using an established method, in which the respiration-competent parental cells were chronically exposed to a low concentration of ethidium bromide (50 ng/ml), and the resulting ρ- cells were subcloned by a serial dilution method (King and Attardi, 1996; Pelicano et al., 2003). One subclone of ρ- cells derived from HL-60 cells was designated as HL60-C6F cells, which have been previously characterized (Pelicano et al., 2003). Four other ρ- subclones (C2, C6, C7, and C8) were derived from Raji cells, and their alterations in mtDNA and mitochondrial functions are shown in Fig. 1. All ρ- cells exhibited defects in respiration, as indicated by a lack of oxygen consumption (Fig. 1 A). However, the ρ- cells still retained their mitochondrial mass with apparently normal transmembrane potential (Fig. 1, B–D). PCR analysis of mtDNA revealed multiple deletions of mtDNA at ND1 and the D-loop regions (Fig. 1 E). The PCR products for COXII, ND4, and ATPase6 genes were reduced to various degrees in different ρ- clones, suggesting possible reduction of these gene copy numbers or mutations in the mtDNA regions for the PCR primers, and thus retarded the PCR reactions. Interestingly, all ρ- cells retained the cyt-b gene sequence at levels comparable to the parental cells. The nuclear gene GAPDH remained unchanged in all clones. The respiration-deficient phenotype of the ρ- cells has been stable in long-term culture (over 20 mo) in medium without ethidium bromide.
Figure 1.

Biochemical and molecular characterization of ρ- cell clones. (A) Comparison of oxygen consumption by parental Raji cells and four ρ- clones (C2, C6, C7, and C8). (B) Mitochondrial mass was determined by flow cytometry analysis of Raji cells in comparison with a representative ρ- clone (Raji-C2) using 60 nM MitoTracker Green. (C) Mitochondrial transmembrane potential (ΔΨm) in parental Raji cells and Raji-C2 cells by flow cytometry analysis using 1 μM rhodamine-123. (D) Quantitative comparison of mitochondrial mass and ΔΨm in Raji cells and four ρ- clones. (E) Analysis of mtDNA deletion in ρ- clones. Total DNA isolated from Raji cells and all four ρ- clones was subjected to PCR for the indicated mtDNA and nuclear gene regions, as described in Materials and methods. Reaction products were analyzed on an agarose gel and visualized by ethidium staining. (F and G) Raji cells or Raji-C8 cells were seeded in semisolid medium (Immocult) and cultured under normoxia or hypoxia conditions for colony formation, as described in Materials and methods. Data were from two separate experiments performed in triplicate. (H) Comparison of the colony formation ratio under hypoxia/normoxia in Raji cells and Raji-C8 cells. Colony formation ratio was determined by dividing the colony formation efficiency under hypoxia by that under normoxia conditions. (I) Raji cells and the ρ- cells (Raji-C8) were seeded in semisolid medium and incubated at 37°C with 1.5% oxygen for 2 wk. Colonies were stained and counted (data from two separate experiments performed in triplicate). Results are expressed as the mean ± the SD.
All ρ- cell clones derived from HL-60 and Raji cells were strictly dependent on glycolysis and required supplements of high glucose, pyruvate, and uridine for growth. Importantly, the ρ- cells exhibited a better ability to grow in hypoxic conditions, a common situation cancer cells encounter in vivo. As illustrated in Fig. 1 F, under normoxia conditions, the parental Raji cells showed better colony-forming efficiency (CFE; 76%) than the respiration-deficient Raji-C8 clone (CFE 56%). However, under reduced oxygen conditions (5% O2), the respiration-deficient clone C8 showed a higher CFE compared with the parental Raji cells (Fig. 1 G). We also noticed that the parental Raji cells formed larger colonies in normoxic conditions than in hypoxia, whereas the colony sizes of the Raji-C8 cells were similarly small both in hypoxia and normoxia. The colony formation ratio under hypoxia/normoxia conditions showed that the Raji-C8 cells had a significant survival advantage under hypoxia compared with the parental cells (Fig. 1 H; P = 0.009). Similarly, when the cells were cultured under a more hypoxic condition (1.5% O2), the ρ- C8 cells exhibited a higher colony formation rate than the parental Raji cells (Fig. 1 I).
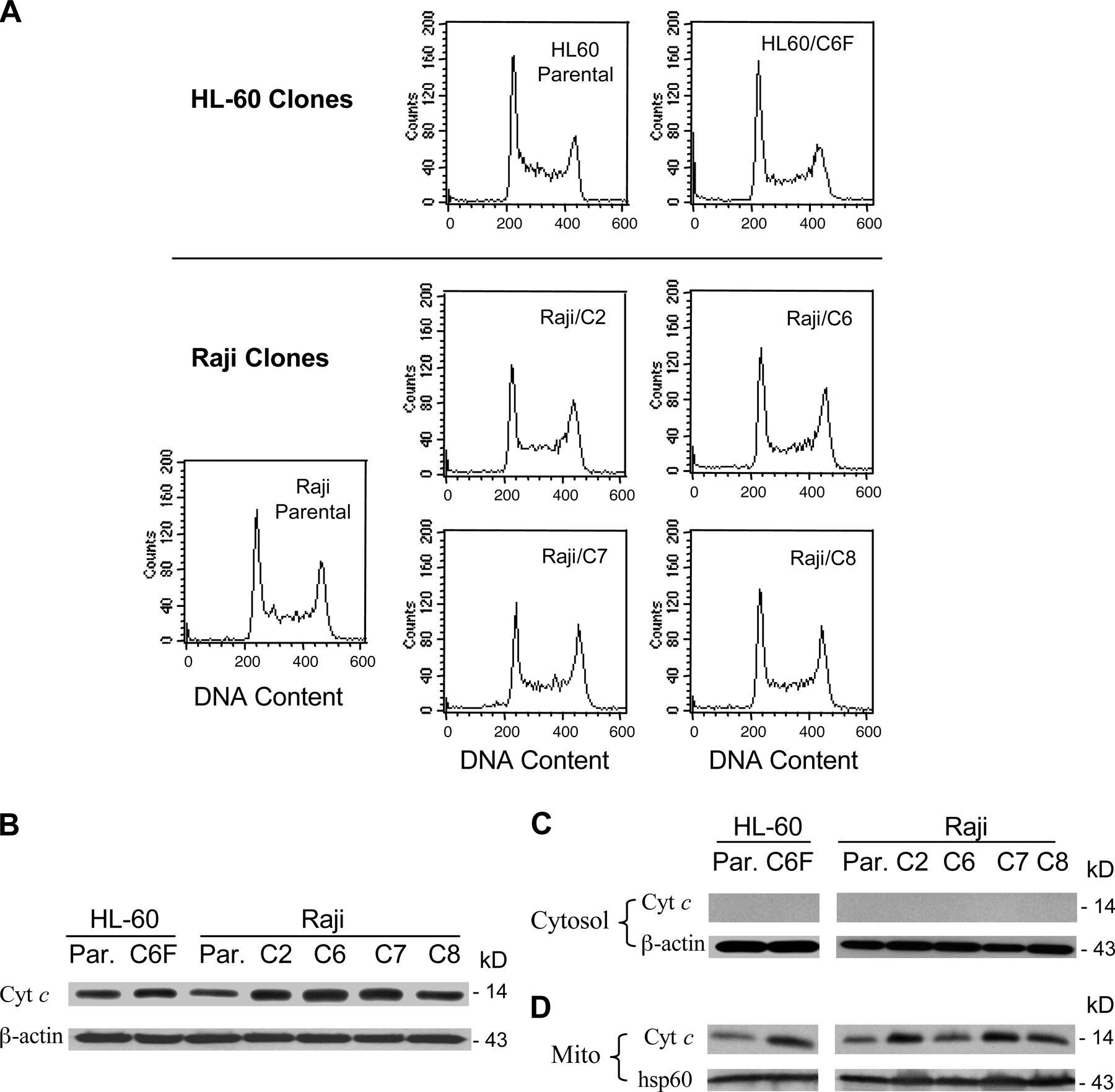
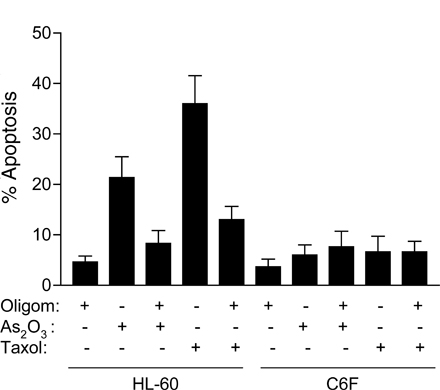
Despite mitochondrial metabolic defects, ρ- cells were not prone to drug-induced cell death, and exhibited reduced sensitivity to common anticancer agents. As shown in Fig. 2 A, HL60-C6F cells were significantly less sensitive to arsenic trioxide (As2O3) and taxol than the parental HL-60 cells, as measured by annexin-V reactivity. Similarly, the Raji ρ- clones also showed less sensitivity to As2O3 (Fig. 2 B), doxorubicin (Fig. 2 C), and vincristine (Fig. 2 D). These findings suggest that the ρ- cells appear to have some survival advantage. Because cell cycle and cell growing rates may also affect drug sensitivity, we compared the cell cycle profiles of the ρ- clones with their respective parental cells (HL-60 and Raji). Analysis of cellular DNA contents by flow cytometry revealed no substantial difference in cell cycle distribution between the ρ- clones and their parental cells (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200512100/DC1). Although the ρ- clones exhibited a moderate decrease in cell growth rate, they retained an active apoptotic response and showed massive cell death when incubated with the glycolytic inhibitor 3-bromopyruvate (Xu et al., 2005). Interestingly, the proapoptotic factor cytochrome c in the ρ- cells was comparable to the parental cells or even slightly increased (Fig. S1 B). Analysis of proteins extracts from mitochondrial and cytosolic fractions showed that cytochrome c was detected only in the mitochondrial fraction in both cell types (Fig. S1, C and D). Thus, the increased expression of this proapoptotic factor in ρ- cells is confined in the mitochondria, which is consistent with a previous observation (Li et al., 1995).
