

Abstract
The physiological role of the mitotic checkpoint protein Bub1 is unknown. To study this role, we generated a series of mutant mice with a gradient of reduced Bub1 expression using wild-type, hypomorphic, and knockout alleles. Bub1 hypomorphic mice are viable, fertile, and overtly normal despite weakened mitotic checkpoint activity and high percentages of aneuploid cells. Bub1 haploinsufficient mice, which have a milder reduction in Bub1 protein than Bub1 hypomorphic mice, also exhibit reduced checkpoint activity and increased aneuploidy, but to a lesser extent. Although cells from Bub1 hypomorphic and haploinsufficient mice have similar rates of chromosome missegregation, cell death after an aberrant separation decreases dramatically with declining Bub1 levels. Importantly, Bub1 hypomorphic mice are highly susceptible to spontaneous tumors, whereas Bub1 haploinsufficient mice are not. These findings demonstrate that loss of Bub1 below a critical threshold drives spontaneous tumorigenesis and suggest that in addition to ensuring proper chromosome segregation, Bub1 is important for mediating cell death when chromosomes missegregate.
Introduction
Accurate segregation of replicated chromosomes during mitosis is essential for the maintenance of genomic integrity. To ensure faithful chromosome segregation, eukaryotic cells have developed a surveillance network called the mitotic checkpoint that delays anaphase onset until sister kinetochores of duplicated chromosomes are properly attached to microtubules emanating from opposite spindle poles (for reviews see Kops et al., 2004; Musacchio and Salmon, 2007). Early in mitosis, various mitotic checkpoint proteins, including Bub1, Bub3, BubR1, Mad1, Mad2, and Mps1, are recruited to unattached kinetochores. These kinetochore-associated checkpoint proteins promote the formation of diffusible Mad2, BubR1, Bub3, and Cdc20 protein complexes that inhibit the anaphase-promoting complex/cyclosome (APC/C), an E3 ubiquitin ligase that drives cells into anaphase by targeting securin and cyclin B for destruction by the 26S proteasome (for reviews see Kops et al., 2005; Peters, 2006; Musacchio and Salmon, 2007). After all chromosome pairs are properly attached to the spindle and aligned in the metaphase plate, mitotic checkpoint proteins dissociate from the APC/C, thus triggering the ubiquitin-mediated destruction of securin and cyclin B. Separase, a protease that is inhibited by securin binding and cyclin B/Cdk1-mediated phosphorylation, then cleaves the kleisin subunit Scc1 of cohesin, thereby allowing sister chromatid disjunction and anaphase onset (for reviews see Nasmyth and Haering, 2005; Peters, 2006).
The discovery of the mitotic checkpoint led to speculation that mutations in mitotic checkpoint genes might play a role in the development of aneuploidy in human cancers (Jallepalli and Lengauer, 2001; Draviam et al., 2004). Over recent years, mutant mitotic checkpoint genes have indeed been identified in various human cancers, although at relatively low frequency (Weaver and Cleveland, 2006; for reviews see Kops et al., 2005; Yuen et al., 2005). The Bub1 kinase is mutated in several cancer types, including colorectal, lung and thyroid cancer, and T cell leukemia (Cahill et al., 1998; Ohshima et al., 2000; Ru et al., 2002; Shichiri et al., 2002). In addition, Bub1 expression is frequently reduced in several human cancers, including colorectal, gastric, and esophageal cancers (Shigeishi et al., 2001; Shichiri et al., 2002; Doak et al., 2004).
Bub1 is a serine/threonine protein kinase that targets to unattached kinetochores at the onset of mitosis (Roberts et al., 1994; Taylor and McKeon, 1997; Yu and Tang, 2005). There it is thought to phosphorylate Cdc20, thereby preventing Cdc20 from activating the APC/C (Chung and Chen, 2003; Chen, 2004; Tang et al., 2004a). Additionally, Bub1 is required for Mad1–Mad2 localization to unattached kinetochores. These complexes function to prevent premature APC/C activation by changing the conformation of monomeric Mad2 such that it can efficiently bind to and inhibit the APC/C coactivator Cdc20 (Luo et al., 2002, 2004; Sironi et al., 2002). Besides Mad1 and Mad2, Bub1 also recruits BubR1, Bub3, centromere protein E (CENP-E), and CENP-F to unattached kinetochores (Sharp-Baker and Chen, 2001; Johnson et al., 2004). Several of these proteins are important for microtubule-kinetochore attachment, which may explain why Bub1-depleted cells have chromosome congression defects (Meraldi and Sorger, 2005). In addition, Bub1 contributes to the stability and inner centromere localization of Shugoshin (Sgo1), a protein that functions as an adaptor for phosphatase PP2A (Tang et al., 2004b, 2006; Kitajima et al., 2005). At the inner centromere, PP2A counteracts the Plk1-mediated release of cohesin until anaphase onset, thus preventing the premature separation of sister centromeres (Riedel et al., 2006). Bub1 also controls the stability and correct positioning of the chromosomal passenger complex to the inner centromeric region of sister chromosomes, a function that appears to be critical for the recruitment of Sgo1 to centromeres (Boyarchuk et al., 2007).
Although the molecular mechanisms of Bub1 action are beginning to emerge, the physiological role of Bub1 in higher eukaryotes is still unknown. The most definitive way to address this role would be to generate Bub1 knockout mice by homologous recombination in embryonic stem (ES) cells. However, previous gene knockout studies for Mad1, Mad2, BubR1, and Bub3 revealed that these mitotic checkpoint proteins are essential for cell proliferation, causing mice to die during the early stages of embryonic development (Dobles et al., 2000; Kalitsis et al., 2000; Babu et al., 2003; Baker et al., 2004; Wang et al., 2004; Iwanaga et al., 2007). Anticipating that Bub1-null mice would be embryonically lethal as well, we generated a series of mice in which the expression of Bub1 protein is reduced in a graded fashion from normal to zero. We find that Bub1-null mice are indeed embryonically lethal but that mice with very low levels of Bub1 protein are viable. Here, we show that Bub1 deficiency is associated with aneuploidy and spontaneous tumorigenesis in a dose-dependent fashion. Furthermore, we provide evidence for a novel role of Bub1 in eliminating cells that have undergone chromosome missegregation.
Results
Generation of mutant mice with graded reduction in Bub1 levels
By homologous recombination, we inserted a neomycin-resistance (Neo) gene flanked by a loxP site into intron 8 and a loxP site into intron 9 of the mouse Bub1 gene (Fig. 1 A). This created a hypomorphic allele (called Bub1N) because the Neo gene harbors a cryptic exon that is known to reduce the level of normally spliced messenger RNA (Jacks et al., 1994; Meyers et al., 1998; Baker et al., 2004). Correctly targeted ES clones were injected into blastocysts, and Bub1+/N offspring were obtained from the resulting chimeras (Fig. 1 C). Bub1+/− mice were established by crossing Bub1+/N males with transgenic females that express Cre recombinase in the germline (Fig. 1 C). Both Bub1+/N and Bub1+/− mice were healthy and indistinguishable from wild-type littermates. Subsequent intercrosses of Bub1+/− mice produced no Bub1−/− newborn mice. Further analysis revealed that Bub1−/− embryos died between days 4.5 and 6.5 of development (unpublished data), which is in agreement with other mitotic checkpoint gene knockout mice. Also, no Bub1N/N pups were born from intercrosses of Bub1+/N mice, implying that the level of wild-type Bub1 protein produced by these hypomorphic alleles was not sufficient for successful embryonic development.
Figure 1.

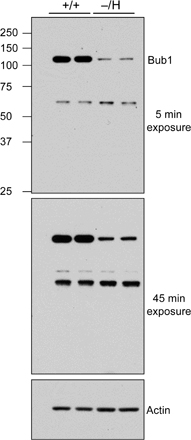
Generation of mice with graded reduction in Bub1 dosage. (A) Schematic representation of the primary Bub1 gene–targeting strategy. Part of the Bub1 locus (+), the first targeting vector with loxP sites (gray triangles), the Neo hypomorphic allele, the knockout allele generated by the expression of Cre recombinase (−), BamHI (B) restriction sites, and the Southern probe are indicated. (B) Schematic representation of the second Bub1 gene–targeting strategy. The second targeting vector, the Hyg hypomorphic allele, and the BamHI (B) and XhoI (X) restriction sites for Southern blotting are indicated. (C) Southern blot analysis of mice with the indicated Bub1 genotypes. The 21-kb, 9.8-kb, 8.4-kb, and 9.5-kb fragments represent the wild-type, Neo hypomorphic, knockout, and Hyg hypomorphic alleles, respectively. (D) Western blot analysis of MEFs isolated from mice carrying the indicated Bub1 alleles with a Bub1-specific antibody (actin was used as a loading control). The – and H alleles can produce truncated protein products of 267 amino acids and 318 amino acids, respectively. However, we were unable to detect these truncated products with our polyclonal antibody against Bub1(25–165) (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200706015/DC1), sugesting that the products are rapidly degraded and/or that their messengers are unstable. (E) Quantitation of the level of Bub1 reduction in Bub1−/H MEFs as compared with Bub1+/+ MEFs.
