

Abstract
The endosomal sorting complexes required for transport (ESCRTs) are required to sort integral membrane proteins into intralumenal vesicles of the multivesicular body (MVB). Mutations in the ESCRT-III subunit CHMP2B were recently associated with frontotemporal dementia and amyotrophic lateral sclerosis (ALS), neurodegenerative diseases characterized by abnormal ubiquitin-positive protein deposits in affected neurons. We show here that autophagic degradation is inhibited in cells depleted of ESCRT subunits and in cells expressing CHMP2B mutants, leading to accumulation of protein aggregates containing ubiquitinated proteins, p62 and Alfy. Moreover, we find that functional MVBs are required for clearance of TDP-43 (identified as the major ubiquitinated protein in ALS and frontotemporal lobar degeneration with ubiquitin deposits), and of expanded polyglutamine aggregates associated with Huntington's disease. Together, our data indicate that efficient autophagic degradation requires functional MVBs and provide a possible explanation to the observed neurodegenerative phenotype seen in patients with CHMP2B mutations.
Introduction
Neurodegenerative disorders such as Huntington's disease (HD) and Parkinson's disease are characterized by the accumulation of intracellular ubiquitin-containing protein aggregates. Although polyubiquitin is a well-known signal for degradation by the ubiquitin–proteasome system, several aggregation-prone proteins have been shown to disrupt the function of the proteasome (Bence et al., 2001; Jana et al., 2001; Urushitani et al., 2002; Snyder et al., 2003). Activation of an alternative lysosomal mechanism of protein degradation, known as macroautophagy (hereafter referred to as autophagy), is often observed in protein aggregation diseases (Ravikumar and Rubinsztein, 2004; Iwata et al., 2005; Yamamoto et al., 2006). Autophagy is a dynamic process whereby cytoplasmic material is sequestered within double membrane-enclosed vesicles (autophagosomes), which eventually fuse with lysosomes where the encapsulated material is degraded (Klionsky and Ohsumi, 1999). This pathway is known to be important in developmental processes and human disease and for meeting amino acid poor growth conditions (Klionsky and Emr, 2000). Recently, it was found that loss of autophagy causes neurodegeneration even in the absence of any disease-associated mutant proteins (Hara et al., 2006; Komatsu et al., 2006), suggesting that the continuous clearance of cellular proteins through basal autophagy prevents their accumulation, and in turn prevents the disruption of neural function and subsequent neurodegeneration.
Autophagy is generally considered a ubiquitous bulk degradation mechanism for long-lived proteins and organelles. The molecular mechanisms underlying the autophagic process have been extensively studied in yeast, using genetic screens to identify autophagy-defective (atg) and vacuolar protein sorting (vps) mutants (Klionsky et al., 2003). Subsequent inactivation of atg orthologues in higher eukaryotes has revealed that the autophagic machinery is highly conserved. Nonetheless, the molecular mechanisms and signals involved in the recognition of autophagic substrates and trafficking of autophagosomes are poorly understood.
The endosomal sorting complexes required for transport (ESCRTs), first identified by characterization of yeast vps class E mutants, have proven important for recognition of ubiquitinated endocytosed integral membrane proteins, their sorting into the intralumenal vesicles (ILVs) of the multivesicular body (MVB) and subsequent degradation in the lysosome/vacuole. Ubiquitinated cargo is first recognized by the Vps27p/Hrs (hepatocyte growth factor–regulated tyrosine kinase substrate)–Hse1p/STAM (signal transducing adaptor molecule) complex. Vps27p/Hrs then recruits the ESCRT-I complex (Vps23p/Tsg101, Vps28p, Vps37p, Mvb12p) to the endosome membrane by binding Vps23p/Tsg101 (tumor susceptibility gene 101). The ubiquitinated cargo is further assumed to be delivered to ESCRT-II (Vps22p, Vps25p, Vps36p) before it gets internalized into MVBs through the activity of ESCRT-III (Vps2p, Vps20p, Vps24p, Vps32p). Ubiquitin is removed proteolytically and the ESCRT machinery is finally dissociated from the endosomal membrane by activity of the ATPase Vps4p/SKD1 (for review see Hurley and Emr, 2006; Slagsvold et al., 2006; Williams and Urbe, 2007).