Figure 2.

Comparison of parental HL-60 and Raji cells with the ρ- clones for their apoptotic response to anticancer agents. (A) Quantitative analysis of apoptosis induced by 1 μg/ml As2O3 and 0.1 μM taxol for 24 h in parental HL-60 cells (Par) and respiration-deficient ρ- cells (HL60-C6F). Apoptosis was assayed by annexin-V/PI staining and quantitated by flow cytometry analysis (n = 4 separate assays). The respiration-deficient HL60-C6F cells were significantly less sensitive to anticancer agents than the parental cells (*, P < 0.05 compared with the parental HL-60 cells). (B–D) Comparison of parental Raji cells with four respiration-deficient ρ- cells for their apoptotic response to 1 μg/ml As2O3 for 24 h (B; n = 7), 0.1 μM doxorubicin for 36 h (C; n = 5), and 5 ng/ml vincristine for 36 h (D; n = 5). Apoptosis was assayed by annexin-V/PI staining and quantitated by flow cytometry analysis. Results are expressed as the mean ± the SD (*, P < 0.05 compared with the parental Raji cells).
Activation of Akt in mitochondrial-deficient cells
All four clones of ρ- cells were then used for examination of changes in gene expression in comparison with their parental Raji cells, using oligonucleotide microarray produced at the University of Texas MD Anderson Cancer Center Genomic Core Facility. Among the genes that consistently exhibited changes in all four ρ- clones, we identified two key molecules, PIK3CA and PTEN, involved in regulation of the Akt survival pathway. This suggested a possibility that Akt might be involved in promoting the survival of ρ- cells. To determine whether Akt was indeed activated in the ρ- cells, the expression of Akt protein level and its phosphorylation status were first assessed by immunoblotting. As shown in Fig. 3 A, Akt phosphorylation at both Ser-473 and Thr-308 increased in all ρ- cells. The total Akt protein did not increase in HL60-C6F cells, and moderately increased in the Raji ρ- cells. Among the ρ- clones, HL60-C6F, Raji-C2, -C6, and -C8 showed a significant increase in Akt phosphorylation, whereas only a slight increase was detected in Raji-C7 cells. Direct analysis of Akt enzyme activity in vitro using glycogen synthase kinase 3 (GSK-3) as the Akt substrate further confirmed the increase of Akt kinase activity in the ρ- cells, albeit with some individual variation (Fig. 3 B). The reason for such variation among individual clones is likely caused by other changes induced by ethidium. To confirm that Akt activation is a general phenomenon in ρ- cells, we analyzed an additional three ρ- clones derived from HL-60 cells, and revealed that these ρ- cells also exhibited a consistent increase of Akt activation (Fig. 3 C).
Figure 3.

The Akt pathway is constitutively activated in ρ- cells. (A) Akt protein expression and its phosphorylation status were determined using anti-Akt and anti–phospho-Akt (Ser-473 and Thr-308) antibodies. The numbers below each lane indicate the relative density. (B) Comparison of in vitro kinase activity of protein extracts from ρ- clones cells and the parental cells. Akt protein was first immunoprecipitated with an anti-Akt antibody, and Akt enzyme activity was determined using an Akt kinase assay kit with GSK-3α/β fusion protein as the assay substrate, as described in Materials and methods. (C) Activation of Akt in ρ- cells from HL-60. Akt protein expression and its active phosphorylation were determined using anti-Akt and anti–phospho-Akt (Ser-473) antibodies. Whole-cell extracts from the parental HL-60 cells and four ρ- clones were prepared and subjected to immunoblotting analysis. (D) Reduced PTEN activity in the respiration-deficient ρ- clones. Expression of both activated PTEN (phosphorylated; phospho-PTEN) and total PTEN protein were determined using anti–phospho-PTEN antibodies (Ser-380 and Thr-382/383) and anti-PTEN antibody. The numbers below each lane show the relative band density normalized by β-actin. The value for the parental sample was expressed as 100%.
We then tested if the increase of Akt activation in ρ- cells might be caused by an increased expression of PI3 kinase, which is a positive regulator of Akt. Western blot analysis showed that the protein expression of PI3Kp110α (catalytic subunit) and PI3Kp85 (regulatory subunit), as well as the tyrosine phosphorylation levels, were similar in the parental and ρ- cells (unpublished data). Thus, Akt activation was unlikely because of an increase in PI3K. Because the phosphatase PTEN is a negative regulator of Akt pathway and the loss of PTEN activity has been correlated with increased Akt activity in cancer cells (Dahia et al., 1999), we compared the PTEN protein levels and its phosphorylation in ρ- cells and their parental cells. PTEN protein and its phosphorylation at Ser-380 and Thr-382/-383 were reduced in most ρ- clones (Fig. 3 D). The PTEN protein and its phosphorylation state were inversely correlated with the degree of Akt phosphorylation (Fig. 3 A), which is consistent with the negative regulatory role of PTEN in Akt signaling.