To bypass this problem, we created a Bub1 hypomorphic allele by the use of an alternative method. This method takes advantage of a hygromycin B phosphotransferase expression (Hyg) cassette that causes a high incidence of premature transcriptional termination when inserted into intronic sequences (van Deursen et al., 1994). We constructed a targeting vector to introduce this Hyg cassette into intron 9 of the endogenous Bub1 gene (Fig. 1 B). Properly targeted ES clones were used to produce Bub1+/H mice. Intercrosses of Bub1+/H mice yielded viable Bub1H/H offspring at the expected Mendelian frequency. Furthermore, interbreeding of Bub1+/H and Bub1+/− mice yielded viable Bub1−/H offspring with normal Mendelian frequency (Fig. 1 C). Like Bub1+/− mice, Bub1H/H and Bub1−/H mice exhibited no changes in development or appearance when compared with wild-type mice. We performed Western blotting to measure the level of wild-type Bub1 protein in mouse embryonic fibroblasts (MEFs) derived from Bub1+/+, Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H mice (Fig. 1, D and E). We assessed that Bub1 signals from Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H MEFs were ∼75%, 50%, 30%, and 20% of those from Bub1+/+ MEFs, respectively. Truncated forms of Bub1 encoded by the – and H alleles were undetectable even after overexposure of the Western blots (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200706015/DC1). Together, these results demonstrated that we had produced a series of mice with decreasing Bub1 protein dosage.
Mice with low amounts of Bub1 have a high percentage of aneuploid cells
To determine whether the reduced expression of Bub1 protein affects the accuracy of chromosome segregation, we collected splenocytes from Bub1+/+, Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H mice at 5 mo of age and prepared metaphase spreads for karyotype analyses. Chromosome counts showed that <1% of wild-type splenocytes were aneuploid (Table I). In contrast, splenocytes from Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H mice had a 6%, 16%, 35%, and 39% incidence of aneuploidy, respectively, revealing an inverse correlation between the level of Bub1 protein and the percentage of aneuploidy in this cell type. Moreover, the range of abnormal chromosome numbers broadened with the decreasing expression of Bub1 protein (Table I). We observed the premature separation of sister chromatids (PMSCS) in 14 and 15% of the mitotic figures from Bub1+/− and Bub1−/H splenocytes but only in 4% of the mitotic figures from Bub1H/H splenocytes (Table I). Thus, there seems to be no clear link between PMSCS and Bub1 dosage in splenocytes.
Table I.
Inverse correlation between Bub1 expression and aneuploidy in mouse splenocytes
| Mouse genotype |
Age (n) |
Mitotic figures inspected |
Aneuploid figures (SD) |
Karyotypes with indicated chromosome number |
Mitotic figures with PMSCS (SD) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 37 | 38 | 39 | 40 | 41 | 42 | 43 | 44 | |||||
| mo | % | % | ||||||||||
| Bub1+/+ | 5 (3) | 150 | 1 (1) | 1 | 149 | 0 (0) | ||||||
| Bub1+/H | 5 (5) | 250 | 6 (1) | 2 | 6 | 236 | 4 | 2 | 2 (1) | |||
| Bub1+/− | 5 (3) | 150 | 16 (2) | 1 | 4 | 6 | 126 | 7 | 6 | 14 (2) | ||
| Bub1H/H | 5 (3) | 150 | 35 (2) | 3 | 8 | 9 | 97 | 16 | 12 | 5 | 4 (1) | |
| Bub1−/H | 5 (3) | 150 | 39 (2) | 1 | 8 | 15 | 91 | 12 | 15 | 7 | 1 | 15 (2) |
Empty spaces mean that there were no karyotypes with the indicated chromosome number.
We further investigated the effect of Bub1 insufficiency on chromosome number stability by performing chromosome counts on metaphase spreads from Bub1+/+, Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H MEFs at passage 5. We found that the percentage of aneuploid metaphases was much higher in Bub1H/H and Bub1−/H MEFs than in Bub1+/− and Bub1+/H MEFs, which, in turn, had a higher percentage than Bub1+/+ MEFs (Table II). PMSCS was not increased in Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H MEFs compared with Bub1+/+ MEFs (Table II). These data confirm that a high percentage of cells with low levels of Bub1 become aneuploid without the apparent requirement of PMSCS.
Table II.
Gradual reduction in Bub1 causes progressively more aneuploidy in MEFs
| Mitotic MEF genotype (n) |
Mitotic figures inspected |
Aneuploid figures (SD) |
SD | Karyotypes with indicated chromosome number
|
Mitotic figures with PMSCS (SD) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 37 | 38 | 39 | 40 | 41 | 42 | 43 | 44 | 45 | 46 | 80 | |||||
| % | % | ||||||||||||||
| Bub1+/+ (3) | 150 | 7 (1) | 1 | 2 | 3 | 132 | 3 | 2 | 8 | 2 (1) | |||||
| Bub1+/H (3) | 150 | 11 (1) | 1 | 5 | 123 | 6 | 6 | 10 | 2 (1) | ||||||
| Bub1+/− (3) | 150 | 14 (0) | 0 | 3 | 8 | 120 | 10 | 9 | 3 (0) | ||||||
| Bub1H/H (3) | 150 | 35 (1) | 1 | 4 | 10 | 88 | 21 | 10 | 8 | 9 | 2 (1) | ||||
| Bub1−/H (3) | 150 | 36 (5) | 5 | 3 | 4 | 7 | 83 | 16 | 7 | 9 | 5 | 3 | 2 | 11 | 3 (0) |
Empty spaces mean that there were no karyotypes with the indicated chromosome number. Karyotyping was performed at passage 5.
Kinetochore-associated proteins require distinct Bub1 levels for proper localization
Many of Bub1's critical functions during mitosis occur at the kinetochore. Therefore, we tested whether the graded reduction of Bub1 expression corresponds to a graded reduction in Bub1 levels at kinetochores. Immunostaining of Bub1+/+, Bub1+/−, and Bub1−/H MEFs with affinity-purified Bub1-specific antibody showed that fluorescence signals at kinetochores progressively declined with decreasing cellular levels of Bub1 expression (Fig. 2 A). To examine how this graded reduction in kinetochore-bound Bub1 affected the localization of mitotic checkpoint proteins whose targeting to kinetochores is Bub1 dependent, we immunostained Bub1+/+, Bub1+/−, and Bub1−/H MEFs with antibodies against the mitotic checkpoint proteins Mad1, Mad2, BubR1, and CENP-E. In Bub1+/+ prometaphase cells, Mad1 staining was concentrated on kinetochores visualized by antikinetochore antibody (Fig. 2 B). However, kinetochore-associated Mad1 signals were much less abundant in the corresponding Bub1+/− and Bub1−/H cells. As Mad1 is required for the kinetochore localization of Mad2 (Chen et al., 1998, 1999), we anticipated that Mad2 staining patterns would also be reduced in Bub1+/− and Bub1−/H prometaphase cells. We tested this prediction, but despite numerous attempts, we were unsuccessful in obtaining kinetochore-associated Mad2 signals in Bub1+/+ MEFs with antibodies that are known to detect Mad2 at kinetochores of human prometaphase cells (see Materials and methods for details). Unlike Mad1, kinetochore-associated BubR1 and CENP-E signals were unaffected in Bub1+/− cells during prometaphase (Fig. 2, C and D). However, kinetochore signals for both of these proteins were dramatically reduced in prometaphase Bub1−/H cells. Western blot analysis showed that Mad1, BubR1, and CENP-E protein levels were similar in Bub1+/+ and Bub1−/H cells (Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200706015/DC1), excluding the possibility that the reduction in kinetochore localization of these proteins in Bub1−/H cells is caused by reduced protein stability.
Figure 2.

