

Abstract
Mammalian mismatch repair (MMR) plays a prominent role in genomic stability and toxicity induced by some DNA damaging agents. Advance in the appreciation of regulation mechanisms of the key MMR protein hMSH2 would certainly lead to valuable information on its role and to a better understanding of MMR system dysfunctions with respect to their consequences in cells. We have previously reported that, in myeloid leukemic U937 cell line, the expression of hMSH2 MMR protein is regulated by protein kinase C (PKC) activity. Here we show that the increase of protein level following PKC activation by phorbol ester (TPA) treatment parallels that of hMSH2 mRNA. Our results support the view that the hMSH2 gene is prone to transcriptional regulation upon TPA induction, and that AP-1 is a factor implicated in the transactivation. When losing the AP-1-dependent hMSH2 promoter activity, either by mutating the AP-1 binding sites of the hMSH2 promoter or by using a dominant negative c-Jun factor, the hMSH2 overexpression induced by TPA is abolished both in vitro and in vivo. Thus the control of hMSH2 expression by PKC appears to be dependent, at least partially, on an up-regulation mediated by AP-1 transactivation.
INTRODUCTION
DNA mismatch repair (MMR) has long been known to be involved in two important cellular processes: (i) the repair of mismatches generated by misincorporation of nucleotides during DNA replication and (ii) the processing of recombination intermediates (reviewed in 1,2). Mammalian MMR system functions as a combination of at least five proteins that recognise mismatches and repair DNA via excision and replacement with the correct nucleotide (for reviews see 2–4). Repair is initiated by either one of two heterodimers, both including the hMSH2 protein. The hMSH2-hMSH6 (hMutSα) and the hMSH2-hMSH3 (hMutSβ) complexes differ on their recognition specificity. The former is directed on base–base substitution or small loops, while the latter binds mainly insertion–deletion of a larger number of nucleotides. The recognition complex bound to DNA recruits hMutLα, a heterodimer of hMLH1 and hPMS2, to perform the repair reaction.
Loss of MMR activity led to a mutator phenotype, consisting of a high mutation rate and an associated instability of genomic microsatellite sequences (MSI). Inactivation of the repair pathway is associated with cancer development, as illustrated by the inherited familial cancer syndrome of hereditary non-polyposis colon cancers (HNPCC) (5,6). Additionally, sporadic malignancies of gastrointestinal, gynaecological and genitourinary tracts are in part attributable to defective repair proteins (7). In addition to the MSI+ phenotype, deficiency in MMR activity leads to different phenotype (8), such as the resistance to genotoxics, exemplified by the mechanism of tolerance to methylating agents (9), or conversely, sensitivity to topoisomerase inhibitors via an indirect mechanism (10).
Alterations of the MMR machinery are mainly documented as point mutations throughout the coding sequence of the components of the MMR system (6) for the most part in the hMSH2 and hMLH1 genes (11). Despite the extensive biochemical and genetic studies, only limited information is available concerning the regulation of the MMR repair system. Non-mutational events can also be responsible for the variation of the MMR capacity. Treatment of cells with alkylating agents leads to an increase of the nuclear level of hMSH2 and hMSH6, and thus the binding activity, due to translocation of the proteins from cytoplasm (12). Post-translational modification has also been described to take place following an in vivo phosphorylation of the hMutα complex, resulting in an improved capability of mismatch binding (13). Phosphorylation is mediated in vitro by casein kinase II (CKII) and protein kinase C (PKC). Moreover, MMR can be modulated by changes in MMR gene expression. In methotrexate-resistant human cells, a gene amplification of hMSH3 leads to an overproduction of the corresponding protein related to the depletion of the hMutSα complex, and thus to a partial mutator phenotype (14,15). Conversely, an epigenetic process marked by promoter region hypermethylation is associated with transcription loss of the hMLH1 gene (16). Likewise the hMSH6 promoter can also be silenced by cytosine methylation (17). And the identification of two elements controlling the transcriptional activity of hMLH1 has been mentioned elsewhere (18). An additional case of transcriptional regulation concerns the hMSH2 gene upon P53 activation (19,20).
Recently, we initiated a study intending to identify internal or external signals that would affect the MMR expression and/or function (21). We found that PKC stimulation was responsible for increased expression and activity of proteins belonging to the MMR system in the U937 leukaemia cells after phorbol-ester treatment. In addition, increase in hMSH2 expression was inhibited by PKC antagonists, and our results suggested that activation of PKC isoforms by phorbol esters or DAG were able to modulate MMR activity.
In this study, we focused on further characterising the mechanism that sustains the increasing of hMSH2 expression. We brought here evidence that phorbol-ester-induced PKC stimulation favoured an induction of the hMSH2 gene transcription. Treatment of the cells with tetradecanoylphorbol-13-acetate (TPA) specifically increases the amount of hMSH2 mRNA. We carried out a functional analysis of the hMSH2 promoter region, in vitro and in vivo, demonstrating a direct participation of the Activator Protein-1 (AP-1) c-Jun factor in the up-regulation. We highlight here the existence of a cascade of positive regulation that occurs by successive activations of PKC, AP-1 transcription factor, hMSH2 gene expression and consequently MMR function. We hypothesise that increased hMSH2 expression could play a protective role in the terminal differentiation pathway in the leukaemia model.
MATERIALS AND METHODS
Cell lines and chemicals
HeLa cells were obtained from the European Molecular Biology Laboratory (Heidelberg, Germany). Monocytic U937 cells were from the ATCC (Rockville, MD). U937 cells, stably transfected with a dominant negative of c-Jun (22) or with the empty vector, were kindly provided by Dr K. Nakaya (Showa University, Tokyo, Japan). Cells were cultured in RPMI 1640 media supplemented with 10% fetal calf serum, 2 mM l-glutamine, 100 µg/ml streptomycin and 200 U/ml of penicillin. For stable transfectants, 200 µg/ml of G418 are added to the culture medium. Cells were maintained at 37°C in a humidified atmosphere with 5% CO2, and were routinely tested for mycoplasmal infection. Treatment with TPA (50 nM, except when notified) were done on serum-starved cells. All the chemicals were purchased from Sigma (St Quentin-Fallavier, France).