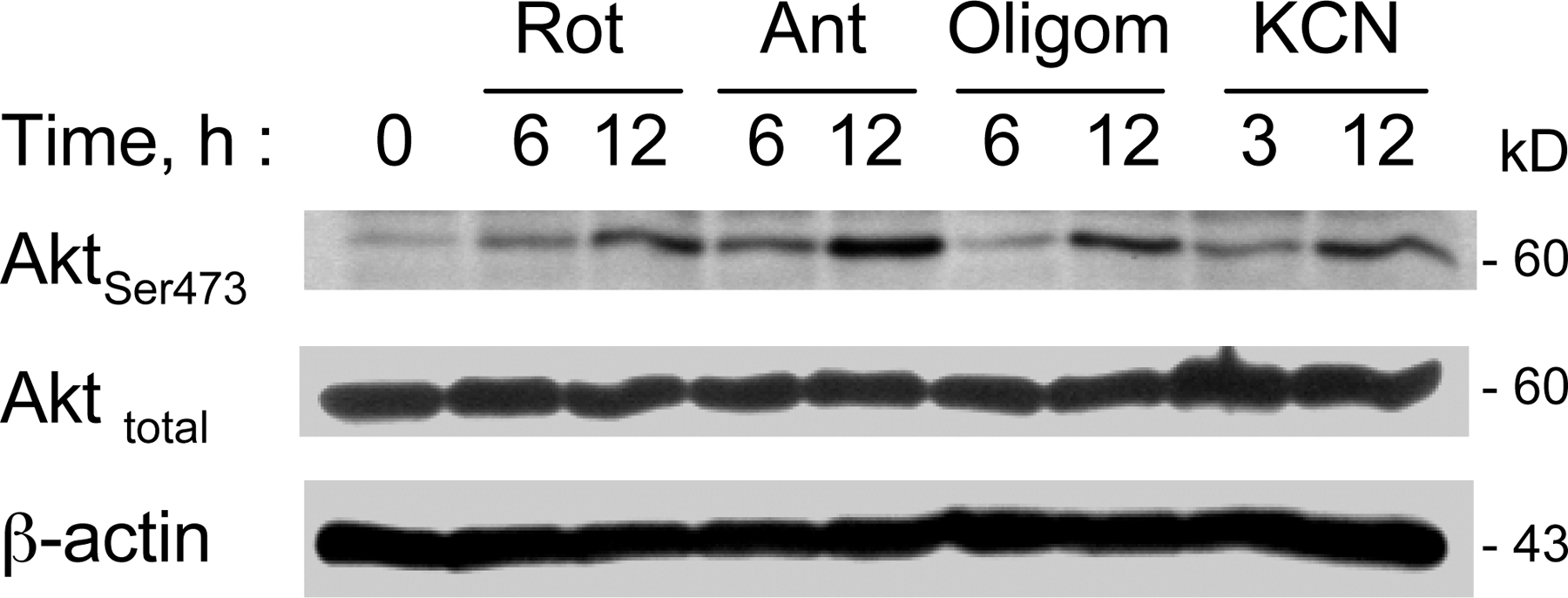
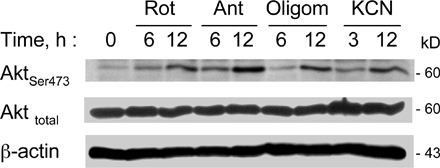
We reasoned that if deficiency in mitochondrial respiration was a key event that suppresses PTEN and causes Akt activation, inhibition of mitochondrial respiratory function in the parental cells should also cause PTEN suppression and Akt activation. To test this cause–effect relationship, we used rotenone, a specific inhibitor of the mitochondrial electron transport complex I, to block respiration in Raji cells. At 100 nM, rotenone effectively blocked respiration as early as 5 min after drug exposure (Fig. 4 A), but did not cause significant cell death during a 24-h incubation, as assessed by annexin-V/propidium iodide (PI) staining (Fig. 4 B). Inhibition of respiration by rotenone caused a time-dependent activation of Akt, indicated by an increase in Akt phosphorylation, which elevated significantly at 3 h and remained active for at least 24 h (Fig. 4 C). Concurrently, there was a time-dependent decrease in phospho-PTEN revealed by Western blotting using phospho-PTEN antibodies (Fig. 4 D). These observations suggest that the inhibition of respiration by rotenone may either cause dephosphorylation of PTEN, or mask the phosphorylated epitope. The overall PTEN protein appeared unchanged when respiration was acutely blocked. Consistently, other inhibitors of mitochondrial respiratory complexes, including antimycin A, cyanide, and oligomycin, also caused Akt activation (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200512100/DC1).
Figure 4.

Inhibition of mitochondrial respiration by rotenone led to Akt activation associated with PTEN inactivation. (A) Inhibition of respiration by rotenone in Raji cells. Cells were pretreated with 100 nM rotenone for 5 min, and oxygen consumption was measured as an indication of mitochondrial respiration. (B) Treatment of Raji cells with 100 nM rotenone (Rot) for 24 h did not cause significant apoptosis, as measured by annexin-V/PI staining. (C) Time-dependent activation of Akt in Raji cells treated with 100 nM rotenone for up to 24 h, and protein extracts were blotted using anti–phospho-Akt (Ser-473 and Thr-308) and anti-Akt antibodies. (D) Time-dependent inactivation of PTEN in respiration-blocked Raji cells treated with rotenone. Raji cells were incubated with 100 nM rotenone for up to 24 h as indicated, and protein extracts were blotted using anti–phospho-PTEN (Ser-380/Thr-382/-383), anti-PTEN, and anti-β–actin antibodies. (E) Antioxidant NAC did not prevent Akt activation induced by rotenone. Raji cells were treated with 100 nM rotenone (Rot) for 24 h in the presence or absence of 5 mM NAC (added 60 min before rotenone). The protein extracts were blotted using anti–phospho-Akt (Ser-473) and anti-Akt antibodies. (F) Comparison of superoxide ( ) content in parental Raji cells and ρ- cells (Raji-C2 clone). Cellular
) content in parental Raji cells and ρ- cells (Raji-C2 clone). Cellular  was analyzed by flow cytometric analysis as previously described (Pelicano et al., 2003). (G) Raji cells were treated with 500 μM H2O2 for 1 h in the presence or absence of 5 mM NAC (added 60 min before H2O2). Cellular ROS was measured by flow cytometry using CM-H2DCFDA. (H) Activation of Akt by exogenous H2O2 detected by Western blotting.
was analyzed by flow cytometric analysis as previously described (Pelicano et al., 2003). (G) Raji cells were treated with 500 μM H2O2 for 1 h in the presence or absence of 5 mM NAC (added 60 min before H2O2). Cellular ROS was measured by flow cytometry using CM-H2DCFDA. (H) Activation of Akt by exogenous H2O2 detected by Western blotting.
Because ROS are known to cause Akt activation, we tested the potential involvement of ROS in Akt activation when mitochondrial respiration was inhibited by rotenone. The addition of the antioxidant N-acetylcysteine (NAC) did not affect rotenone-induced Akt activation, suggesting that ROS was not a major factor contributing to Akt activation in respiration-suppressed cells (Fig. 4 E). Furthermore, analysis of superoxide in ρ- cells showed that they contained lower basal levels of superoxide than the parental cells (Fig. 4 F). Thus, these data suggest that ROS may not be a critical mediator to activate Akt in cells lacking mitochondrial respiration. It should be noted, however, that exogenous ROS could induce Akt activation in Raji cells, as indicated by a significant Akt phosphorylation in the presence of 0.5 mM H2O2 (Fig. 4 H). As expected, the H2O2-induced Akt activation could be suppressed by NAC (Fig. 4, G and H). This was different from the Akt activation in respiration-deficient cells.