Proper targeting of Mad1 to kinetochores is highly sensitive to Bub1 down-regulation. MEFs with various levels of Bub1 expression were analyzed for the proper localization of proteins whose association with kinetochores or centromeres is known to be Bub1 dependent. (A) Fluorescent images of Bub1+/+, Bub1+/−, and Bub1−/H prophase cells stained for kinetochores, Bub1, and DNA showing that the gradual reduction of cellular Bub1 protein levels corresponds with a gradual decline in kinetochore-associated Bub1 protein. (B) Images of Bub1+/+, Bub1+/−, and Bub1−/H prometaphase cells stained for kinetochores, Mad1, and DNA demonstrating that kinetochore targeting of Mad1 is highly sensitive to Bub1 down-regulation. (C) Fluorescent images of prometaphase cells of the indicated genotypes stained for kinetochores, BubR1, and DNA showing that BubR1 localization to kinetochores is severely perturbed in Bub1−/H MEFs but not in Bub1+/− MEFs. (D) Fluorescent images of prometaphase cells of the indicated genotypes stained for kinetochores, CENP-E, and DNA demonstrating that CENP-E localization to kinetochores is impaired in Bub1−/H MEFs but not in Bub1+/− MEFs. (E) Fluorescent images of Bub1+/+ and Bub1−/H prometaphase cells stained for kinetochores and Sgo1 showing that considerably fewer Sgo1-positive centromeres are present in Bub1−/H MEFs. Bar, 10 μM.
Next, we tested how the graded reduction in kinetochore-bound Bub1 levels affected the subcellular localization of Sgo1 and Aurora B, both of which have been reported to require Bub1 for their proper localization to the centromeres (Tang et al., 2004b, 2006; Kitajima et al., 2005; Boyarchuk et al., 2007). Fewer Sgo1-positive centromeres were observed in Bub1−/H prometaphases than in Bub1+/+ prometaphases (Fig. 2 E). In contrast, no such decrease was observed in Bub1+/− prometaphase cells (unpublished data). Immunostainings for Aurora B revealed that the localization of this protein was normal in both Bub1+/− and Bub1−/H prometaphase cells (Fig. S2 B). Thus, whereas most proteins that require Bub1 for proper localization to kinetochores/centromeres are mislocalized in Bub1−/H MEFs, only Mad1 is mislocalized in Bub1+/− cells.
Bub1 insufficient cells have a weakened mitotic checkpoint
To analyze the activity of the mitotic checkpoint in MEFs with a graded reduction in Bub1 expression, we performed a nocodazole challenge assay (Jeganathan et al., 2005; Baker et al., 2006). In this assay, MEFs were first transduced with a retrovirus encoding a YFP-tagged H2B fusion protein to allow the visualization of chromosomes by fluorescence microscopy (Fig. 3 A). MEFs were then challenged with nocodazole, and 20–30 cells undergoing nuclear envelope breakdown (NEBD) were marked and monitored at 15-min intervals to determine when their chromatin decondenses. The duration of arrest in mitosis, which is defined as the interval between NEBD (onset of mitosis) and chromatin decondensation (exit from mitosis without cytokinesis), was calculated and plotted. The time at which 50% of the cells have exited mitosis was used for comparison. Nocodazole- challenged Bub1+/+ MEFs typically remained arrested in prometaphase for 7.2 h (Fig. 3 B). Bub1+/H and Bub1+/− MEFs were impaired in their ability to maintain this arrest, with 50% of the cells exiting around 5.4 h. However, Bub1H/H and Bub1−/H MEFs exhibited a more profound defect, with 50% of the cells exiting mitosis at 3.8 h and 3.5 h, respectively. Thus, the mitotic checkpoint appears to be considerably weaker in Bub1H/H and Bub1−/H MEFs than in Bub1+/H and Bub1+/− MEFs.
Figure 3.

Mitotic checkpoint activity analysis. (A) Schematic of the experimental design (for details see the first two paragraphs of Results). (B) Analysis of mitotic checkpoint activity of MEFs of the indicated genotypes (n = 3 for each genotype). Error bars represent the SEM. Dotted arrows mark the times at which 50% of cells had exited from mitosis. Asterisks indicate a statistical difference from Bub1+/+, Bub1+/H, and Bub1+/− MEFs using the logrank test (*, P < 0.001).
Bub1 insufficiency causes various chromosome segregation defects
Next, MEFs with graded reductions in Bub1 expression were screened for chromosome segregation defects. In essence, we followed YFP-H2B–positive MEFs through an unchallenged mitosis by live cell imaging and determined the fraction of mitotic cells with chromosome segregation abnormalities. Two known defects underlying chromosome missegregation, congression failure and chromosome lagging (Fig. 4, A and B), were observed at higher rates in Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H MEFs than in Bub1+/+ MEFs (Table III). The combined incidence of the aforementioned defects was remarkably similar in Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H MEFs (Table III). Furthermore, anaphases with centrophilic chromosomes that segregate faster than the other chromosomes (Fig. 4 C) were observed at an approximately two- to fivefold higher frequency in Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H MEFs than in Bub1+/+ MEFs (Table III). Whether this type of abnormality leads to chromosome missegregation is unclear, but even with the inclusion of this defect, the overall incidence of chromosome segregation abnormalities remains very similar in Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H MEFs (Table III). Irrespective of Bub1 genotype, most cells with abnormal chromosome segregation events involved a single chromosome (or a duplicated chromosome). Occasionally, two or three chromosomes were implicated (Table IV and Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200706015/DC1). Thus, the aforementioned analyses suggest that the accuracy of chromosome segregation is highly dependent on a full complement of Bub1 protein and that both small and large reductions in Bub1 cause chromosome missegregation at comparable rates.
Figure 4.

Bub1 insufficiency causes various chromosome segregation errors. (A) Examples of metaphases with misaligned chromosomes (arrows). (B) Anaphases with lagging chromosomes (arrows). (C) Anaphase with a centrophilic chromosome (arrow).
Table III.
Analysis of chromosome segregation abnormalities in Bub1-insufficient MEFs
| MEF genotype (n) | Mitotic cells inspected |
Metaphases with misaligned chromosomes |
Anaphases with lagging chromosomes |
Anaphases with centrophilic chromosomes |
Cells with segregation defectsa |
|---|---|---|---|---|---|
| % | % | % | % | ||
| Bub1+/+ (5) | 106 | 0.9 | 3.8 | 0.9 | 4.7 (5.6) |
| Bub1+/H (4) | 91 | 3.3 | 6.6 | 3.3 | 9.9 (13.2) |
| Bub1+/− (6) | 142 | 7.7 | 6.3 | 2.1 | 12 (14.1) |
| Bub1H/H (4) | 122 | 6.5 | 4.9 | 4.8 | 11.6 (16.4) |
| Bub1−/H (5) | 168 | 7.1 | 6.6 | 2.4 | 13.1 (15.5) |
All cells scored as metaphases with misaligned chromosomes displayed congression failure at anaphase onset.
Percentage of cells with misaligned and/or lagging chromosomes. The percentage of cells with chromosome segregation abnormalities is presented in parentheses.
Table IV.
Most chromosome missegregation events involve a single chromosome irrespective of the level of Bub1 expression
| Mitotic figures inspected (n) |
Number of cells with segregation defectsa |
Number of cells with one abnormally segregated chromosome |
Number of cells with two or three abnormally segregated chromosomesb |
|---|---|---|---|
| Bub1+/+ (106) | 6 | 6 (100%) | 0 (0%) |
| Bub1+/H (91) | 12 | 11 (92%) | 1 (8%) |
| Bub1+/− (142) | 20 | 15 (75%) | 5 (25%) |
| Bub1H/H (122) | 20 | 17 (85%) | 3 (15%) |
| Bub1−/H (168) | 26 | 21 (81%) | 5 (19%) |
The chromosomes involved were either single chromosomes or duplicated chromosomes.
None of the cells had more than three abnormally segregated chromosomes (see Table S1).