Semi-quantitative reverse transcription
Total RNA was extracted from cell cultures using TRIZOL reagent (Gibco BRL) and resuspended in H2O-DEPC. Quantity and quality of RNAs were evaluated by capillar electrophoresis on Agilent 2000 Bioanalyser (Agilent Technologies, Massy, France).
RT–PCR experiments were realised with the ‘Access RT–PCR system’ kit (Promega, Madison, WI) to amplify fragments of mRNA of hMSH2 (450 bp), of the human β2 microglobuline (HB2, 167 bp) and the Escherichia coli kanamycin resistance gene (323 bp), using three sets of oligonucleotides. Sequences were the following: for hMSH2, 5′-TTGGCATATAAGGCTTCTCCTGGC-3′ and 5′-TGC AACCTGATTCTCCATTTCTGG-3′; for HB2, 5′-CATCC AGCGTACTCCAAAGA-3′ and 5′-GACAAGTCTGAATG CTCCAC-3′; for the prokaryotic gene, 5′-GCCATTCTCAC CGGATTCAGTCGTC-3′ and 5′-AGCCGCCGTCCCGTC AAGTCAG-3′. Reactions consisted of co-amplification of both the target RNA and the standard prokaryotic RNA in the same tube, in a final volume of 75 µl containing 2.35 pg of control RNA, 2.25 µg of total RNA, 1.5 µM of the two corresponding sets of primers, 20 µCi of [α-32P]dCTP (ICN, 3000 Ci/mmol) added to the other components of the kit. During completion of the cDNA synthesis, samplings of 5 µl were brought out at cycles 15, 20, 25, 30, 35, for electrophoresis analysis on 5% polyacrylamide gel. Reaction products were detected and quantified on a PhosphorImager (Storm System, Molecular Dynamics). The standard prokaryotic molecule did not interfere with the target. Signal from the bacterial exogenous control template coamplified in the same RT–PCR reaction served to normalise and to compare the hMSH2 sample data.
Western blot analysis
Total cell extracts were prepared from 2 × 107 cells following the method described by Andrews and Faller (23). After separation of the proteins (30 µg) on a 8% SDS–polyacrylamide gel and transfer onto PVDF membrane (Hybond-P, Amersham Pharmacia Biotech), immunoblotting was performed using mouse monoclonal hMSH2 antibody (1 µg/ml, cloneG219-1129, Pharmingen). Mouse monoclonal PCNA antibody (clone PC10, Dako) was used as a loading control. After washing the blots, horseradish peroxidase- conjugated anti-mouse antibody was added to perform a chemoluminescent detection using the Super Signal kit (Pierce).
Electrophoretic mobility shift assay (EMSA)
Protein extracts used in EMSA were the same as described above. Substrates were duplex 32P-labeled oligonucleotides. Sequences of the oligonucleotide used in the mismatch band shift experiments have been published (24). Top strand sequences of the oligonucleotides used in the AP-1 band shift experiments are: AP-1/Coll., 5′-ACACACGCTGA CTCAGCATAAGCC-; AP-1/612, 5′-CGAATGAGTGAAT CATCAACGAGT-; 612mut, 5′- CGAATGAGTTAACC ATCAACGAGT-; 291mut, 5′-CTTGCAGCTTAAGAAA CACAGAAA-. A random 34mer was used as non-specific oligonucleotide. Band shift assays were performed as previously reported (25). Briefly, extracts (30 µg) were incubated for 5 min at room temperature with 2 pmol of non-radioactive duplex in 15 µl of reaction mixture (25 mM HEPES–KOH pH 8.0, 0.5 mM EDTA, 1 mM dithiothreitol, 50 mM KCl, 50 µg/ml BSA, 5% glycerol). Incubation with the added radiolabeled oligonucleotide (20 fmol) continued for 20 min. Products of the reaction were separated on 6% polyacrylamide gels, and detected on the PhosphorImager (Storm System, Molecular Dynamics). When testing the effect of c-Jun antibody (rabbit polyclonal antibody, clone sc-44, Santa Cruz Biotechnology, CA) on the mobility complex, 0.5 µg of the antibody was added to the extracts prior to the radiolabeled oligonucleotide.
Expression vectors for transient transfection:
Constructs for transfection were derivatives of pGL3 vector (Promega). The pGL3-Promoter vector was used as control. The hMSH2 luciferase reporter plasmid was kindly provided by Dr K. A. Strait (LDS Hospital, Salt Lake City, UT) and consisted of a 1.88 kb fragment of the upstream promoter region of the hMSH2 gene fused to the luciferase gene on the pGL3-Basic vector (20). Mutations in the AP-1 sites of the hMSH2 promoter were introduced using the Quick change Mutagenesis kit (Stratagene, La Jolla, CA) with the mutant oligonucleotides designed for the EMSA experiments (612mut and 291mut, compare with ‘Electrophoretic mobility shift assay’, above). A pEGF plasmid (Clontech laboratories, Palo Alto, CA) served as reporter for transfection efficiencies by immunofluorescence analysis on FACS Calibur flow cytometer (Becton Dickinson, USA).