NADH accumulation in respiration-deficient cells leads to inactivation of PTEN
Because NADH is an essential substrate (electron donor) for the mitochondrial electron transport chain, defects in the respiratory chain function could lead to an accumulation of NADH. Indeed, chemical inhibition of respiration by rotenone caused a substantial increase in cellular NADH, from 81 to 130 arbitrary units (Fig. 5 A). Consistent with this observation, mitochondrial genetic defects also led to a significant increase of NADH in HL60-C6F cell and all ρ- clones from Raji cells (Fig. 5 B). Interestingly, incubation of cell extracts from sonicated Raji cells with NADH (0.01–1 mM) in the presence of 1 mM ATP led to a significant increase in Akt phosphorylation without changing Akt protein levels (Fig. 5 C). Collectively, these observations suggest the possibility that the accumulation of NADH might be directly involved in Akt activation in the ρ- cells.
Figure 5.

Blockage of mitochondrial respiration caused an increase of cellular NADH and activation of Akt. (A) Inhibition of mitochondrial respiration by rotenone (Rot) caused a substantial increase in cellular NADH. The parental Raji cells were treated with 100 nM rotenone for 2 h. Cellular NADH was determined by measuring the relative NADH fluorescent intensity using flow cytometry analysis, as described in Materials and methods. (B) Quantification of NADH levels in parental HL-60 and Raji cells in comparison with their respective ρ- cell clones. (C) NADH enhanced Akt phosphorylation at Ser-473 in vitro. Protein lysates of Raji cells were prepared by sonication, as described in Materials and methods. The lysates containing cellular proteins and plasma lipid membranes were incubated for 20 min with the indicated concentrations of NADH and 1 mM ATP. Akt phosphorylation and total Akt protein were assayed by Western blotting.
Analysis of lactate (product of glycolysis) in culture medium showed that ρ- cells produced significantly more lactate (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200512100/DC1). The lack of oxygen consumption and increased lactate accumulation in ρ- cells indicate that the glycolytic pathway is highly active in these cells. We speculated that the increase in glycolysis for ATP generation in ρ- cells might divert the glucose metabolic flow away from the pentose phosphate pathway (PPP) and lead to a decrease of NADPH because PPP is the major pathway for NADPH generation. To test this possibility, HPLC analysis was performed to determine the ratios of NADH/NADPH in ρ- cells and parental cells. Under normal culture conditions, the ratios of NADH/NADPH in parental HL-60 and Raji cells were 0.50 and 0.36, respectively, suggesting an active NADPH generation in parental cells (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200512100/DC1). In contrast, all ρ- clones exhibited reversed NADH/NADPH ratios (1.41–2.46), which is consistent with the accumulation of NADH and the decrease in NADPH generation (Fig. S4 B).
To investigate the mechanism by which changes in cellular NADH and NADPH promote Akt activation, we tested the possibility that the increase in NADH/NADPH ratio might modify the redox status and function of PTEN, which is known to be redox-sensitive and dependent on NADPH/thioredoxin (Trx) to maintain its enzyme activity (Lee et al., 2002). Protein extracts from Raji cells were incubated with NADH, NADPH, Trx, or a combination of the three. The reaction products were divided into two portions for Western blot analysis under reducing and nonreducing conditions. As illustrated in Fig. 6 A, similar phospho-PTEN signals were detected in all samples under reducing condition, suggesting that there was no significant difference in PTEN phosphorylation. However, when the same samples were analyzed under nonreducing condition, striking differences in phospho-PTEN signals were observed. NADH caused a significant decrease of the phospho-PTEN signal detectable under nonreducing condition (Fig. 6 A, lane 1), suggesting that PTEN was likely in an oxidized state, which masked the epitope for antibody binding caused by disulfide bond formation. Oxidation of PTEN by hydrogen peroxide has been shown to cause such a conformational change (Lee et al., 2002). Incubation of cell extracts with NADPH/Trx kept the PTEN protein in a reduced state, and the phosphoepitopes were readily detected (Fig. 6 A, lanes 2–3). Combination of NADPH and Trx also kept PTEN in a reduced state (Fig. 6 A, lane 5). Surprisingly, NADH + Trx did not reduce PTEN, and thus the accessibility to epitopes at Ser-380 and Thr-382/-383 was limited (Fig. 6 A, lane 4). These data suggest that NADH may compete with NADPH/Trx to affect the redox state of PTEN. To test this possibility, protein extracts were incubated with NADPH/Trx in the presence of various concentrations of NADH, and the accessibility to epitopes at Ser-380 and Thr-382/-383 was determined under nonreducing conditions. As shown in Fig. 6 B, NADH competed with NADPH, and caused a concentration-dependent decrease of phospho-PTEN signal (lanes 3–5).
Figure 6.

Role of NADH in Akt activation through inactivation of PTEN by a redox-regulated mechanism. (A) Effect of NADH, NADPH, and Trx on the redox state of PTEN. Protein extracts were prepared from Raji cells by sonication, dialyzed with PBS for 60 min at 4°C, and then incubated with NADH, NADPH, and Trx in the presence of 1 mM ATP as indicated (37°C, 30 min). The redox status of PTEN was measured by assaying the accessibility of phosphorylated Ser-380 and Thr-382/383 epitopes by specific antibodies under reducing (2.5% β-mercaptoethanol in loading buffer, indicated by “R”) and nonreducing conditions (without β-mercaptoethanol, indicated by “NR”). Oxidation of PTEN masks the epitopes because of the formation of an intramolecular disulfide bond, which was broken by β-mercaptoethanol. (B) Competition between NADH and NADPH and their effects on the redox status of PTEN. Protein lysates of Raji cells were incubated at 37°C for 30 min with NADH, NADPH, and Trx (10 μg/60 μl reaction) in the presence of 1 mM ATP. The redox status of PTEN was measured by Western blot analysis under nonreducing condition, as described for A. The number below each lane indicates the relative density of the respective protein band normalized by β-actin. The relative band density of the reduced PTEN in the presence of NADPH and Trx (lane 2) is given an arbitrary value of 1. (C) Cellular localization of PTEN protein in Raji cells treated with or without 100 nM rotenone for 20 h and in Raji-C8 ρ- cells. Cells were fixed, immunostained for PTEN, and photographed, as described in Materials and methods. Arrows indicate the portion of PTEN protein associated with plasma membrane in parental Raji cells.
Fluorescent confocal microscopy analysis was then used to evaluate the localization of PTEN protein in Raji cells under various conditions. As illustrated in Fig. 6 C, a portion of the PTEN protein was localized in cellular membrane region under normal culture conditions. Incubation of Raji cells with rotenone decreased the membrane localization of PTEN, which exhibited defused intracellular distribution. The decrease in PTEN membrane localization was also observed in the respiration-deficient Raji-C8 cells (Fig. 6 C). These data suggest that mitochondrial defects or respiration inhibition could decrease PTEN membrane localization.
Akt activation in respiration-deficient cells is PTEN dependent
To further evaluate the role of PTEN inactivation in mediating NADH-induced Akt activation, we used four cancer cell lines with either wild-type PTEN (HCT116 and LN-229 cells) or PTEN-null (Jurkat and U87-MG cells) and tested their Akt activation in response to modulation of mitochondrial respiration. Incubation of HCT116 and LN-229 cells (wt PTEN) with rotenone led to a time-dependent Akt activation, which was indicated by an increase in Ser-473 phosphorylation (Fig. 7 A). In contrast, rotenone failed to further activate Akt in the PTEN-null cell lines (Jurkat and U87-MG, Fig. 7 B). It should be noted that although Akt phosphorylation was constitutively high in the PTEN-null cells, Akt could still be further activated in these cells by other stimuli, such as TRAIL (Zauli et al., 2005) and T cell antigen receptor stimulation with anti-CD3 (Kwon et al., 2003).