Depletion of ESCRT subunits results in MVBs with abnormal morphology, called the “class E compartment” in yeast. The effects of depleting different ESCRT subunits on internalization and degradation of integral membrane proteins such as the epidermal growth factor receptor (EGFR) have been well characterized (Slagsvold et al., 2006). However, little is known about the role of functional MVBs and ESCRT subunits for autophagic degradation. The classical view has been that the autophagic and endocytic pathways converge at the lysosomal level, but autophagosomes have also been found to undergo fusions with earlier parts of the endocytic pathway (Tooze et al., 1990; Punnonen et al., 1993; Liou et al., 1997; Berg et al., 1998). The term “amphisome” is used to describe pre-autolysosomal compartments containing both autophagic and endocytic material (Gordon and Seglen, 1988), but the specificity of amphisome formation and the molecular mechanism involved are poorly understood.
Recently, the ESCRT-III subunit CHMP2B (charged multivesicular body protein 2B, also known as chromatin-modifying protein 2B)/Vps2B was found to be mutated in a large Danish pedigree with frontotemporal dementia (FTD) (Skibinski et al., 2005), and in patients with amyotrophic lateral sclerosis (ALS) (Parkinson et al., 2006). FTD is the second most common form of presenile dementia after Alzheimer's disease (Ratnavalli et al., 2002; Harvey et al., 2003) and is characterized neuropathologically by the presence of either tau pathology or ubiquitin pathology, which is termed frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions (FTLD-U) (Neary et al., 2005). The cellular pathologies of both FTLD-U and ALS demonstrate accumulation of ubiquitin-positive protein deposits that are also positive for p62/Sequestosome-1, a common component of protein inclusions associated with neurodegenerative disease (Talbot and Ansorge, 2006). p62 can bind polyubiquitin through its UBA domain (Vadlamudi et al., 1996) and interacts with the autophagic protein Atg8/LC3 (Bjorkoy et al., 2005; Pankiv et al., 2007), thus providing a possible link between protein accumulation and aggregation with autophagy-mediated clearance.
In light of the mutations found in CHMP2B, we asked whether the ESCRT machinery is required for autophagic degradation and prevention of formation of protein aggregates associated with neurodegenerative disease. In this study we show that autophagic degradation is inhibited in cells depleted of ESCRT subunits and in cells overexpressing CHMP2B mutants, leading to accumulation of protein aggregates containing ubiquitinated proteins, p62 and Alfy (autophagy-linked FYVE protein). Moreover, we show that TAR DNA-binding protein 43 (TDP-43), recently identified as the major ubiquitinated protein in FTLD-U and ALS, accumulates in ubiquitin-positive inclusions in ESCRT-depleted cells. Using conditional cell-based systems for HD, we find that functional MVBs are also required for efficient clearance of the expanded polyglutamine aggregates. Collectively, our data indicate that efficient autophagic degradation requires functional MVBs and provide a possible explanation to the observed neurodegenerative phenotype seen in patients with CHMP2B mutations.
Results
Non-endosomal ubiquitin-positive structures accumulate in ESCRT-depleted cells
To investigate whether depletion of ESCRT subunits leads to accumulation of ubiquitinated proteins, HeLa cells transfected with siRNA against Hrs, ESCRT-I (Tsg101), -II (Vps22), or -III (Vps24) were labeled with antibodies recognizing ubiquitin (Ub), the early endosome antigen 1 (EEA1) and the late endosomal/lysosomal marker Lamp2 and analyzed by confocal immunofluorescence microscopy. In contrast to siRNA controls (Fig. 1, A and B; and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200702115/DC1), depletion of Hrs resulted in accumulation of ubiquitin on EEA1-positive early endosomes (Fig. 1 C and Fig. S1). This was also the case in cells depleted of Tsg101, but in addition large ubiquitin-positive EEA1-negative structures were seen in close proximity to Lamp2-positive membranes (Fig. 1 D and Fig. S1). Cells depleted of Vps22 generally had a similar phenotype as Tsg101-depleted cells (Fig. 1 E and Fig. S1), but the penetrance of the Vps22-depleted phenotype was weaker than in cells lacking Tsg101. This could be due to different siRNA-mediated knockdown efficiency (Fig. 1 G). There was no accumulation of ubiquitin on early endosomes in Vps24-depleted cells, but large ubiquitin-positive structures that either were devoid of endosomal membranes or localized close to Lamp2-positive structures could be detected (Fig. 1 F and Fig. S1). Similar results were obtained using at least two different siRNA oligonucleotides for each protein. We used siRNA against the Vps24 subunit of the ESCRT-III complex, found to form a sub-complex with Vps2/CHMP2 (Babst et al., 2002; von Schwedler et al., 2003), as two isoforms of Vps2/CHMP2 are expressed in HeLa cells (unpublished data). Depletion of one ESCRT subunit did not affect the expression levels or stability of subunits of other ESCRT complexes (unpublished data).