Transient transfection and luciferase activity assays
Conditions for transient transfections of U937 cells were set up using the pEGF vector. Briefly, 106 cells were electroporated at 1 Hz, 300 V for 10 × 5 ms (CNRS Cell eletropulsator, Jouan, St Herblain, France) in 100 µl of an isoosmotic solution (10 mM K2HPO4/KH2PO4, 1 mM MgCl2, 250 mM saccharose) with 10 µg of the Firefly luciferase vectors (pGL3 plasmids), and 10 µg of the pEGF plasmid or the Renilla luciferase vectors (pRL-TK, Promega) as internal controls. Electroporated cells were resuspended in 2 ml of 0.5 % serum-containing medium for 24 h incubation at 37°C, 5% CO2, and then stimulated by TPA (100 nM) for 6 h. Luciferase assays were performed using the Dual-Luciferase reporter assay system (Promega) that allows sequential determination of the Firefly luciferase activity from the hMSH2 construct vectors (pGL3) and the Renilla luciferase activity from the internal control vector (pRL-TK) in the same cell lysate sample. Luminescence was recorded on a TopCount.NXT luminometer (Packard Bioscience Company) for 5 s. All the data are expressed as relative luciferase activity corresponding to the normalisation with the Renilla luciferase values.
RESULTS
Increase of hMSH2 mRNA level through PKC activation
Exposure of U937 cells to TPA stimulates the global PKC activity as a function of time and dose (26). In a previous article, we reported an induction of hMSH2 expression in response to PKC stimulation (21). When U937 cells were TPA-treated at various times (3, 6 and 24 h), the amount of hMSH2 protein gradually increased as the incubation time with TPA progressed (Fig. 1A). The elevation of hMSH2 level paralleled that of mismatch binding activity determined by EMSA experiment (Fig. 1B).
Figure 1.
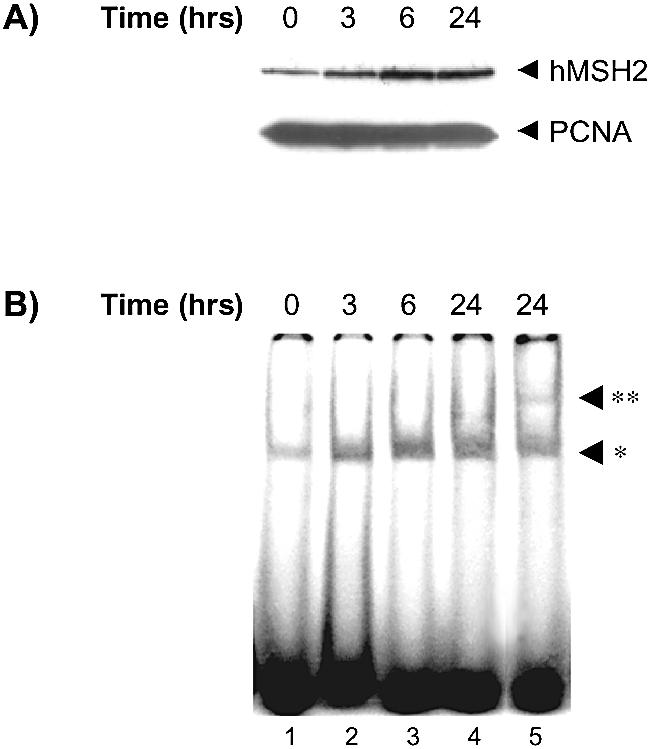
Kinetics of induction of hMSH2 expression and binding activity through TPA activation. U937 cells were TPA-treated (50 nM) for various times (0, 3, 6 and 24 h, as indicated) and protein extracts were prepared for the following two sets of experiments. (A) On the top panel, hMSH2 expression was observed as a 105 kDa band, by western blot analysis with 30 µg of extracts. On the bottom panel, detection of the PCNA protein (36 kDa) serves as a loading control. Immunoreactive proteins were detected as described in Materials and Methods. (B) Binding activity of the hMSH2-hMSH6 complex was evaluated on a 34mer duplex containing a TG mispair (lanes 1–4) with 30 µg of extracts. Supershift assays were performed incubating hMSH6-antibodies with the 24-h treated cell extracts (lane 5). The single and double asterisks designate the positions of the bound oligonucleotides and the supershift complex respectively.
To clarify the contribution of the PKC stimulation to the upregulation of hMSH2 expression, we evaluated a possible transcriptional activation of hMSH2. This possibility ought to show a concomitant increase of the hMSH2 mRNA with the protein levels in response to PKC induction. In RT–PCR experiments, we observed that the yield of hMSH2 transcripts greatly rose as a consequence of TPA treatment (Fig. 2A). The hMSH2 mRNA increase was not associated with changes in the steady-state levels of the HB2 control mRNA (Fig. 2A).
Figure 2.

Measurement of hMSH2 mRNA level in TPA-treated or not treated U937 cells by RT–PCR analysis. Radioactive RT–PCR experiments were carried out on total RNAs (2.25 µg) from U937 cultures previously incubated or not with TPA (50 nM) for 24 h, as indicated. Amplification with specific primer sets for hMSH2 or HB2 mRNAs jointly included a procaryotic standard RNA in the reaction tube for both experiments. (A) Products from RT–PCR experiments were fractionated by polyacrylamide gel electrophoresis and co-amplified radioactive bands for HB2 (167 bp) and procaryotic control (323 bp) on the left panel, and hMSH2 (450 bp) and procaryotic control on the right panel, are designated. (B) The amount of radioactivity present in the hMSH2 (solid line) and control (dotted line) cDNA bands of the same tube were quantified by densitometric scanning for a range of amplification cycles from 15 to 35. Values are shown graphically, for conditions of U937 cultures with (closed square) or without TPA (open square). (C) Kinetic of the hMSH2 mRNA induction following a TPA treatment of 3, 6 or 24 h, reported as ratios of the amount of radioactivity quantified at the end of the amplification cycles relative to the corresponding value obtained without TPA treatment.
A semi-quantitative analysis at various amplification cycles specified the difference in the relative efficiency of cDNA synthesis from TPA-treated and non-treated cells (Fig. 2B). The two curves related to the coming out of the PCR products separated at the 15th cycle, and the divergence range from 2- to 5- fold for the later cycles in favour of the TPA-treated cells. Measurements were made within the linear range of amplification. The extent of hMSH2 mRNA was increased by ∼5-fold when comparing the values after 24 h of incubation time with TPA (Fig. 2C). From these data, it appears that the induction of hMSH2 mRNA content and protein levels, following TPA treatment, is reached by means of similar extent and kinetics (Figs 1 and 2).