Reduced cell death after chromosome missegregation as Bub1 levels decline
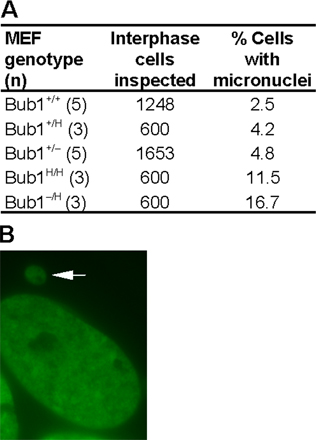
Initially, we were surprised that chromosome missegregation rates were similar in Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H MEFs because the percentage of aneuploid cells was much higher in Bub1H/H and Bub1−/H cultures than in Bub1+/H and Bub1+/− cultures (Table II). One explanation could be that cell survival after chromosome missegregation increases with decreasing Bub1 levels. To explore this possibility, we infected Bub1+/+, Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H MEFs with the YFP-H2B virus and monitored the fate of cells undergoing chromosome missegregation for up to 12 h by live cell imaging. Typically, 95% of Bub1+/+ MEFs died within several hours after chromosome missegregation (Table V and Videos 5 and 6, available at http://www.jcb.org/cgi/content/full/jcb.200706015/DC1). This percentage declined progressively and sharply as Bub1 expression decreased, with only 32% of Bub1−/H MEFs dying after a missegregation event (Table V, Fig. 5 A, and Videos 1 and 2). Cells dying after chromosome missegregation consistently showed nuclear fragmentation and/or cytoplasmic blebbing (Fig. 5, B and C; and Videos 3, 4, 7, and 8). Cells with accurate segregation rarely died during the 12-h observation period, irrespective of Bub1 genotype (Table V and Videos 9 and 10). From this, we conclude that although the rates of chromosome missegregation are comparable at various levels of Bub1 reduction, aneuploid cells accumulate to higher steady-state levels in cultures with low amounts of the protein because cells in these cultures are more likely to survive after chromosome missegregation. Consistent with this interpretation, we found that micronuclei, which we observed by live cell imaging to result from misaligned, centrophilic, or lagging chromosomes, accumulated steadily with decreasing Bub1 levels (Fig. S3).
Table V.
Cell death after chromosome missegregation decreases as Bub1 expression declines
| MEF genotype (n) | Cells with missegregation monitoreda |
Apoptosis incidence |
Apoptosis (1 cell) |
Apoptosis (2 cells) |
Cells with normal mitosis monitored |
Apoptosis incidence |
|---|---|---|---|---|---|---|
| % | % | % | % | |||
| Bub1+/+ (3) | 16 | 94 | 0 | 100 | 38 | 3 |
| Bub1+/H (3) | 28 | 68 | 26 | 74 | 17 | 6 |
| Bub1+/− (3) | 23 | 65 | 9 | 91 | 7 | 0 |
| Bub1H/H (3) | 26 | 42 | 12 | 88 | 16 | 0 |
| Bub1−/H (3) | 31 | 32 | 14 | 86 | 14 | 0 |
Cells analyzed had either one or a few missegregated chromosomes.
Figure 5.

Analysis of cell fate after chromosome missegregation by live cell imaging. MEF cultures expressing YFP-tagged H2B were screened for cells entering mitosis by live cell imaging. Cells undergoing chromosome missegregation were monitored for up to 12 h after the missegregation occurred to determine cell fate. (A) Time-lapse sequence of a Bub1−/H cell undergoing chromosome missegregation and whose daughter cells survived for at least 12 h. t = 0 is the time at which missegregation occurred. (B) Time-lapse sequence of a Bub1+/− cell undergoing chromosome missegregation whose daughter cells died ∼7 h after the defect occurred. (A and B) Arrows mark the locations of the missegregated chromosomes. (C) High resolution images of two Bub1+/− daughter cells undergoing cell death ∼5 h after they underwent chromosome missegregation.
To explore whether Bub1 plays a more general role in cell death signaling, we measured cell survival to various kinds of DNA-damaging agents. MEFs with graded reduction in Bub1 protein levels were exposed to increasing concentrations of doxorubicin, mitomycin C, or paraquat for 48 h. Cell survival was then determined by using the MTS assay. Cell survival in these agents was similar for Bub1+/+, Bub1+/H, Bub1+/−, Bub1H/H, and Bub1−/H MEFs (Fig. S4, A–C; available at http://www.jcb.org/cgi/content/full/jcb.200706015/DC1). In addition, decreased Bub1 expression also did not increase survival to prolonged exposure to nocodazole, a spindle poison that induces tetraploidization by driving prometaphase cells into G1 without chromosome segregation (Fig. S4 D). These experiments suggest a rather specific role for Bub1 in mediating cell death after the missegregation of one or a few chromosomes.
Spontaneous tumorigenesis is increased in Bub1 hypomorphic mice
To determine the long-term consequences of Bub1 down- regulation, we created and monitored cohorts of Bub1+/+ (n = 160), Bub1+/− (n = 142), Bub1H/H (n = 137), and Bub1−/H (n = 238) mice on a mixed 129 × C57BL/6 background. Earlier, we reported that BubR1 hypomorphic mice have a short lifespan, are infertile, and develop various early aging–associated phenotypes (Baker et al., 2004). We note that no such phenotypes were observed in any of our Bub1 mutant mice (unpublished data). However, we found that Bub1−/H and Bub1H/H mice were significantly more prone to spontaneous tumors than Bub1+/+ mice (Fig. 6, A and B). Bub1−/H mice had a significantly shorter median tumor-free survival (530 d) than Bub1H/H mice (676 d), which, in turn, had a significantly shorter median tumor-free survival than Bub1+/+ mice (772 d; Fig. 6 B). Moreover, Bub1−/H and Bub1H/H mice developed a different spectrum of tumors than did Bub1+/+ mice (Fig. 6, C–F′). Bub1−/H mice developed significantly more sarcomas, lymphomas, and lung tumors. Bub1H/H mice were also prone to develop sarcomas but not lymphomas and lung tumors. Bub1H/H mice were highly susceptible to hepatocellular carcinomas, a tumor type that was not significantly increased in Bub1−/H mice. In contrast to Bub1−/H and Bub1H/H mice, Bub1+/− mice showed a trend toward decreased tumor formation, particularly in liver and lung tissue (Fig. 6, B and C). Collectively, these data establish a causal relationship between the down-regulation of Bub1 expression and cancer development and suggest that there is a threshold level of Bub1 below which the incidence of neoplastic transformation progressively increases. Our data further imply that Bub1 reductions above the threshold may slightly inhibit tumor formation in particular tissues.
Figure 6.

Bub1−/H and Bub1H/H mice are prone to spontaneous tumors. (A) Tumor-free survival curves of Bub1+/+, Bub1+/−, Bub1H/H, and Bub1−/H mice. The asterisks mark curves that are significantly different from wild-type using a logrank test (P < 0.0001). We note that the tumor-free survival of a small cohort of Bub1+/H mice (n = 10) was similar to that of Bub1+/+ mice (not depicted). Furthermore, the median tumor-free survival of Bub1−/H mice was significantly shorter than that of Bub1H/H mice (P < 0.01). (B) Spontaneous tumor incidence and tumor latency of Bub1+/+, Bub1+/−, Bub1H/H, and Bub1−/H mice. (C) Tumor spectrum of Bub1+/+, Bub1+/−, Bub1H/H, and Bub1−/H mice. Asterisks mark values that are significantly different from wild type using a Fisher exact Chi-square test. (D) An overt hepatocellular carcinoma is indicated by the dashed line. (D′) Hematoxylin and eosin stained well-differentiated hepatocellular carcinoma, showing a proliferation of mildly atypical hepatocytes with abundant vascular channels, a lack of normal portal tracts, and a nodular focus (arrowheads) with mildly thickened trabeculae. (E) Thymic lymphoma (dashed line). (F) Overt lung adenocarcinoma (dashed circle). (F′) Hematoxylin and eosin–stained low-power magnification of a typical lung adenocarcinoma (dashed circle).
Increased incidence of DMBA-induced tumors in Bub1 mutant mice
Based on the aforementioned data, we conclude that Bub1+/− mice have enough Bub1 protein to protect themselves against spontaneous tumorigenesis. To determine whether this level is sufficient to guard against carcinogen-induced tumors, we administered a single dose of 0.5% DMBA (9,10-dimethylbenz-A-athracene) in acetone to the dorsal skin of 3–5-d-old pups generated from Bub1+/− × Bub1+/+ intercrosses. 5 mo after treatment, we killed the mice and screened for tumors. Irrespective of the genotype, tumors were exclusively detectable in the lungs. Bub1+/− mice exhibited a two- to threefold higher incidence of lung tumors than in Bub1+/+ mice (Fig. 7 A). Moreover, the tumor burden of Bub1+/− mice was increased approximately threefold (Fig. 7 B). From this experiment, we conclude that Bub1 heterozygous knockout mice are prone to carcinogen-induced tumorigenesis.