Figure 1.

Non-endosomal ubiquitin-positive structures accumulate in ESCRT depleted cells. HeLa cells were left untransfected (A) or transfected with control (B), Hrs (C), Tsg101 (D), Vps22 (E), or Vps24 (F) siRNA for 5 d and processed for immunofluorescence microscopy. Cells were labeled with antibodies against EEA1 (blue), ubiquitin (mono and poly-Ub, red), and Lamp2 (green). Colocalization between EEA1 and Ub is indicated in purple and between Ub and Lamp2 in yellow. Bar, 10 μm. Single channel images of the insets are shown in Fig. S1 (available at http://www.jcb.org/cgi/content/full/jcb.200702115/DC1). (G) Knock-down efficiencies were analyzed by Western blotting. Equal loading was verified by anti-α-tubulin immunoblotting.
Together, our data show that depletions of Hrs or the ESCRT-I and -II subunits, Tsg101 and Vps22, respectively, but not of the ESCRT-III subunit Vps24, result in accumulation of ubiquitinated proteins on early endosomes, probably representing internalized mono-ubiquitinated plasma membrane proteins or components of the sorting machinery. In addition, large ubiquitin-positive structures, often found in close proximity to the lysosomal marker Lamp2, accumulate in cells depleted of Tsg101, Vps22, and Vps24.
p62-positive structures accumulate in cells depleted of Tsg101 and Vps24
To further investigate the nature of the large ubiquitin-positive EEA1-negative structures found in ESCRT-depleted cells, we used cells treated with siRNA against Tsg101 and Vps24 for further studies. We first asked whether p62 and Alfy, proteins known to associate with cytoplasmic ubiquitin-positive structures (Simonsen et al., 2004; Bjorkoy et al., 2005), accumulate in cells depleted of Tsg101 or Vps24. As can be seen in Fig. 2, both p62 (Fig. 2 A) and Alfy (Fig. 2 B) were found to associate with the ubiquitin-positive structures that accumulate in ESCRT-depleted cells. Both the number and size of p62-positive structures were dramatically increased in Tsg101- and Vps24-depleted cells, as quantified from more than 300 cells using ImageJ software. The p62-positive particles were grouped according to their size, and the numbers per 100 cells are presented in Fig. 2 C. Control cells had an average of 355p62-positive structures per 100 cells, with only 29 having an area above 2 μm2. In contrast, cells depleted of Tsg101 or Vps24 had on average 1,251 and 1,605 p62-positive structures, with 263 and 332 above 2 μm2, respectively. Thus, depletion of ESCRT subunits leads to both increased numbers and sizes of ubiquitin-, p62- and Alfy-positive aggregates or inclusions.
Figure 2.

p62- and Alfy-positive structures accumulate in cells depleted of Tsg101 or Vps24. HeLa cells transfected with siRNA against Tsg101 or Vps24 were fixed, permeabilized, and stained with antibodies against ubiquitin (red) and p62 (green) (A) or against ubiquitin (green) and Alfy (red) (B). Colocalization is indicated in yellow. Bar, 10 μm. (C) The number and size (area) of p62-positive particles were quantified using ImageJ software. Approximately 100 cells from three different experiments (n = 334 (control), 309 (Tsg101), and 246 (Vps24)) were used for the quantification and the data is presented as average number per 100 cells. Error bars = SEM.