Presence of putative binding sites for AP-1 transcriptional activators in MSH2 promoter
One consequence of the activation of PKC is a downstream activation of one of the mitogen-activated protein kinase (MAPK) pathways, and the activation of AP-1 transcriptional factors (27–30). The 5′ regulatory promoter region of the hMSH2 gene has been characterised (31,32), and the sequence analysis shows up the presence of two consensus AP-1 binding sites indicative of a potential regulation of hMSH2 expression. These putative binding sites, also called TRE (TPA response element), are located at –612 and –291 from the start of transcription (schematic representation in Fig. 3A).
Figure 3.

Specific formation of complexes on the TRE like site of hMSH2 promoter. (A) Schematic representation of the 5′ regulatory promoter region of the hMSH2 gene picturing the position and sequence of the two putative AP-1 like sites (filled boxes) at –612 and –291. Base changes in the two mutated sites are noted. In between presence of two consensus P53 sites (open boxes) is mentioned. (B) The hMSH2 AP-1 site (AP-1/612) carried on a 24mer duplex was assayed for recognition by factors provided by HeLa cells extracts (30 µg) (lanes 5 and 6), in parallel to the defined AP-1 site of the collagenase gene promoter (AP-1/Coll) (lanes 2 and 3). A 100-fold molar excess of duplex competitors was added, either non-specific (N.S.) or containing an AP-1 box, as indicated. Lane 1 shows the migration of the AP-1 probe when missing HeLa cell extracts in the reaction. (C) Same experiments were realised with mutated AP-1 binding site (612mut and 291mut) used either as competitors for AP-1/612 substrate (lanes 3 and 4) or directly as substrates for binding (lanes 6 and 7). (D) The effect of inhibitory c-Jun antibodies (0.5 µg) (lanes 2 and 4) was tested for the binding on AP-1/Coll or AP-1/612 oligonucleotides, in the presence of non-specific competitors. Arrows denote the position of the specific protein/AP-1 DNA complex.
To assign an explicit role to the hMSH2 TRE sites, we evaluated the capability of AP-1 binding to these promoter sequences by EMSA. With oligonucleotides containing the –612 AP-1 binding site of hMSH2 (AP-1/612), a retardation mobility band was detected (Fig. 3B, lane 5) which migrated similarly to that obtained with a duplex of the consensus AP-1 collagenase sequence (AP-1/Coll, Fig. 3B, lane 2). Specificity of binding was confirmed by competition experiments (Fig. 3B). The presence in excess of AP-1/612 duplex was able to prevent bandshift of the collagenase AP-1. In contrast, introduction of a mutation into the –612 sequence rendered it ineffective in the competition experiments (Fig. 3C). The same results were obtained with alteration in the second hMSH2 AP-1 binding site (AP-1/291, Fig. 3C, lane 4). Neither one of these two mutated sites, when used as a substrate, showed any significant complex (Fig. 3C, lanes 6 and 7). These results indicated that the –612 duplex, and potentially the –291 duplex, carried the classical TRE sequence target for the AP-1 transcription factors.
We further confirmed that the protein complex bound to the hMSH2 promoter sequences contained the AP-1 factor c-Jun. Bandshift experiments were realised in the presence of particular c-Jun-antibodies that inhibit the binding of AP-1 complex to its specific site. Inclusion of this antibody in the reaction mixture effectively caused a large reduction of the slow migrating band with both the AP-1/Coll and the AP-1/612 duplexes (Fig. 3D).
Enhancement of the AP-1 binding to the hMSH2 promoter sequence by TPA treatment
We hypothesised that AP-1 might be involved in the regulation of hMSH2 transcription. Treatment with TPA induces a stimulation of AP-1 activity that could be mediated via a PKC signal in different cellular contexts. In this respect, we would expect a better efficiency in AP-1 recognition of the promoter for TPA-treated U937 cells than for non-treated controls. We found a faint binding activity on the hMSH2 promoter sequence with U937 cells extracts (Fig. 4). However, the binding activity appeared really sharper when U937 cells were TPA-treated. The specificity of recognition was confirmed by competition with AP-1/Coll duplexes. In parallel, same U937 extracts, treated and not, were assayed for a separate binding activity driven by the Ku heterodimer as a control of the protein extracts quality. As expected, same extent of retardation mobility band was observed for both extracts (Fig. 4, lanes 5 and 6). These results show that the low AP-1 binding activity present in the U937 cells was enhanced by TPA effect.
Figure 4.
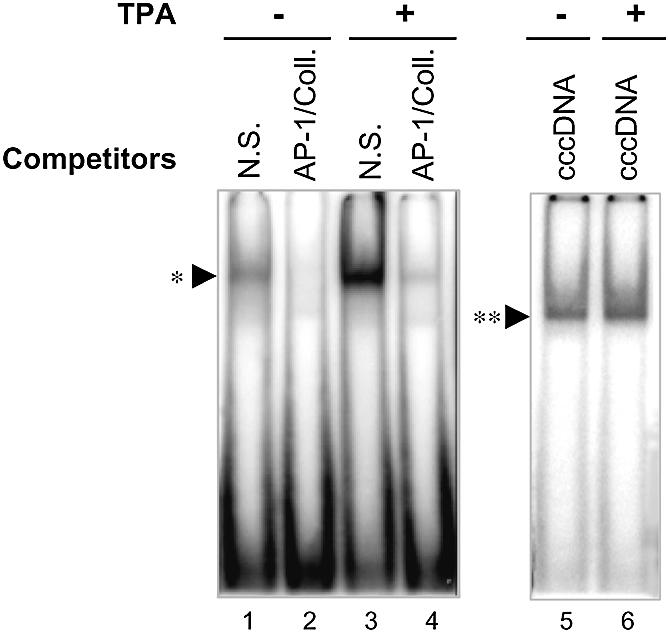
Modulation of AP-1 binding activity in U937 cells depending on TPA treatment. Left, extracts from untreated or TPA-induced U937cells (as indicated) were assayed for AP-1 DNA-binding activity using the TRE site of the collagenase promoter (AP-1/Coll, lanes 1–4). Right, same cell extracts were assessed for an alternate binding capacity of Ku heterodimer on a 18mer linear dsDNA probe (lanes 5 and 6). Added competitors are mentioned in the figure as non-specific oligonucleotides (N.S.), AP-1/Coll. oligonucleotides (AP-1/Coll.) or covalently closed circular DNA (cccDNA). Conditions for band shift reaction were identical to those used in Figure 3. TPA-induced AP-1 complex and Ku–DNA complex are respectively identified by single and double asterisks.