Figure 7.
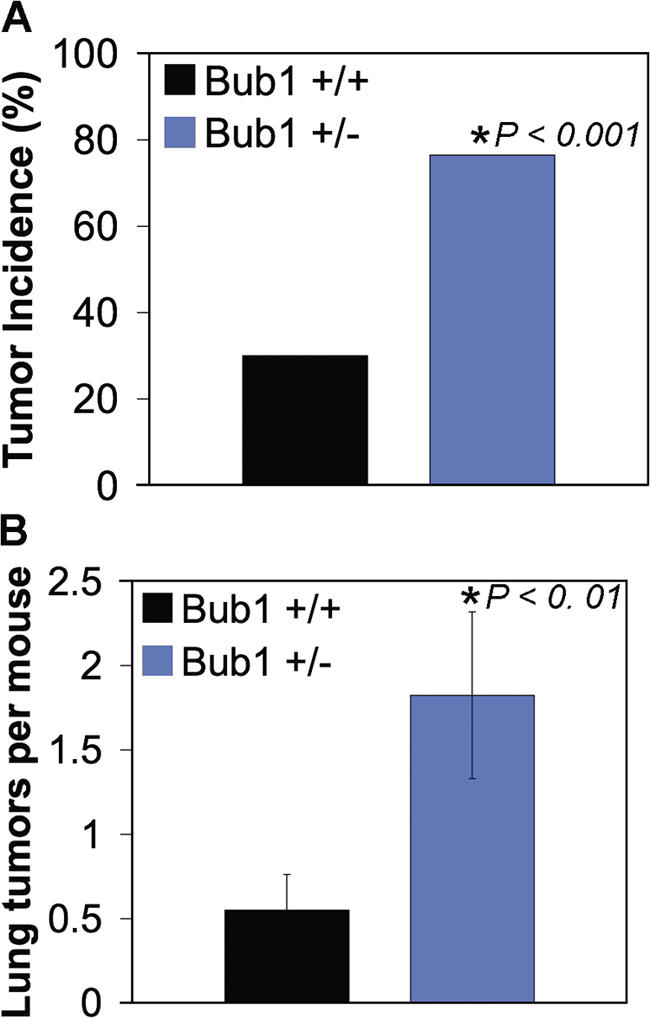
DMBA-induced tumor formation in Bub1 haploinsufficient mice. (A) The occurrence of lung tumors in 5-mo-old mice plotted as the percentage of incidence. (B) The mean number of lung adenomas per mouse ± SEM (error bars). (A and B) The asterisk marks a value that is significantly different from wild type using a chi-squared test (A) and a Wilcoxon rank sum test (B).
Discussion
In this study, we produced a series of mutant mice in which the expression of Bub1 is reduced in a graded fashion from normal to zero by the use of wild-type, hypomorphic, and knockout alleles to determine the physiological role of Bub1. As anticipated, we find that the complete loss of Bub1 leads to embryonic lethality. Strongly reduced Bub1 expression (up to approximately fivefold reduction) does not interfere with embryogenesis and allows for the development of adult mice that are overtly indistinguishable from their wild-type littermates. However, the reduction of Bub1 levels does have adverse consequences on genomic stability in these cells. Karyotyping of splenocytes and MEFs from our series of mutant mice established an inverse correlation between Bub1 expression level and aneuploidy. Failure of chromosome congression during metaphase is the main chromosome segregation defect resulting from Bub1 insufficiency. Although small and large reductions in Bub1 levels cause similar rates of chromosome missegregation, rates of cell survival after aberrant chromosome segregation increase considerably with declining Bub1 levels, providing a plausible explanation for why large reductions cause more aneuploidy than small ones. Furthermore, the reduction of Bub1 protein affected the strength of the mitotic checkpoint and loading of certain proteins onto centromeres or kinetochores. Bub1 haploinsufficiency in mice does not cause spontaneous tumors, but, as Bub1 levels drop further, animals become highly susceptible to a variety of spontaneous tumors, with the highest rate of tumorigenesis seen at the lowest level of Bub1 expression.
Bub1 is known to be required for the binding of several other mitotic checkpoint proteins to kinetochores (Sharp-Baker and Chen, 2001; Johnson et al., 2004; Meraldi et al., 2004). Our analysis of MEFs with graded reduction in Bub1 expression now reveals that these proteins require different levels of Bub1 protein for their normal recruitment to kinetochores. In particular, the recruitment of Mad1 to kinetochores is dramatically reduced when Bub1 is down-regulated. We speculate that Mad2, which forms a complex with Mad1 at kinetochores (Yu, 2006), is similarly sensitive to Bub1 down-regulation, although we were unable to confirm this because of the lack of an antibody that detects mouse Mad2 at kinetochores. Besides Mad1, the recruitment of BubR1 and CENP-E to kinetochores is also sensitive to Bub1 down-regulation, but not as sensitive as Mad1, as their localization is normal in Bub1 heterozygous MEFs. Recent studies have presented evidence that Bub1 functions to recruit Sgo1 to centromeres to prevent the precocious separation of sister kinetochores (Tang et al., 2004b; Kitajima et al., 2005). Consistent with these studies, we find that centromeric Sgo1 levels are reduced in Bub1 mutant MEFs, but only when Bub1 expression is strongly down-regulated. However, this did not result in premature sister kinetochore separation, implying that an even further drop in centromeric Sgo1 is required to trigger the cleavage of cohesin molecules that link sister centromeres. A recent study has implicated Bub1 in the targeting of chromosomal passenger complexes to centromeres in early mitosis (Boyarchuk et al., 2007). Our finding that very low amounts of Bub1 are sufficient for directing these complexes to centromeres suggests that near complete Bub1 depletion is required to dislocate the passenger complex from mitotic centromeres.
One of our more surprising findings is the observation that a relatively small reduction in Bub1 expression has a major impact on the accuracy of chromosome congression. What could be the explanation for this observation? Although the precise role of Bub1 in chromosome congression is currently not known, it is believed that this role involves kinetochore assembly (Meraldi and Sorger, 2005). Of the mitotic checkpoint proteins whose recruitment is Bub1 dependent, only CENP-E has so far been implicated in chromosome congression. Thus, one explanation for the congression failure in Bub1 mutant MEFs might be a CENP-E recruitment defect. Consistent with this, we find that the targeting of CENP-E to kinetochores is perturbed in Bub1 hypomorphic MEFs. On the other hand, Bub1 haploinsufficient MEFs, which display similar rates of congression failure as Bub1 hypomorphic MEFs, exhibit normal CENP-E recruitment to kinetochores, implying that the mechanism of congression failure is CENP-E independent. This conclusion is further supported by data of Meraldi and Sorger (2005) demonstrating that the depletion of Bub1 in HeLa cells by RNA interference causes chromosome congression defects in the absence of CENP-E mislocalization. Therefore, it remains unclear how Bub1 promotes proper chromosome congression. Nonetheless, we suspect that it involves a known or novel kinetochore- associated protein that functions in microtubule capture and whose recruitment to kinetochores is highly dependent on a full complement of Bub1.
Our analysis of MEFs with graded reduction in Bub1 expression indicates that relatively small shortages in Bub1, such as those seen in Bub1+/H and Bub1+/− MEFs, weaken the mitotic checkpoint considerably. It is plausible that the impaired recruitment of Mad1 (and presumably Mad2) to kinetochores undermines the mitotic checkpoint in these cells, as kinetochore-associated Mad1–Mad2 complexes generate soluble Mad2–Cdc20 complexes that bind to and inactivate APC/C. Larger reductions in Bub1, as present in Bub1H/H and Bub1−/H MEFs, had an even more profound impact on mitotic checkpoint activity. We propose that this is caused, at least in part, by the added loss of CENP-E and BubR1 from kinetochores, as kinetochore-bound CENP-E and BubR1 molecules have been implicated in the assembly of various inhibitory protein complexes that target APC/C (Mao et al., 2003, 2005). The Bub1 kinase also can inhibit APC/C directly through the phosphorylation of Cdc20 (Chung and Chen, 2003; Chen, 2004; Tang et al., 2004a). We have not addressed whether the phosphorylation of Cdc20 is affected in our mutant series of MEFs as a result of the current lack of antibodies that recognize phosphorylated mouse Cdc20.