Figure 7.

PTEN-dependent activation of Akt in whole cells by inhibition of mitochondrial respiration. (A and B) PTEN-dependent activation of Akt by rotenone in whole cells. The wild-type PTEN cells (HCT116 and LN-229) and PTEN-null cells (Jurkat and U87-MG) were incubated with 100 nM rotenone (Rot) for the indicated times, and Akt activation was measured by Western blot analysis of Ser-473 phosphorylation. Total Akt was also blotted for comparison. (C and D) PTEN-dependent induction of Akt activation by hypoxia. The wild-type PTEN cells (HCT116 and LN-229) and PTEN-null cells (Jurkat and U87-MG) were incubated under hypoxic conditions for the indicated times, and protein extracts were immediately prepared for analysis of total Akt protein and its phosphorylation at Ser-473, as described in Materials and methods.
Hypoxia is frequently seen in the tumor microenvironment. Cancer cells under hypoxic conditions mainly use glycolysis to generate ATP and are metabolically similar to ρ- cells. We further evaluate the role of PTEN inactivation in mediating Akt activation under hypoxic conditions. When HCT116 and LN-229 cells (wild-type PTEN) were incubated under hypoxic conditions, there was a significant increase in Akt activation as early as 2 h; this increase was indicated by increased phosphorylation at Ser-473 (Fig. 7 C). In contrast, the PTEN-null cells (Jurkat and U87-GM) showed little change in their Akt phosphorylation (Fig. 7 D), further supporting the important role of PTEN in hypoxia-mediated Akt activation.
Inhibition of Akt activation sensitizes ρ- cells to anticancer agents
To evaluate the contribution of Akt activation to drug resistance in ρ- cells, we tested the effect of inhibiting Akt pathway on drug-induced apoptosis in the ρ- cells. Wortmannin, which is an inhibitor of the PI3K–Akt pathway, caused a concentration-dependent decrease in Akt phosphorylation at Ser-473 and Thr-308 in HL60-C6F cells (Fig. 8 A). Wortmannin also caused dephosphorylation of Akt in all Raji ρ- clones (Fig. 8 A). Another PI3K–Akt inhibitor, LY249002, produced similar results (unpublished data). Inhibition of PI3K/Akt by wortmannin significantly increased the sensitivity of ρ- cells to apoptosis induction by As2O3, as assessed by annexin-V reactivity (Fig. 8 B) and cleavage of procaspase-3 (Fig. 8, C and D). We also used SH-6, which is a selective Akt inhibitor that does not affect the upstream kinases (Kozikowski et al., 2003), to further test sensitization of ρ- cells. SH-6 was effective in enhancing apoptotic response to As2O3 in ρ- cells (Fig. 8 E). Thus, inhibition of the Akt pathway seems effective in enhancing the activity of anticancer agents against cancer cells with respiration defects. In contrast, inhibition of the mitochondrial ATP synthase (complex V) by oligomycin decreased cellular sensitivity to As2O3 and Taxol in parental HL-60 cells (Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200512100/DC1). Consistently, oligomycin did not significantly affect drug sensitivity in ρ- cells (HL60-C6F), which exhibited Akt activation and resistance to drug-induced apoptosis (Fig. S5). The insensitivity of ρ- cells to oligomycin is consistent with previous observations that the sensitivity of ATP-synthase (complex V) to oligomycin depends on the mitochondrial DNA-encoded subunit 6 of F1-ATPase (Avner and Griffiths, 1973), and that cells lacking mtDNA possess normal levels of the nuclear DNA- encoded α and β subunits of F1-ATPase, which is functional and sensitive to azide or aurovertin, but insensitive to oligomycin (Buchet and Godinot, 1998).
Figure 8.

Inhibition of PI3K–Akt pathway enhances drug sensitivity in respiration-deficient cells. (A) Inhibition of Akt phosphorylation by wortmannin in HL60-C6F cells and Raji ρ- clones. Cells were incubated for 24 h with the indicated concentrations of wortmannin (Wortm), and protein extracts were blotted using anti–phospho-Akt (Ser-473 and Thr-308) and anti-Akt antibodies. (B) Inhibition of PI3K–Akt pathway enhanced drug sensitivity in ρ- cells. All five ρ- clones were pretreated with or without wortmannin for 1 h and incubated with 1 μg/ml As2O3 for 24 h. Apoptotic cells were measured by flow cytometry analysis using annexin-V/PI staining. Results are the mean ± the SD of four separate experiments. (C) HL60-C6F cells were pretreated with or without 10 μM wortmannin for 1 h and incubated with 1 μg/ml As2O3 for up to 24 h. Cell lysates were probed for pro–caspase-3 by immunoblotting. A decrease in pro–caspase-3 band intensity indicates its cleavage during apoptosis. (D) Raji ρ- cells (C2) were pretreated with or without 1 μM wortmannin for 1 h, and incubated with 1 μg/ml As2O3 for up to 24 h. Cell lysates were probed for pro–caspase-3 by immunoblotting. (E) Five clones of ρ- cells were pretreated for 1 h with the Akt inhibitor SH-6, as indicated (3–20 μM), and incubated with 1 μg/ml As2O3 for 24 h. Apoptotic cells were then quantified by flow cytometry analysis using annexin-V/PI staining. Results are the mean ± the SD of four separate experiments.
Discussion
Mitochondrial respiration malfunction and increased glycolysis are frequently observed in cancer cells. This metabolic alteration, known as the Warburg effect, is caused by complex biochemical and molecular mechanisms. mtDNA mutations and tissue hypoxia represent genetic and environmental factors contributing to the Warburg effect. Cells deficient in respiration because of mtDNA alterations or hypoxic conditions are forced to produce ATP through glycolysis, which is much less efficient than oxidative phosphorylation. Nevertheless, cancer cells manage to overcome such an apparent metabolic disadvantage, survive in vivo, and eventually emerge as a malignant cell population resistant to anticancer agents at the late stages of the disease progression. Thus, understanding the mechanisms underlying the increased survival capacity in cancer cells with compromised mitochondrial respiratory function is an important research area.
Our study suggests that mitochondrial respiration deficiency leads to activation of the Akt survival pathway through NADH-mediated inactivation of PTEN. This is a novel mechanism contributing to increased survival and drug resistance in cancer cells with compromised mitochondrial respiration. Several lines of evidences support this conclusion, as follows: (a) Cells that lack mitochondrial respiration because of mtDNA deletion, chemical inhibition of the electron transport chain, or exposure to hypoxia all exhibited significant Akt activation. (b) The cellular NADH/NADPH ratio abnormally increased when mitochondrial respiration was suppressed, and this was associated with a decrease in plasma membrane–associated PTEN. (c) Exogenous NADH led to inactivation of PTEN and activation of Akt in vitro. The inactivation of PTEN seems to be caused by redox modulation because NADH competed with NADPH/Trx to keep PTEN in an oxidized (inactive) state. These findings are consistent with previous studies showing inactivation of PTEN by oxidation using hydrogen peroxide (Lee et al., 2002; Kwon et al., 2004). (d) Cells lacking functional PTEN did not respond to respiratory inhibition or hypoxia, and exhibited no further Akt activation, indicating the important role of PTEN in this process.