Effect of AP-1 on the expression and regulation of hMSH2
The above results were in agreement with the proposition of a cascade of regulation where TPA-stimulated PKC would lead to an activation of AP-1 factors. We then intended to verify the next step of this cascade, i.e. the implication of AP-1 activation in the induction of hMSH2 expression. For this purpose, we used U937 cells that expressed a dominant negative form of c-Jun (dn-Jun) (22). The dominant negative mutant, lacking the transactivating domain of c-Jun, retains the ability to bind DNA, but is devoid of AP-1 activity. Extracts from dn-Jun cells, either TPA-treated or not, did not show any difference in the hMSH2 expression level (Fig. 5A), in comparison to the augmentation of the hMSH2 content detected in wild type U937 cells following the same TPA treatment (Fig. 1A). The presence of a non-functional AP-1 transcription factor in U937 cells abolished the effect of overexpression of hMSH2 induced by TPA.
Figure 5.

Deficit of TPA-induction of the hMSH2 expression associated to the loss of c-Jun transactivation activity. U937 cells carrying a dominant negative form of c-Jun (dnJun) were cultured in the presence (+) or absence (–) of 50 nM of TPA. (A) Cell extracts were analysed for hMSH2 and, as a control, PCNA protein expression by western blot analysis. (B) RT–PCR experiments were completed on total RNAs extrated from untreated or treated U937 cells, carrying the dn-Jun plasmid (dnJun) or the vector alone (U937), with procedures similar to those described in Figure 2. Left, co-amplification of HB2 (HB2) and procaryotic control (control) mRNAs. Right, co-amplification of hMSH2 (hMSH2) and procaryotic control mRNAs.
Beyond that, we checked whether the presence of dominant negative c-Jun would directly affect the transcriptional regulation of hMSH2. Total RNA extracted from TPA-treated dn-Jun cells was analysed in RT–PCR experiments. Whereas the levels of control HB2 mRNA were identical, the number of specific hMSH2 transcripts was strictly below that observed in TPA-treated U937 WT cells (Fig. 5B), and it correlated better with the level of hMSH2 mRNAs from non-treated U937 cells. These data assign to c-Jun a functional role in the regulation of expression of hMSH2 through its activation by TPA.
Functional role of the hMSH2 TRE sequences in vivo
Next, we investigated in vivo the effects of the AP-1 binding sites on the hMSH2 promoter activity. For this purpose, we transiently transfected TPA-treated or untreated U937 cells with luciferase reporter gene whose expression was dependent on the hMSH2 promoter. The constructs contained 1.88 kb of the 5′ upstream proximal region of the hMSH2 gene (20), either wild type or specifically mutated in the two (–612 and –291) TRE sites (Fig. 6A). The base substitutions introduced were the same as those used in band shift assays (Fig. 3C). When comparing the wild type promoter (wt) and the mutated form (mut), the mutant promoter showed a 2-fold reduction in the luciferase activity (Fig. 6B). The same observation was made with the two independently single mutants in the –612 or –291 boxes (data not shown). These results validated an effect of mutations in the AP-1 sequences on the transactivation of the hMSH2 gene. For further experiments, we generated a deletion of the wild type and mutated promoter to –950 bp (Δwt and Δmut, Fig. 6A). This deletion produced a higher decrease (3.4-fold) in the ratio of luciferase activity than the mutation previously examined (compare mut and Δmut in Fig. 6B). Similarly, a 2-fold reduction of luciferase activity was observed in the presence of curcumin, an inhibitor of c-Jun synthesis (33), while a higher reduction was detected in presence of the dominant negative c-Jun (Fig. 6B). The effect obtained with the dominant negative c-Jun mutant (6-fold reduction) was more pronounced than that with the mutated AP-1 sites. This could suggest that mutations in the binding sites did not completely abolish the binding capacity of AP-1 in vivo, in spite of the absence of detectable binding on these mutated sequences in vitro (Fig. 3C). Finally the co-transfection experiments established that transactivation of hMSH2 promoter is likely to require the intervention of AP-1.
Figure 6.

TPA-enhancement of the hMSH2 promoter is dependent on functional AP-1 binding sites. U937 cells were transiently cotransfected with derivatives of luciferase reporter vectors according to the procedure outlined in Materials and Methods, and afterwards treated or not by TPA (100 nM for 6 h), as indicated. (A) A schematic structure of the constructs in pGL3 plasmid represents the luciferase gene (open box) fused to various forms of the hMSH2 promoter sequence (bold line). A 1.88 kb fragment of the hMSH2 promoter region carrying the AP-1 binding sites, either wild type (wt) or mutated (mut), was inserted upstream of the luciferase gene. Constructs ‘Δwt’ and ‘Δmut’ correspond to the 5′ truncated sequence of the wild type and mutated promoter region of hMSH2 due to a 930 bp deletion. (B) TPA-stimulated U937 cells were transfected with either wild type or mut hMSH2 promoter, or with the deleted forms of the plasmids. ‘Wt:cur’ means that U937 cells transfected with the wild type hMSH2 promoter were treated with curcumin (50 µM) for 1 h prior to addition of TPA. For ‘dn-Jun’, transfections with wild type hMSH2 promoter were performed on TPA-treated U937 cells containing the dominant negative mutant of c-Jun. Results are shown as the relative luciferase activity reported to that of TPA-treated U937 transfected with the wild type hMSH2 promoter, except for transfections with the Δmut deleted plasmid where the ratio is normalised to transfections with the Δwt plasmid. Data are the means of three independent experiments.