Although it has been well established that gross abnormalities of chromosome segregation (frequently referred to as mitotic catastrophe) often cause cell death (Castedo et al., 2004), the fate of cells undergoing the random missegregation of only one or a few chromosomes has been unknown. Here, we show by the use of live cell imaging that wild-type primary MEFs die at very high rates after minor abnormalities in chromosome segregation. The implication of this finding is that aneuploidy rates in cultured wild-type MEFs are substantially higher than metaphase spread karyotypes reveal. Our discovery that cell death rates after chromosome missegregation dramatically decline with decreasing levels of Bub1 creates a molecular entry point for studying the underlying cell death mechanism. Whether Bub1 plays a unique role in this mechanism or whether there is a broader connection between mitotic checkpoint damage and decreased cell death after chromosome missegregation is an important question for future analysis. Bub1's dual function as a guardian of high fidelity chromosome segregation and as a mediator of cell death after aberrant segregation is reminiscent of proteins such as ATM (ataxia telangiectasia mutated) and p53 that function in both DNA repair and apoptosis in response to DNA damage (Sancar et al., 2004). A recent study showed that Bub1-depleted cancer cell lines display increased mitotic cell death when they are exposed to agents that perturb kinetochore-microtubule attachment, such as nocodazole (Niikura et al., 2007). We observed no such effect in nocodazole-treated MEFs with graded reduction in Bub1 expression (Fig. S4 D), suggesting that the impact of the Bub1 level of expression on mitotic cell death induced by spindle poisons is cell type and/or transformation status dependent.
Bub1 expression is reduced in several human cancers, including colorectal, gastric, and esophageal tumors (Shigeishi et al., 2001; Shichiri et al., 2002; Doak et al., 2004); however, it was unknown whether the reduced expression of this mitotic checkpoint protein is causally implicated in tumorigenesis. Analysis of our series of Bub1 mutant mice firmly establishes that the reduced expression of Bub1 leads to the development of spontaneous tumors in mice, but only when Bub1 levels fall below a threshold level. Those with the most drastic reductions of Bub1 expression have the shortest tumor latency and the highest incidence of tumors. The level of Bub1 required to prevent spontaneous tumorigenesis appears to vary per tissue, as illustrated by the fact that only mice with the most profound reduction in Bub1 are predisposed to lymphomas and lung tumors. In the liver, the optimal level of Bub1 down-regulation is not the lowest level, as Bub1H/H mice but not Bub1−/H mice are prone to hepatocellular carcinomas. Adding even more complexity is the discovery that Bub1 haploinsufficiency exerts a slight tumor-suppressive effect in both liver and lung tissue. This finding is consistent with the recent discovery that CENP-E haploinsufficiency inhibits tumorigenesis in certain mouse tissues (Weaver et al., 2007). However, unlike CENP-E insufficiency, Bub1 haploinsufficiency does not inhibit DMBA-induced tumorigenesis. In fact, Bub1 haploinsufficient mice are highly susceptible to lung tumors when challenged with this carcinogen. This observation implies that the loss of one Bub1 gene copy acts to accelerate the development of tumors initiated by particular cancer gene mutations.
Because Bub1 hypomorphic mice have a high percentage of aneuploid cells and are predisposed to spontaneous tumors, whereas Bub1 haploinsufficient mice have a relatively low percentage of aneuploid cells and are not tumor prone, it is tempting to speculate that it is the increase in aneuploidy that drives tumorigenesis in Bub1 hypomorphic mice. However, the fact that both Rae1/Bub3 and Rae1/Nup98 double-haploinsufficient mice develop aneuploidy at rates very similar to that of Bub1 hypomorphic mice but are not prone to spontaneous tumors argues against this idea (Babu et al., 2003; Baker et al., 2006; Jeganathan et al., 2005, 2006). One possible explanation for this discrepancy could be that as a result of the decreased cell death in response to chromosome missegregation, Bub1 hypomorphic mice may develop a wider variety of abnormal karyotypes than Rae1/Bub3 and Rae1/Nup98 double-haploinsufficient mice, thereby perhaps increasing the incidence of karyotypes that have the ability to drive tumorigenesis. However, the role of aneuploidy in tumorigenesis is clearly highly complex, and it will be necessary to carefully examine each individual regulator of chromosome segregation for its involvement in tumorigenesis through the use of animal models. We expect these efforts to allow the identification of a subset of mitotic regulators that are particularly important for tumor prevention. Among them may be mitotic regulators that serve as molecular hubs within the mitotic checkpoint or other networks that regulate proper chromosome segregation or mitotic regulators with connectivity to other pathways that guard against neoplastic transformation.
In this study, we have used a series of mutant mice to demonstrate that only after reducing Bub1 levels beyond a threshold level do mice start to develop spontaneous tumors. Had we used only Bub1 haploinsufficient mice rather than a series of mice with graded reduction in Bub1 expression, our conclusions would have been dramatically different in that we would conclude that Bub1 does not act as a tumor suppressor itself. Heterozygous knockout models for several other mitotic checkpoint genes are also not predisposed to spontaneous tumorigenesis. For a more definitive understanding of the roles these genes have in tumor prevention, it will be useful to use hypomorphic alleles to further reduce their level of expression in mice.
Materials and methods
Generation of Bub1 mutant mice and analyses of tumorigenesis
An 8.5-kb Bub1 129Sv/J genomic DNA fragment was used to generate both targeting vectors used. Gene-targeting procedures were performed as previously described (van Deursen et al., 1996). We identified targeted ES cell clones by Southern blot analysis using a 3′ probe on BamHI-cut genomic DNA (Fig. 1 A). Mutant mice were derived from targeted ES cell clones through standard procedures. These mice were maintained on a mixed 129Sv/E × C57BL/6 genetic background. Mice in tumor susceptibility experiments were observed daily for the development of overt tumors or signs of ill health. Moribund mice were killed, and all major organs were screened for overt tumors using a dissection microscope (SZX12; Olympus). Tumors that were collected were processed by standard procedures for histopathology. Prism software (GraphPad Software, Inc.) was used for the generation of tumor-free survival curves and for statistical analyses. DMBA treatment was performed as previously described (Serrano et al., 1996; Babu et al., 2003). All major organs were screened for overt tumors using a dissection microscope (SZX12; Olympus). Harvested tumors were routinely processed for histopathological confirmation. We note that all mice were housed in a pathogen-free barrier environment.
Western blot analysis and indirect immunofluorescence
Western blot analyses and indirect immunofluorescence were performed as previously described (Kasper et al., 1999). A laser-scanning microscope (LSM 510 v3.2SP2; Carl Zeiss MicroImaging, Inc.) as well as a microscope (Axiovert 100M; Carl Zeiss MicroImaging, Inc.) with a c-Apochromat 100× oil immersion objective (Carl Zeiss MicroImaging, Inc.) was used to analyze immunostained cells and to capture representative images. Primary antibodies were visualized with appropriate secondary antibodies conjugated to AlexaFluor594, -488. or -647 (Invitrogen). Primary antibodies used for Western blotting and indirect immunofluorescence were as follows: rabbit anti–human Bub1(25–165), rabbit anti–human BubR1(382–420) (Baker et al., 2004), rabbit anti–human Mad1 (provided by T. Yen, Fox Chase Cancer Center, Philadelphia, PA), rabbit anti–human SgoI([1–262][177–351]) (provided by H. Yu, University of Texas Southwestern, Houston, TX; Tang et al., 2004b), mouse anti–Aurora B (BD Biosciences), human anticentromeric antibody (Antibodies, Inc.), and rabbit anti–CENP-E (provided by D. Cleveland, Ludwig Institute for Cancer Research, La Jolla, CA). Mad2 antibodies tested were as follows: rabbit anti–human Mad2(FL-205) (Santa Cruz Biotechnology, Inc.), mouse anti–human Mad2 (BD Biosciences), and rabbit anti–human Mad2 (Covance). None of these Mad2 antibodies detects Mad2 at kinetochores of mitotic MEFs.
Karyotyping of MEFs and splenocytes
Chromosome counts on metaphase spreads were performed as previously described (Babu et al., 2003). We note that cells were scored as diploid (n = 40 chromosomes), tetraploid (n = 80 chromosomes), or aneuploid (Weaver et al., 2007).