Under physiological conditions, NADH is generated through glycolysis and the tricarboxylic acid cycle, whereas NADPH is produced mainly via the PPP (shunt). The proportion of glucose directed to each pathway is regulated by the cellular energy metabolic state. Mitochondrial defects render cancer cells dependent on glycolysis for ATP supply, and the NADH generated from the tricarboxylic acid cycle is not used effectively because of the decrease in oxidative phosphorylation. These metabolic alterations lead to an accumulation of NADH. At the same time, NADPH production from the PPP decreases because of increased utilization of glucose for glycolysis. Indeed, we consistently observed that the NADH/NADPH ratio was significantly increased in all eight clones of ρ- cells (Fig. S4). Because NADH competes with NADPH and compromises the ability of NADPH/Trx to keep PTEN in a reduced state, the metabolic changes in cancer cells with mitochondrial defects would lead to inactivation of PTEN and activation of Akt. Interestingly, NAC suppressed Akt activation induced by H2O2, but did not decrease rotenone-induced Akt phosphorylation. The likely explanation is that the redox-sensitive PTEN was inactivated when the ratio of NADH/NADPH was significantly increased. This elevated ratio could not be modulated by NAC when cells were treated with rotenone. In contrast, the antioxidant NAC effectively decreased H2O2 and reduced its direct effect on PTEN.
The PI3K–Akt pathway is critical for cell survival (Cantley, 2002; Vivanco et al., 2002). Activation of PI3K results in generation of PIP3, which leads to activation of phosphoinositide-dependent kinase-1 (PDK-1) and phosphorylation of Akt. In contrast, the lipid phosphatase PTEN removes a phosphate from PIP3, and thus acts as a negative regulator of Akt. Loss of PTEN leads to Akt activation in cancer cells (Wu et al., 1998). Thus, it is likely that oxidation of PTEN suppresses its phosphatase activity and subsequently leads to Akt activation. Indeed, PTEN is sensitive to oxidative inactivation by H2O2 (Connor et al., 2005). The demonstration that respiration defects lead to activation of the Akt pathway caused by the accumulation of NADH and inactivation of PTEN reveals a novel mechanism by which cancer cells survive under respiration-compromised conditions. Fig. 9 illustrates a model of this cell survival mechanism.
Figure 9.

Schematic illustration of the mechanisms by which Akt is activated in cancer cells with mitochondrial respiration injury. Genetic factors (mtDNA mutations/deletions) and environmental factors (hypoxic conditions) cause defects in mitochondrial respiration and render the cancer cells highly dependent on glycolysis for ATP generation. This metabolic alteration leads to an increase in NADH caused by the lack of its utilization by the respiratory chain and a decrease in NADPH from the PPP. The increased NADH effectively competes with NADPH and compromises the ability of NADPH/Trx to keep PTEN protein in the reduced state, leading to inactivation of PTEN and activation of Akt. The solid arrows indicate increased activity in the respective steps; the dashed arrows indicate decreased activity.
The degree of Akt activation among the ρ- clones appeared somewhat heterogeneous. It is possible that during the process of establishing the ρ- clones, the use of ethidium bromide to deplete mtDNA might also cause nuclear DNA mutations, which might affect PTEN function and/or Akt activation. This could also explain the heterogeneous colony formation efficiencies observed among the ρ- clones. Although this heterogeneity reflects the complexity of the experimental systems, the conclusion that mitochondrial respiration defects lead to NADH-mediated PTEN inactivation and Akt activation remains valid. This argument is supported by the observations that Akt activation was observed in all eight ρ- clones, in cells treated with respiratory chain inhibitor rotenone, and in cells under hypoxia in a PTEN-dependent manner.
Because mitochondrial DNA mutations and hypoxia with subsequently increased glycolysis are prevalent in cancer cells (Polyak et al., 1998; Wallace, 1999; Fliss et al., 2000; Copeland et al., 2002; Gatenby and Gillies, 2004), activation of the Akt pathway through NADH-mediated PTEN inactivation is likely an important survival mechanism for cancer cells with such metabolic alterations. Additionally, the ability of Akt to promote glucose uptake may also contribute to cell survival (Rathmell et al., 2003). Interestingly, a recent study showed that Akt activation stimulates cells to use the glycolytic pathway to generate ATP (Elstrom et al., 2004). The observations that hypoxia caused Akt activation in both HCT116 and LN-229 cells and that respiratory-deficient cells exhibited certain growth advantage in hypoxia conditions further illustrate the clinical relevance of this mechanism in cancer cell survival and growth in vivo. Furthermore, if Akt activation is an important mechanism contributing to decreased drug sensitivity associated with the Warburg effect, it is possible to overcome such drug resistance by inhibition of Akt activation. In fact, our data suggest that this is possible. Further investigation is warranted to evaluate the clinical implications of this therapeutic strategy.
Materials and methods
Cell and cell culture
Human leukemia cells lines (HL-60 and Jurkat) and lymphoma cell line (Raji) were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS and 2 mM l-glutamine at 37°C with 5% CO2. Human malignant glioma cells (U87-MG and LN-229; S. Kondo, MD Anderson Cancer Center, Houston, TX) were cultured in DMEM medium supplemented with 10% FBS and 2 mM l-glutamine. HCT116 human colon cancer cells from B. Vogelstein (Johns Hopkins University, Baltimore, MD) were cultured in McCoy's 5A medium supplemented with 10% FBS and 2 mM l-glutamine. The respiration-deficient ρ- cells were established using an established method with ethidium bromide (King and Attardi, 1996), and maintained in RPMI 1640 medium containing 0.47% glucose, 10% FBS, 2 mM l-glutamine, 50 μg/ml uridine, and 1 mM pyruvate without ethidium bromide as previously described (Pelicano et al., 2003). To compare colony formation capacity under normoxia and hypoxia conditions, the parental Raji cells and the respiration-deficient Raji-C8 cells were seeded onto 6-well plates (300 cells/well for normoxia and 400 cells/well for hypoxia) in semisolid medium (MethoCult; StemCell Technologies, Inc.) containing 0.47% glucose, 10% FBS, 2 mM l-glutamine, 50 μg/ml uridine, and 1 mM pyruvate, and incubated at 37°C under normoxia (21% oxygen) or hypoxia (1.5–5% oxygen) conditions for 2 wk. Colonies were stained, photographed, and counted.
Antibodies and reagents
The following antibodies were used for immunoblotting analyses using standard Western blotting procedures: phospho-Akt antibodies, anti–GSK-3, anti–phospho-GSK-3α/β, anti-PI3Kp110α, anti-PI3Kp85, anti–phospho-Tyr of PI3Kp85, anti-PTEN, and phospho-PTEN antibodies (all purchased from Cell Signaling Technology); anti–β-actin (Sigma-Aldrich); anti-Akt1/2 antibodies (Santa Cruz Biotechnology, Inc); anti–pro-caspase-3 (BD Biosciences); LY294002, wortmannin, rotenone, NADH, NADPH, doxorubicin, taxol, As2O3, and vincristine (Sigma-Aldrich); SH-6 (AG Scientific, Inc.); Trx (Promega); MitoTracker Green, dihydroethidium, and rhodamine-123 (Invitrogen); and Methocult H4230 (StemCell Technologies, Inc.).