DISCUSSION
Various lines of evidence indicate that DNA repair activities could be regulated at the transcriptional and/or post-transcriptional level leading to phenotypes such as cell resistance or sensitivity to drugs. Among internal or external controls, the activation of PKC has been reported as one of them (34). Our recent results on MMR activity in myeloid cell lines provided an additional observation on the up-regulation of hMSH2 protein expression after PKC activation by TPA (21). However, the increased protein expression could lie on various non-exclusive mechanisms. We present here evidence that phorbol-ester stimulation induces a transcriptional up-regulation of the hMSH2 gene. By combinations of in vitro and in vivo experiments, we correlated the TPA transactivation of the hMSH2 promoter both to a functional role of the AP-1 binding sites present in the promoter sequence and to the activity of the c-Jun protein.
Following a 24 h-TPA treatment, we found a degree of induction of hMSH2 transcripts that correlated with the 3- to 5-fold increase in protein levels previously detected (21). Induction of mRNA seemed to precede the augmentation of protein level. RT–PCR analysis revealed a similar extent of mRNA content for 6 and 24 h of TPA stimulation (data not shown), when the kinetic of protein increase is still linear between these two times (Fig. 1). However, we do not exclude that a post-transcriptional regulation of hMSH2 can superimpose to the transcriptional positive regulation. In this view, PKC phosphorylation of the hMutSα complex has been recently described (13 and H. Hernandez-Pigeon, unpublished observations) leading to an increased protein activity (13) and/or stability (Hernandez, unpublished observations). An example of dual check of protein expression, i.e. pre- and post-transcriptional control, driven by the PKC, has already been described for another repair gene encoding the alkyl guanine DNA methyl-transferase (MGMT). The MGMT gene transcription is up regulated by PKC depending on AP-1 sequence element (34). However, in this case, in vivo or in vitro phosphorylation of the protein resulted in a reduced MGMT activity (35). The significance of the apparent contradictory intervention of the PKC in the regulation of expression/function of the MGMT is still unclear but could be concerned by the particular mechanism of the MGMT reaction, which is stoichiometric and irreversible.
We produce here two lines of evidence clearly establishing a role of AP-1 in the transactivation of hMSH2 gene. First, we show that the two AP-1 sequences in the promoter region are functional. One (–612) matches exactly the consensus sequence, while the other is slightly degenerated (–291). Both related oligonucleotides were able to bind the AP-1 factor (c-Jun) in band shift experiments. Most importantly, mutations in these sites significantly prevented in vivo the induction of promoter activity associated with phorbol-ester stimulation. Second, we present a set of evidence implicating the participation of c-Jun in the protein complexes formed on the hMSH2 AP-1 sequences. Up regulation of hMSH2 gene seems to depend on a major part on the presence of c-Jun, as demonstrated by experiments carried out using either a dominant negative mutant or specific inhibitor (Figs 5 and 6).
We already had an indication that the AP-1 binding sites present in the hMSH2 promoter were potentially functional, even if under specific conditions, since these sites were involved in the P53 regulation of hMSH2 associated with UV exposure (19). The synergism between P53 and c-Jun in response to UV irradiation can be explained on the basis of the presence of binding sites for both P53 and AP-1 in the hMSH2 promoter sequence. However, the type of positive up regulation that we describe here for the hMSH2 transcription is clearly independent of P53, since the U937 cell line used in the study is P53 negative (36 and data not shown). In that case, the P53 independence could be related to the nature of the regulation stimulus, i.e. TPA instead of a DNA damaging agent. Nevertheless, we cannot exclude that the presence of a functional P53 in the U937 cells would even increase the transactivation of hMSH2.
Additional transcriptional regulatory elements could be involved in the TPA-induced mRNA increase. In transient expression assays, when deleting the 5′ distal sequence of the hMSH2 promoter, we could amplify the difference in relative luciferase activity between wild type and AP-1 mutated promoter (Fig. 6), despite the fact that a similar deletion has already been described as producing no change in the basal promoter activity (20). This result suggests that the region corresponding to the deletion could specifically perturb the AP-1-dependent transcriptional activation of hMSH2 gene. The hMSH2 promoter displays characteristic features of housekeeping genes (31), and, in addition to AP-1 binding sites, SP1 (stimulating protein 1), and degenerated NF-κB transcriptional activator sequences are also present in the promoter region. However, it should be noted that data regarding the expression vector carrying a deleted promoter do not constitute a proof for the intervention of other regulatory factors, but could reflect the artificial nature of this kind of experiment since structure of the promoter region on a DNA plasmid cannot be strictly compared to the endogenous promoter on the chromosome. More accurate analysis of the functionality of the hMSH2 promoter should clarify this point.
The transcriptional regulation of hMSH2 described here may represent a case of regulation fitting both a particular cell line context and the action of a defined inducing agent. Our study concerned leukaemia cells in which the TPA stimulation of PKC is known to play a critical role in signal transduction. Exposure to phorbol-ester is well known to induce the differentiation of immature cells into more mature monocytes/macrophages. However, the conditions of TPA-treatment used in our study do not allow U937 cells to reach the final stage of differentiation as attested by the absence of characteristic features, i.e. cell adherence and monocytic morphology (data not shown). From this point of view, significance of the hMSH2 TPA-induced activation could be related to the shift of transcription associated with the initiation of differentiation, given that hMSH2 would be a candidate for participating in the transcription coupled repair pathway (37). Alternatively, since the hMSH2 protein has been implicated in the repair of oxidative damage (38,39), the AP-1 induction of the hMSH2 gene could then represent a preventive response to the higher ambient O2 tension to which cells can be exposed from the differentiation pathway.