Live cell imaging experiments
To allow the visualization of chromosomes by fluorescent microscopy on living cells, we used a retrovirus expressing YFP-tagged H2B (Jeganathan et al., 2005). Passage 2 MEFs were seeded in T25 flasks at 75% confluence and cultured in DME/10% FBS at 3% oxygen. 12 h after seeding and again every 12 h for at least three times, the medium was replaced with medium harvested from EcoPACK pMSCV-puro-H2B-YFP viral producer cell lines. Cells were then seeded onto 35-mm glass-bottomed culture dishes (MatTek Corp.) and cultured in DME/10% FBS. Approximately 24 h later, experiments were performed using a microscope system (Axio Observer; Carl Zeiss MicroImaging, Inc.) with CO2 Module S, TempModule S, Heating Unit XL S, a plan Apo 63× NA 1.4 oil differential interference contrast III objective (Carl Zeiss MicroImaging, Inc.), camera (AxioCam MRm; Carl Zeiss MicroImaging, Inc.), and AxioVision 4.6 software (Carl Zeiss MicroImaging, Inc.). The imaging medium was DME/10% FBS. The temperature of the imaging medium was kept at 37°C. The exposure times in nocodazole challenge experiments were 100 ms at 2 × 2 binning. Time of arrest in mitosis was defined as the interval between NEBD (onset of mitosis) and chromatin decondensation (exit from mitosis without cytokinesis). Interframe intervals were 15 min for nocodazole challenge. Analysis of mitotic defects was performed as previously described (Baker et al., 2006). For analysis of the incidence of cell death after chromosome missegregation, MEFs undergoing abnormal chromosome segregation were marked and followed with an interframe interval of 30 min for up to 12 h. Cell death was preceded by severe nuclear blebbing and cytoplasmic fragmentation. For each of the aforementioned experiments, we examined at least three independent clones per genotype unless otherwise noted. Prism software (GraphPad Software, Inc.) was used for statistical analyses. To evaluate the incidence of micronuclei formation, at least 600 YFP-H2B–expressing interphase MEFs were screened for the presence of micronuclei by live cell microscopy.
Cell survival assays
Analyses of cell survival in response to doxorubicin, mitomycin C, and paraquat were performed as described previously (Baker et al., 2004) with the exception that passage 3 MEFs were used instead of passage 2 MEFs. For analysis of cell death in response to nocodazole treatment, 105 passage 3 MEFs were seeded in duplicate for three independent cell lines of each genotype. After ∼12 h, normal medium was replaced with medium containing 100 ng/ml nocodazole, and cells were cultured for an additional 72 h. All cells were collected after this time, and tryptan blue exclusion was used to count living cells.
Online supplemental material
Fig. S1 shows that truncated proteins encoded by the Bub1 knockout and hypomorphic alleles are undetectable by immunoblotting. Fig. S2 shows that Mad1, BubR1, and CENP-E protein levels were similar in Bub1+/+ and Bub1−/H cells and that Aurora B is not mislocalized in Bub1−/H cells. Fig. S3 shows that the incidence of micronuclei increases with declining levels of Bub1. Fig. S4 shows that the Bub1 level of expression has no impact on MEF cell survival to DNA-damaging agents and prolonged nocodazole exposure. Videos 1 and 2 show videos of the Bub1−/H MEF presented in Fig. 5 A. Videos 3 and 4 show videos of the Bub1+/− MEF presented in Fig. 5 B. Videos 5 and 6 show a Bub1+/+ MEF undergoing chromosome missegregation in mitosis. Both daughter cells die after exit from mitosis. Videos 7 and 8 show a Bub1+/H MEF undergoing chromosome missegregation. One of the two daughter cells undergoes cell death. Videos 9 and 10 show a Bub1H/H MEF undergoing normal chromosome segregation. Both daughter cells survive. Table S1 presents data about the number of chromosomes that are abnormally segregated in cells with mutliple segregation defects. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200706015/DC1.
Supplementary Material
Acknowledgments
We thank Paul Galardy, Rick Bram, Robin Ricke, and Debbie Pearson for critical reading of the manuscript and helpful discussions. We are grateful to Hongtao Yu, Tim Yen, and Don Cleveland for providing Sgo1, Mad1, and CENP-E antibodies, respectively.
This work was supported by a National Institutes of Health grant to J.M. van Deursen.
K. Jeganathan, L. Malureanu, and D.J. Baker contributed equally to this paper.
Abbreviations used in this paper: APC/C, anaphase-promoting complex/cyclosome; CENP, centromere protein; ES, embryonic stem; MEF, mouse embryonic fibroblast; NEBD, nuclear envelope breakdown; PMSCS, premature separation of sister chromatids.
References
- Babu, J.R., K.B. Jeganathan, D.J. Baker, X. Wu, N. Kang-Decker, and J.M. van Deursen. 2003. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J. Cell Biol. 160:341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, D.J., K.B. Jeganathan, J.D. Cameron, M. Thompson, S. Juneja, A. Kopecka, R. Kumar, R.B. Jenkins, P.C. de Groen, P. Roche, and J.M. van Deursen. 2004. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 36:744–749. [DOI] [PubMed] [Google Scholar]
- Baker, D.J., K.B. Jeganathan, L. Malureanu, C. Perez-Terzic, A. Terzic, and J.M. van Deursen. 2006. Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. J. Cell Biol. 172:529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyarchuk, Y., A. Salic, M. Dasso, and A. Arnaoutov. 2007. Bub1 is essential for assembly of the functional inner centromere. J. Cell Biol. 176:919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill, D.P., C. Lengauer, J. Yu, G.J. Riggins, J.K. Willson, S.D. Markowitz, K.W. Kinzler, and B. Vogelstein. 1998. Mutations of mitotic checkpoint genes in human cancers. Nature. 392:300–303. [DOI] [PubMed] [Google Scholar]
- Castedo, M., J.L. Perfettini, T. Roumier, K. Andreau, R. Medema, and G. Kroemer. 2004. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 23:2825–2837. [DOI] [PubMed] [Google Scholar]
- Chen, R.H. 2004. Phosphorylation and activation of Bub1 on unattached chromosomes facilitate the spindle checkpoint. EMBO J. 23:3113–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R.H., A. Shevchenko, M. Mann, and A.W. Murray. 1998. Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J. Cell Biol. 143:283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R.H., D.M. Brady, D. Smith, A.W. Murray, and K.G. Hardwick. 1999. The spindle checkpoint of budding yeast depends on a tight complex between the Mad1 and Mad2 proteins. Mol. Biol. Cell. 10:2607–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, E., and R.H. Chen. 2003. Phosphorylation of Cdc20 is required for its inhibition by the spindle checkpoint. Nat. Cell Biol. 5:748–753. [DOI] [PubMed] [Google Scholar]
- Doak, S.H., G.J. Jenkins, E.M. Parry, A.P. Griffiths, J.N. Baxter, and J.M. Parry. 2004. Differential expression of the MAD2, BUB1 and HSP27 genes in Barrett's oesophagus-their association with aneuploidy and neoplastic progression. Mutat. Res. 547:133–144. [DOI] [PubMed] [Google Scholar]
- Dobles, M., V. Liberal, M.L. Scott, R. Benezra, and P.K. Sorger. 2000. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 101:635–645. [DOI] [PubMed] [Google Scholar]
- Draviam, V.M., S. Xie, and P.K. Sorger. 2004. Chromosome segregation and genomic stability. Curr. Opin. Genet. Dev. 14:120–125. [DOI] [PubMed] [Google Scholar]
- Iwanaga, Y., Y.H. Chi, A. Miyazato, S. Sheleg, K. Haller, J.M. Peloponese Jr., Y. Li, J.M. Ward, R. Benezra, and K.T. Jeang. 2007. Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res. 67:160–166. [DOI] [PubMed] [Google Scholar]
- Jacks, T., T.S. Shih, E.M. Schmitt, R.T. Bronson, A. Bernards, and R.A. Weinberg. 1994. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 7:353–361. [DOI] [PubMed] [Google Scholar]
- Jallepalli, P.V., and C. Lengauer. 2001. Chromosome segregation and cancer: cutting through the mystery. Nat. Rev. Cancer. 1:109–117. [DOI] [PubMed] [Google Scholar]