Flow cytometry analysis of mitochondria mass, ΔΨm, NADH, and apoptosis
Cells were stained with 60 nM MitoTracker Green (60 min) to measure the mitochondrial mass, with 100 ng/ml dihydroethidium (60 min) to detect superoxide, or with 1 μM rhodamine-123 (60 min) to evaluate the mitochondrial transmembrane potential, as previously described (Pelicano et al., 2003). Analysis was performed using a FACScan flow cytometer (Becton Dickinson). A minimum of 10,000 cells per sample was analyzed. Cellular NADH was measured by quantifying its intrinsic fluorescence under ultraviolet excitation, using a flow cytometer (LSR II; Becton Dickinson) equipped with a 325-nm excitation laser and a 440-nm centered band-pass filter, as previously described (Robinson et al., 2000). To analyze drug-induced apoptosis, cells were stained with annexin-V–FITC and PI, or with a monoclonal FITC-conjugated antiactive caspase-3 antibody according to the manufacturer's instruction (BD Biosciences). Data acquisition and analysis were performed using a FACScan flow cytometer with the CellQuest software (BD Biosciences). Cells that were positively stained by annexin- V–FITC only (early apoptosis) and positive for both annexin-V–FITC and PI (late apoptosis) were quantitated, and both subpopulations were considered as overall death cells.
Assays of Akt kinase activity and phosphorylation in vitro
The Akt enzyme activity was assayed using an Akt kinase kit according to the manufacturer's directions (Cell Signaling Technology). To analyze the effect of NADH and NADPH on Akt activation in vitro, cell lysates were prepared by sonication. Raji cells were suspended in PBS containing a cocktail of protease inhibitors (Roche), sonicated on ice bath, and centrifuged in a refrigerated centrifuge (model 5415R; Eppendorf) at 10,000 rpm for 15 min to remove cell debris. Endogenous NADH and NADPH in the cell lysates were removed by dialysis in cold PBS containing protease inhibitors for 60 min. The cell lysates containing proteins and sonicated plasma lipid membranes were incubated for 20 min with NADH, NADPH, and ATP as specified in the figure legends. Akt phosphorylation at Ser-473 and total Akt protein were assayed by Western blotting.
Measurement of respiration activity
Oxygen consumption in intact cells was measured as an indication of mitochondrial respiration activity. Cells (5 × 106) were suspended in 1 ml of culture medium preequilibrated with 21% oxygen; then they were placed in a sealed respiration chamber to monitor oxygen consumption, as previously described (Pelicano et al., 2003).
PCR analysis of mtDNA
Total DNA containing nuclear and mitochondrial DNA was isolated from 3 × 106 cells, as previously described (Carew et al., 2003). The nucleotide sequences of the PCR primers for mitochondrial D loop (15–484), NDI (3,304–3,836), COXII (7,645–8,215), ATPase 6 (8,539–9,059), ND4 (11,403–11,927), Cytochrome b (15,260–15,774), and GAPDH, and the PCR reaction conditions were previously described (Carew et al., 2003). The PCR products were analyzed by electrophoresis on a 1.2% Agarose gel, stained with ethidium bromide, and photographed.
Measurement of intracellular NADH and NADPH by HPLC
The parental cells (HL-60 and Raji) and ρ- cells in exponentially growing phase were washed twice with PBS, and NADH and NADPH were extracted using 0.4 N HClO4, followed by neutralization with concentrated KOH. The neutralized cell extracts were immediately analyzed for NADH and NADPH, using a HPLC method adapted from previously described procedures (Reiss et al., 1984). In brief, NADH/NADPH standards or cell extracts (550 μl; equivalent to 5.5 × 106 cells) were applied to an anion-exchange column (Partisil-10 SAX; Whatman) and run at a flow rate of 1.5 ml/min using a concave gradient (curve #9) from 100% buffer A (5 mM NaH2PO4, pH 4.0) to 100% buffer B (250 mM NaH2PO4 + 0.5 M NaCl, pH 4.75) over 15 min, followed by another 15-min isocratic 100% buffer B, using a HPLC system (Waters Alliance) equipped with Empower Software. NADH and NADPH were detected by their UV absorbance at 340 nm, with retention times of 17.1 and 20.1 min, respectively, under these conditions. Because auto-oxidation of NADH and NADPH in the samples can decrease the UV absorbance at 340 nm, HPLC analysis was performed immediately after cell extracts were prepared. To minimize the potential effect of auto-oxidation on quantitation, the ratio of NADH/NADPH was calculated from the peak areas of NADH and NADPH of the same HPLC chromatograms. This ratio did not change when the same sample was injected at different times. This normalization allows a quantitative comparison of NADH/NADPH ratios between the parental cells and the ρ- clones.
Immunofluorescence cytochemistry and confocal microscopy
Cells were cytospun onto poly-l-lysine–coated glass slides using a cytospin (Shandon-Elliot) and were immediately fixed with 100% acetone for 5 min. After blocking in PBS containing 1% BSA, the samples were stained with anti-PTEN mouse monoclonal antibody for 1 h (clone PTEN-18; Sigma-Aldrich), followed by a 1-h incubation with Texas red–conjugated anti–mouse secondary antibody (Vector Laboratories; 1:300). The cells were visualized using a laser scanning confocal microscope (FluoView 500; Olympus). Images were captured using a 60× objective with proper filter sets (model IX71FVSF-2; Olympus).
Statistical analysis
A t-test was used to evaluate the statistical differences of the experimental values between two samples to be compared.
Online supplemental material
Fig. S1 shows the comparison of cell cycle profiles and cytochrome c expression in ρ- clones and their parental cells. Fig. S2 shows the activation of Akt in Raji cells treated with respiratory chain inhibitors. Fig. S3 shows the lactate production in ρ- cells in comparison with their parental cells (HL-60 and Raji). Fig. S4 shows the comparison of NADH/NADPH ratios in parental HL-60 and Raji cells and respiration-deficient cell clones. Fig. S5 shows the effect of oligomycin on cellular sensitivity to taxol and As2O3 in HL-60 cells and HL-60–C6F cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200512100/DC1.
Supplementary Material
Acknowledgments
We thank Dr. Bert Vogelstein (Johns Hopkins University) for providing HCT116 cells, Dr. Seiji Kondo (MD Anderson cancer Center) for providing U87-MG and LN-229 cells, and Wendy D. Schober for her expert assistance with flow cytometry analysis.
This work was supported in part by grants CA85563, CA109041, CA100428, CA100632, and CA16672 from the National Cancer Institute at the National Institutes of Health. J.S. Carew is a recipient of a fellowship from the American Legion Auxiliary.
R. Sasaki's current address is Division of Radiology, Kobe Graduate School of Medicine, Kobe City, Hyogo 650-0017, Japan.
Abbreviations used in this paper: GSK, glycogen synthase kinase; mtDNA, mitochondrial DNA; NAC, N-acetylcysteine; PI, propidium iodide; PI3K, phosphatidylinositol 3-kinase; PIP3, phosphatidylinositol-3′-phosphate; PPP, pentose phosphate pathway; ROS, reactive oxygen species; Trx, thioredoxin.