In summary, our results demonstrate the existence of an AP-1 positive control of the hMSH2 gene and open up the possibility of a regulation operating on other contexts. Interestingly, AP-1 has been suggested to play a critical role in the cellular response to genotoxic agents. This role includes the regulation of genetic programs associated with a process fundamental for cellular protection and survival, such as DNA repair. Indeed AP-1 target genes exist among known repair genes, ERCC1 and MGMT (34,40). And for both of them, AP-1 induced transcription arises in response to genotoxic stress mediated by cisplatin (40), or alkylating agents (34). Given this background, we have undertaken studies aimed at determining potential extracellular signals, including genotoxic agents, implicated in the modulation of hMSH2 gene expression, and at examining whether other MMR genes are AP-1 responsive genes.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr K. A. Strait for providing the hMSH2 luciferase reporter plasmid, Dr K. Nakaya for the gift of the dn-Jun cells, and Christine Delteil for technical assistance in the transfection experiments. Giuseppe Villani is gratefully acknowledged for critical reading of the manuscript. This work was supported by grants from Fondation de France.
REFERENCES
- 1.Kolodner R. (1996) Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev., 10, 1433–1442. [DOI] [PubMed] [Google Scholar]
- 2.Modrich P. and Lahue,R. (1996) Mismatch repair in replication fidelity, genetic recombination and cancer biology. Annu. Rev. Biochem., 65, 101–133. [DOI] [PubMed] [Google Scholar]
- 3.Jiricny J. (1998) Replication errors: cha(lle)nging the genome. EMBO J., 17, 6427–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kolodner R.D. and Marsischky,G.T. (1999) Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev., 9, 89–96. [DOI] [PubMed] [Google Scholar]
- 5.Jiricny J. and Nystrom-Lahti,M. (2000) Mismatch repair defects in cancer. Curr. Opin. Genet. Dev., 10, 157–161. [DOI] [PubMed] [Google Scholar]
- 6.de la Chapelle A. and Peltomaki,P. (1998) The genetics of hereditary common cancers. Curr. Opin. Genet. Dev., 8, 298–303. [DOI] [PubMed] [Google Scholar]
- 7.Kinzler K.W. and Vogelstein,B. (1996) Lessons from hereditary colorectal cancer. Cell, 87, 159–170. [DOI] [PubMed] [Google Scholar]
- 8.Jacob S. and Praz,F. (2002) DNA mismatch repair defects: role in colorectal carcinogenesis. Biochimie, 84, 27–47. [DOI] [PubMed] [Google Scholar]
- 9.Karran P. and Bignami,M. (1996) Drug-related killings: a case of mistaken identity. Chem. Biol., 3, 875–879. [DOI] [PubMed] [Google Scholar]
- 10.Jacob S., Aguado,M., Fallik,D. and Praz,F. (2001) The role of the DNA mismatch repair system in the cytotoxicity of the topoisomerase inhibitors camptothecin and etoposide to human colorectal cancer cells. Cancer Res., 61, 6555–6562. [PubMed] [Google Scholar]
- 11.Papadopoulos N. and Lindblom,A. (1997) Molecular basis of HNPCC: mutations of MMR genes. Hum. Mutat., 10, 89–99. [DOI] [PubMed] [Google Scholar]
- 12.Christmann M. and Kaina,B. (2000) Nuclear translocation of mismatch repair proteins MSH2 and MSH6 as a response of cells to alkylating agents. J. Biol. Chem., 275, 36256–36262. [DOI] [PubMed] [Google Scholar]
- 13.Christmann M., Tomicic,M.T. and Kaina,B. (2002) Phosphorylation of mismatch repair proteins MSH2 and MSH6 affecting MutSalpha mismatch-binding activity. Nucleic Acids Res., 30, 1959–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marra G., Iaccarino,I., Lettieri,T., Roscilli,G., Delmastro,P. and Jiricny,J. (1998) Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc. Natl Acad. Sci. USA, 95, 8568–8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drummond J.T., Genschel,J., Wolf,E. and Modrich,P. (1997) DHFR/MSH3 amplification in methotrexate-resistant cells alters the hMutSalpha/hMutSbeta ratio and reduces the efficiency of base-base mismatch repair. Proc. Natl Acad. Sci. USA, 94, 10144–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kane M.F., Loda,M., Gaida,G.M., Lipman,J., Mishra,R., Goldman,H., Jessup,J.M. and Kolodner,R. (1997) Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res., 57, 808–811. [PubMed] [Google Scholar]
- 17.Bearzatto A., Szadkowski,M., Macpherson,P., Jiricny,J. and Karran,P. (2000) Epigenetic regulation of the MGMT and hMSH6 DNA repair genes in cells resistant to methylating agents. Cancer Res., 60, 3262–3270. [PubMed] [Google Scholar]
- 18.Quaresima B., Faniello,M.C., Baudi,F., Cuda,G., Grandinetti,C., Tassone,P., Costanzo,F. and Venuta,S. (2001) Transcriptional regulation of the mismatch repair gene hMLH1. Gene, 275, 261–265. [DOI] [PubMed] [Google Scholar]
- 19.Scherer S.J., Maier,S.M., Seifert,M., Hanselmann,R.G., Zang,K.D., Muller-Hermelink,H.K., Angel,P., Welter,C. and Schartl,M. (2000) p53 and c-Jun functionally synergize in the regulation of the DNA repair gene hMSH2 in response to UV. J. Biol. Chem., 275, 37469–37473. [DOI] [PubMed] [Google Scholar]