- Jeganathan, K.B., L. Malureanu, and J.M. van Deursen. 2005. The Rae1-Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature. 438:1036–1039. [DOI] [PubMed] [Google Scholar]
- Jeganathan, K.B., D.J. Baker, and J.M. van Deursen. 2006. Securin associates with APCCdh1 in prometaphase but its destruction is delayed by Rae1 and Nup98 until the metaphase/anaphase transition. Cell Cycle. 5:366–370. [DOI] [PubMed] [Google Scholar]
- Johnson, V.L., M.I. Scott, S.V. Holt, D. Hussein, and S.S. Taylor. 2004. Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J. Cell Sci. 117:1577–1589. [DOI] [PubMed] [Google Scholar]
- Kalitsis, P., E. Earle, K.J. Fowler, and K.H. Choo. 2000. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 14:2277–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper, L.H., P.K. Brindle, C.A. Schnabel, C.E. Pritchard, M.L. Cleary, and J.M. van Deursen. 1999. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol. Cell. Biol. 19:764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima, T.S., S. Hauf, M. Ohsugi, T. Yamamoto, and Y. Watanabe. 2005. Human Bub1 defines the persistent cohesion site along the mitotic chromosome by affecting Shugoshin localization. Curr. Biol. 15:353–359. [DOI] [PubMed] [Google Scholar]
- Kops, G.J., D.R. Foltz, and D.W. Cleveland. 2004. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. USA. 101:8699–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops, G.J., B.A. Weaver, and D.W. Cleveland. 2005. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer. 5:773–785. [DOI] [PubMed] [Google Scholar]
- Luo, X., Z. Tang, J. Rizo, and H. Yu. 2002. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol. Cell. 9:59–71. [DOI] [PubMed] [Google Scholar]
- Luo, X., Z. Tang, G. Xia, K. Wassmann, T. Matsumoto, J. Rizo, and H. Yu. 2004. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat. Struct. Mol. Biol. 11:338–345. [DOI] [PubMed] [Google Scholar]
- Mao, Y., A. Abrieu, and D.W. Cleveland. 2003. Activating and silencing the mitotic checkpoint through CENP-E-dependent activation/inactivation of BubR1. Cell. 114:87–98. [DOI] [PubMed] [Google Scholar]
- Mao, Y., A. Desai, and D.W. Cleveland. 2005. Microtubule capture by CENP-E silences BubR1-dependent mitotic checkpoint signaling. J. Cell Biol. 170:873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi, P., and P.K. Sorger. 2005. A dual role for Bub1 in the spindle checkpoint and chromosome congression. EMBO J. 24:1621–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi, P., V.M. Draviam, and P.K. Sorger. 2004. Timing and checkpoints in the regulation of mitotic progression. Dev. Cell. 7:45–60. [DOI] [PubMed] [Google Scholar]
- Meyers, E.N., M. Lewandoski, and G.R. Martin. 1998. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat. Genet. 18:136–141. [DOI] [PubMed] [Google Scholar]
- Musacchio, A., and E.D. Salmon. 2007. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8:379–393. [DOI] [PubMed] [Google Scholar]
- Nasmyth, K., and C.H. Haering. 2005. The structure and function of SMC and kleisin complexes. Annu. Rev. Biochem. 74:595–648. [DOI] [PubMed] [Google Scholar]
- Niikura, Y., A. Dixit, R. Scott, G. Perkins, and K. Kitagawa. 2007. BUB1 mediation of caspase-independent mitotic death determines cell fate. J. Cell Biol. 178:283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima, K., S. Haraoka, S. Yoshioka, M. Hamasaki, T. Fujiki, J. Suzumiya, C. Kawasaki, M. Kanda, and M. Kikuchi. 2000. Mutation analysis of mitotic checkpoint genes (hBUB1 and hBUBR1) and microsatellite instability in adult T-cell leukemia/lymphoma. Cancer Lett. 158:141–150. [DOI] [PubMed] [Google Scholar]
- Peters, J.M. 2006. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol. 7:644–656. [DOI] [PubMed] [Google Scholar]
- Riedel, C.G., V.L. Katis, Y. Katou, S. Mori, T. Itoh, W. Helmhart, M. Galova, M. Petronczki, J. Gregan, B. Cetin, et al. 2006. Protein phosphatase 2A protects centromeric sister chromatid cohesion during meiosis I. Nature. 441:53–61. [DOI] [PubMed] [Google Scholar]
- Roberts, B.T., K.A. Farr, and M.A. Hoyt. 1994. The Saccharomyces cerevisiae checkpoint gene BUB1 encodes a novel protein kinase. Mol. Cell. Biol. 14:8282–8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru, H.Y., R.L. Chen, W.C. Lu, and J.H. Chen. 2002. hBUB1 defects in leukemia and lymphoma cells. Oncogene. 21:4673–4679. [DOI] [PubMed] [Google Scholar]
- Sancar, A., L.A. Lindsey-Boltz, K. Unsal-Kacmaz, and S. Linn. 2004. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73:39–85. [DOI] [PubMed] [Google Scholar]
- Serrano, M., H. Lee, L. Chin, C. Cordon-Cardo, D. Beach, and R.A. DePinho. 1996. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 85:27–37. [DOI] [PubMed] [Google Scholar]
- Sharp-Baker, H., and R.H. Chen. 2001. Spindle checkpoint protein Bub1 is required for kinetochore localization of Mad1, Mad2, Bub3, and CENP-E, independently of its kinase activity. J. Cell Biol. 153:1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichiri, M., K. Yoshinaga, H. Hisatomi, K. Sugihara, and Y. Hirata. 2002. Genetic and epigenetic inactivation of mitotic checkpoint genes hBUB1 and hBUBR1 and their relationship to survival. Cancer Res. 62:13–17. [PubMed] [Google Scholar]
- Shigeishi, H., N. Oue, H. Kuniyasu, A. Wakikawa, H. Yokozaki, T. Ishikawa, and W. Yasui. 2001. Expression of Bub1 gene correlates with tumor proliferating activity in human gastric carcinomas. Pathobiology. 69:24–29. [DOI] [PubMed] [Google Scholar]
- Sironi, L., M. Mapelli, S. Knapp, A. De Antoni, K.T. Jeang, and A. Musacchio. 2002. Crystal structure of the tetrameric Mad1-Mad2 core complex: implications of a ‘safety belt’ binding mechanism for the spindle checkpoint. EMBO J. 21:2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z., H. Shu, D. Oncel, S. Chen, and H. Yu. 2004. a. Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol. Cell. 16:387–397. [DOI] [PubMed] [Google Scholar]
- Tang, Z., Y. Sun, S.E. Harley, H. Zou, and H. Yu. 2004. b. Human Bub1 protects centromeric sister-chromatid cohesion through Shugoshin during mitosis. Proc. Natl. Acad. Sci. USA. 101:18012–18017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z., H. Shu, W. Qi, N. Mahmood, M.C. Mumby, and H. Yu. 2006. PP2A is required for centromeric localization of Sgo1 and proper chromosome segregation. Dev. Cell. 10:575–585. [DOI] [PubMed] [Google Scholar]
- Taylor, S.S., and F. McKeon. 1997. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 89:727–735. [DOI] [PubMed] [Google Scholar]
- van Deursen, J., W. Ruitenbeek, A. Heerschap, P. Jap, H. ter Laak, and B. Wieringa. 1994. Creatine kinase (CK) in skeletal muscle energy metabolism: a study of mouse mutants with graded reduction in muscle CK expression. Proc. Natl. Acad. Sci. USA. 91:9091–9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen, J., J. Boer, L. Kasper, and G. Grosveld. 1996. G2 arrest and impaired nucleocytoplasmic transport in mouse embryos lacking the proto-oncogene CAN/Nup214. EMBO J. 15:5574–5583. [PMC free article] [PubMed] [Google Scholar]
- Wang, Q., T. Liu, Y. Fang, S. Xie, X. Huang, R. Mahmood, G. Ramaswamy, K.M. Sakamoto, Z. Darzynkiewicz, M. Xu, and W. Dai. 2004. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood. 103:1278–1285. [DOI] [PubMed] [Google Scholar]
- Weaver, B.A., and D.W. Cleveland. 2006. Does aneuploidy cause cancer? Curr. Opin. Cell Biol. 18:658–667. [DOI] [PubMed] [Google Scholar]
- Weaver, B.A., A.D. Silk, C. Montagna, P. Verdier-Pinard, and D.W. Cleveland. 2007. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 11:25–36. [DOI] [PubMed] [Google Scholar]
- Yu, H., and Z. Tang. 2005. Bub1 multitasking in mitosis. Cell Cycle. 4:262–265. [PubMed] [Google Scholar]
- Yu, H. 2006. Structural activation of Mad2 in the mitotic spindle checkpoint: the two-state Mad2 model versus the Mad2 template model. J. Cell Biol. 173:153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen, K.W., B. Montpetit, and P. Hieter. 2005. The kinetochore and cancer: what's the connection? Curr. Opin. Cell Biol. 17:576–582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}