References
- Aggarwal, B.B. 2004. Nuclear factor-kappa B: the enemy within. Cancer Cell. 3:203–208. [DOI] [PubMed] [Google Scholar]
- Avner, P.R., and D.E. Griffiths. 1973. Studies on energy-linked reactions. Genetic analysis of oligomycin-resistant mutants of Saccharomyces cerevisiae. Eur. J. Biochem. 32:312–321. [DOI] [PubMed] [Google Scholar]
- Buchet, K., and C. Godinot. 1998. Functional F1-ATPase essential in maintaining growth and membrane potential of human mitochondrial DNA-depleted ρ° cells. J. Biol. Chem. 273:22983–22989. [DOI] [PubMed] [Google Scholar]
- Cantley, L.C. 2002. The phosphoinositide 3-kinase pathway. Science. 296:1655–1657. [DOI] [PubMed] [Google Scholar]
- Carew, J.S., Y. Zhou, M. Albitar, J.D. Carew, M.J. Keating, and P. Huang. 2003. Mitochondrial DNA mutations in primary leukemia cells after chemotherapy: clinical significance and therapeutic implications. Leukemia. 17:1437–1447. [DOI] [PubMed] [Google Scholar]
- Chan, S.L., and V.C. Yu. 2004. Proteins of the bcl-2 family in apoptosis signaling: from mechanistic insights to therapeutic opportunities. Clin. Exp. Pharmacol. Physiol. 31:119–128. [DOI] [PubMed] [Google Scholar]
- Connor, K.M., S. Subbaram, K.J. Regan, K.K. Nelson, J.E. Mazurkiewicz, P.J. Bartholomew, A.E. Aplin, Y.-T. Tai, J. Aguirre-Ghiso, S.C. Flores, and J.A. Melendez. 2005. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 280:16916–16924. [DOI] [PubMed] [Google Scholar]
- Copeland, W.C., J.T. Wachsman, F.M. Johnson, and J.S. Penta. 2002. Mitochondrial DNA alterations in cancer. Cancer Invest. 20:557–569. [DOI] [PubMed] [Google Scholar]
- Dahia, P.L., R.C. Aguiar, J. Alberta, J.B. Kum, S. Caron, H. Sill, D.J. Marsh, J. Ritz, A. Freedman, C. Stiles, and C. Eng. 1999. PTEN is inversely correlated with the cell survival factor Akt/PKB and is inactivated via multiple mechanisms in haematological malignancies. Hum. Mol. Genet. 2:185–193. [DOI] [PubMed] [Google Scholar]
- Downward, J. 2004. PI 3-kinase, Akt and cell survival. Semin. Cell Dev. Biol. 15:177–182. [DOI] [PubMed] [Google Scholar]
- Edinger, A.L., and C.B. Thompson. 2002. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell. 13:2276–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstrom, R.L., D.E. Bauer, M. Buzzai, R. Karnauskas, M.H. Harris, D.R. Plas, H. Zhuang, R.M. Cinalli, A. Alavi, C.M. Rudin, and C.B. Thompson. 2004. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 64:3892–3899. [DOI] [PubMed] [Google Scholar]
- Fliss, M.S., H. Usadel, O.L. Caballero, L. Wu, M.R. Buta, S.M. Eleff, J. Jen, and D. Sidransky. 2000. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. 287:2017–2019. [DOI] [PubMed] [Google Scholar]
- Gatenby, R.A., and R.J. Gillies. 2004. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer. 4:891–899. [DOI] [PubMed] [Google Scholar]
- King, M.P., and G. Attardi. 1996. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol. 264:304–313. [DOI] [PubMed] [Google Scholar]
- Kozikowski, A.P., H. Sun, J. Brognard, and P.A. Dennis. 2003. Novel PI analogues selectively block activation of the pro-survival serine/threonine kinase Akt. J. Am. Chem. Soc. 125:1144–1145. [DOI] [PubMed] [Google Scholar]
- Kwon, J., S. Devadas, and M.S. Williams. 2003. T cell receptor-stimulated generation of hydrogen peroxide inhibits MEK-ERK activation and lck serine phosphorylation. Free Radic. Biol. Med. 35:406–417. [DOI] [PubMed] [Google Scholar]
- Kwon, J., S.R. Lee, K.S. Yang, Y. Ahn, Y.J. Kim, E.R. Stadtman, and S.G. Rhee. 2004. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA. 101:16419–16424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K., P.D. Neufer, and R.S. Williams. 1995. Nuclear responses to depletion of mitochondrial DNA in human cells. Am. J. Physiol. Cell Physiol. 269:C1265–C1270. [DOI] [PubMed] [Google Scholar]
- Lee, S.R., K.S. Yang, J. Kwon, C. Lee, W. Jeong, and S.G. Rhee. 2002. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 277:20336–20342. [DOI] [PubMed] [Google Scholar]
- Nomoto, S., M. Sanchez-Cespedes, and D. Sidransky. 2002. Identification of mtDNA mutations in human cancer. Methods Mol. Biol. 197:107–117. [DOI] [PubMed] [Google Scholar]
- Pelicano, H., L. Feng, Y. Zhou, J.S. Carew, E.O. Hileman, W. Plunkett, M.J. Keating, and P. Huang. 2003. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J. Biol. Chem. 278:37832–37839. [DOI] [PubMed] [Google Scholar]
- Polyak, K., Y. Li, H. Zhu, C. Lengauer, J.K. Willson, S.D. Markowitz, M.A. Trush, K.W. Kinzler, and B. Vogelstein. 1998. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 20:291–293. [DOI] [PubMed] [Google Scholar]
- Rathmell, J.C., C.J. Fox, D.R. Plas, P.S. Hammerman, R.M. Cinalli, and C.B. Thompson. 2003. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell. Biol. 23:7315–7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss, P.D., P.F. Zuurendonk, and R.L. Veech. 1984. Measurement of tissue purine, pyrimidine, and other nucleotides by radial compression high-performance liquid chromatography. Anal. Biochem. 140:162–171. [DOI] [PubMed] [Google Scholar]
- Robinson, J.P., Z. Darzynkiewicz, P.N. Dean, L.G. Dressler, P.S. Rabinovitch, C.C. Stewart, H.J. Tanke, and L.L. Wheeless. 2000. Current Protocols in Cytometry. Supplement 14. John Wiley & Sons, Inc. New York.
- Simonnet, H., N. Alazard, K. Pfeiffer, C. Gallou, C. Beroud, J. Demont, R. Bouvier, H. Schagger, and C. Godinot. 2002. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis. 23:759–768. [DOI] [PubMed] [Google Scholar]
- Stambolic, V., A. Suzuki, J.L. de la Pompa, G.M. Brothers, C. Mirtsos, T. Sasaki, J. Ruland, J.M. Penninger, D.P. Siderovski, and T.W. Mak. 1998. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 95:29–39. [DOI] [PubMed] [Google Scholar]
- Vivanco, I., L. Charles, and C.L. Sawyers. 2002. The phosphatidylinositol 3-Kinase–Akt pathway in human cancer. Nat. Rev. Cancer. 2:489–501. [DOI] [PubMed] [Google Scholar]
- Wallace, D.C. 1999. Mitochondrial diseases in man and mouse. Science. 283:1482–1488. [DOI] [PubMed] [Google Scholar]
- Warburg, O. 1930. The Metabolism of Tumors. Constable and Company, Ltd. London. 327 pp.
- Warburg, O. 1956. On the origin of cancer cells. Science. 123:309–314. [DOI] [PubMed] [Google Scholar]
- Wu, X., K. Senechal, M.S. Neshat, Y.E. Whang, and C.L. Sawyers. 1998. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA. 95:15587–15591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, R., H. Pelicano, Y. Zhou, J.S. Carew, L. Feng, K.N. Bhalla, M.J. Keating, and P. Huang. 2005. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 65:613–621. [PubMed] [Google Scholar]
- Zauli, G., S. Sancilio, A. Cataldi, N. Sabatini, D. Bosco, and R. Di Pietro. 2005. PI-3K/Akt and NF-kappaB/IkappaBalpha pathways are activated in Jurkat T cells in response to TRAIL treatment. J. Cell. Physiol. 202:900–911. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}