- 20.Warnick C.T., Dabbas,B., Ford,C.D. and Strait,K.A. (2001) Identification of a p53 response element in the promoter region of the hMSH2 gene required for expression in A2780 ovarian cancer cells. J. Biol. Chem., 276, 27363–27370. [DOI] [PubMed] [Google Scholar]
- 21.Humbert O., Hermine,T., Hernandez,H., Bouget,T., Selves,J., Laurent,G., Salles,B. and Lautier,D. (2002) Implication of protein kinase C in the regulation of DNA mismatch repair protein expression and function. J. Biol. Chem., 277, 18061–18068. [DOI] [PubMed] [Google Scholar]
- 22.Watabe M., Ito,K., Masuda,Y., Nakajo,S. and Nakaya,K. (1998) Activation of AP-1 is required for bufalin-induced apoptosis in human leukemia U937 cells. Oncogene, 16, 779–787. [DOI] [PubMed] [Google Scholar]
- 23.Andrews N.C. and Faller,D.V. (1991) A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res., 19, 2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephenson C. and Karran,P. (1989) Selective binding to DNA base pair mismatches by proteins from human cells. J. Biol. Chem., 264, 21177–21182. [PubMed] [Google Scholar]
- 25.Macpherson P., Humbert,O. and Karran,P. (1998) Frameshift mismatch recognition by the human MutS alpha complex. Mutat. Res., 408, 55–66. [DOI] [PubMed] [Google Scholar]
- 26.Mansat V., Laurent,G., Levade,T., Bettaieb,A. and Jaffrezou,J.P. (1997) The protein kinase C activators phorbol esters and phosphatidylserine inhibit neutral sphingomyelinase activation, ceramide generation and apoptosis triggered by daunorubicin. Cancer Res., 57, 5300–5304. [PubMed] [Google Scholar]
- 27.Karin M. (1995) The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem., 270, 16483–16486. [DOI] [PubMed] [Google Scholar]
- 28.Efimova T., LaCelle,P., Welter,J.F. and Eckert,R.L. (1998) Regulation of human involucrin promoter activity by a protein kinase C, Ras, MEKK1, MEK3, p38/RK, AP1 signal transduction pathway. J. Biol. Chem., 273, 24387–24395. [DOI] [PubMed] [Google Scholar]
- 29.Trejo J., Massamiri,T., Deng,T., Dewji,N.N., Bayney,R.M. and Brown,J.H. (1994) A direct role for protein kinase C and the transcription factor Jun/AP-1 in the regulation of the Alzheimer’s beta-amyloid precursor protein gene. J. Biol. Chem., 269, 21682–21690. [PubMed] [Google Scholar]
- 30.Genot E.M., Parker,P.J. and Cantrell,D.A. (1995) Analysis of the role of protein kinase C-alpha, -epsilon and -zeta in T cell activation. J. Biol. Chem., 270, 9833–9839. [DOI] [PubMed] [Google Scholar]
- 31.Iwahashi Y., Ito,E., Yanagisawa,Y., Akiyama,Y., Yuasa,Y., Onodera,T. and Maruyama,K. (1998) Promoter analysis of the human mismatch repair gene hMSH2. Gene, 213, 141–147. [DOI] [PubMed] [Google Scholar]
- 32.Scherer S.J., Seib,T., Seitz,G., Dooley,S. and Welter,C. (1996) Isolation and characterization of the human mismatch repair gene hMSH2 promoter region. Hum. Genet., 97, 114–116. [DOI] [PubMed] [Google Scholar]
- 33.Huang T.S., Lee,S.C. and Lin,J.K. (1991) Suppression of c-Jun/AP-1 activation by an inhibitor of tumor promotion in mouse fibroblast cells. Proc. Natl Acad. Sci. USA, 88, 5292–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boldogh I., Ramana,C.V., Chen,Z., Biswas,T., Hazra,T.K., Grosch,S., Grombacher,T., Mitra,S. and Kaina,B. (1998) Regulation of expression of the DNA repair gene O6-methylguanine-DNA methyltransferase via protein kinase C-mediated signaling. Cancer Res., 58, 3950–3956. [PubMed] [Google Scholar]
- 35.Srivenugopal K.S., Mullapudi,S.R., Shou,J., Hazra,T.K. and Ali-Osman,F. (2000) Protein phosphorylation is a regulatory mechanism for O6-alkylguanine-DNA alkyltransferase in human brain tumor cells. Cancer Res., 60, 282–287. [PubMed] [Google Scholar]
- 36.Clave E., Carosella,E.D., Gluckman,E., Dubray,B. and Socie,G. (1996) Ionizing radiation effects on the KG1a primitive hematopoietic cell line. Int. J. Radiat. Oncol. Biol. Phys., 35, 709–719. [DOI] [PubMed] [Google Scholar]
- 37.Mellon I., Rajpal,D.K., Koi,M., Boland,C.R. and Champe,G.N. (1996) Transcription-coupled repair deficiency and mutations in human mismatch repair genes. Science, 272, 557–560. [DOI] [PubMed] [Google Scholar]
- 38.Colussi C., Parlanti,E., Degan,P., Aquilina,G., Barnes,D., Macpherson,P., Karran,P., Crescenzi,M., Dogliotti,E. and Bignami,M. (2002) The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr. Biol., 12, 912–918. [DOI] [PubMed] [Google Scholar]
- 39.DeWeese T.L., Shipman,J.M., Larrier,N.A., Buckley,N.M., Kidd,L.R., Groopman,J.D., Cutler,R.G., te Riele,H. and Nelson,W.G. (1998) Mouse embryonic stem cells carrying one or two defective Msh2 alleles respond abnormally to oxidative stress inflicted by low-level radiation. Proc. Natl Acad. Sci. USA, 95, 11915–11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Q., Gardner,K., Zhang,L., Tsang,B., Bostick-Bruton,F. and Reed,E. (1998) Cisplatin induction of ERCC-1 mRNA expression in A2780/CP70 human ovarian cancer cells. J. Biol. Chem., 273, 23419–23425. [DOI] [PubMed] [Google Scholar]