Autophagic degradation is impeded in Tsg101- and Vps24-depleted cells
Formation of ubiquitin-positive aggregates, as seen in Tsg101- and Vps24-depleted cells, could be caused either by increased protein synthesis or decreased protein degradation. The ubiquitin–proteasome system is the principal means of eliminating polyubiquitinated damaged or misfolded proteins, but also loss of autophagy was recently found to lead to accumulation of ubiquitin-positive inclusions and cause neurodegeneration in mouse models (Hara et al., 2006; Komatsu et al., 2006). We therefore asked whether protein synthesis, proteasome activity, or autophagic degradation was affected by knockdown of ESCRT subunits. The total level of protein synthesis, as measured by incorporation of [3H]-leucine, was not considerably changed in ESCRT-depleted cells (Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200702115/DC1). Neither the relative mRNA levels of p62 nor LC3-B were significantly increased in ESCRT-depleted cells, as measured by quantitative real-time PCR (qRT-PCR) (Fig. S2 B). Proteasome activity was analyzed in the absence or presence of the proteasome inhibitor PSI in control and ESCRT-depleted cells, and, interestingly, proteasome activity was increased rather than decreased in ESCRT-depleted cells (Fig. S2 C). This could be due to proteolytic cross-talk, induced by inhibition of autophagic degradation in ESCRT knockdown cells, as described in the next section.
LC3/Atg8 is a widely used marker for autophagy, as it binds specifically to autophagic membranes and remains bound throughout the pathway (Kabeya et al., 2000; Mizushima et al., 2001). p62, known to interact with LC3 and to be degraded by autophagy (Bjorkoy et al., 2005; Pankiv et al., 2007), is another commonly used autophagy marker. HeLa cells stably expressing LC3 fused to GFP (Bampton et al., 2005) were transfected with control, Tsg101, or Vps24 siRNA, and the levels of GFP-LC3 and p62 were analyzed by confocal microscopy. Although p62 and GFP-LC3 colocalized on small cytoplasmic structures in control cells (Fig. 3 A, inset), a massive accumulation of these two proteins were seen in cells depleted of Tsg101 or Vps24 (Fig. 3, B and C, insets). Both the overall levels of p62 and GFP-LC3 and their degree of colocalization were increased in Tsg101- and Vps24-depleted cells, as quantified from three independent experiments (Fig. 3 D). p62 accumulation was also seen upon immunoblotting of cell lysates from ESCRT-depleted HeLa (Fig. 3 E), HepII, and U2OS cells (unpublished data). Using differential detergent extraction, a technique often used as a quantitative measure of protein inclusion formation (Scherzinger et al., 1997; Wanker et al., 1999), we observed a shift in p62 from less stringent extraction buffers (1% Triton-X-100, soluble) into 2% SDS (insoluble) in ESCRT-depleted cells, representing a shift into a more aggregated conformation (Fig. 3 E). p62 accumulation was also seen in cells depleted of Atg5 (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200702115/DC1), and although the mechanism responsible for p62 accumulation is clearly different in Atg5- and ESCRT-depleted cells, these data further demonstrate that p62-positive aggregates form when autophagic turnover is inhibited HeLa cells.
Figure 3.

Autophagic degradation is impeded in Tsg101- and Vps24-depleted cells. HeLa cells stably expressing GFP-LC3 (green) were transfected with control (A), Vps24 (B), or Tsg101 (C) siRNA for 5 d and processed for immunofluorescence analysis. Cells were labeled with antibodies against p62 (red) and analyzed by confocal microscopy. Colocalization is indicated in yellow. Bar, 10 μm. (D) The total level of p62 and GFP-LC3 (normalized to control cells) and their degree of colocalization (% of total) in control and Tsg101- and Vps24-depleted cells were quantified using the Zeiss LSM 510 Meta software. 30 cells from three independent experiments were used for quantification. Error bars = SEM. (E) Western blot showing accumulation of LC3-II and p62 in HeLa cells depleted of Vps24 and Tsg101. p62 also accumulates in the insoluble fraction (bottom picture), indicating a shift to a more aggregated form upon depleted of Vps24 and Tsg101. (F) Western blot showing that LC3-II levels were similar in control and Vps24-depleted cells treated with Bafilomycin A.
As GFP-LC3 has the potential to associate with membrane-free aggregates (Kuma et al., 2007), probably due to its complex formation with p62 (Bjorkoy et al., 2005; Pankiv et al., 2007), a better way to analyze cellular autophagy levels is to measure the ratio between the 18-kD cytosolic and 16-kD lipidated autophagosome-bound form of LC3, LC3-I, and LC3-II, respectively (Kabeya et al., 2000, 2004). As can be seen in Fig. 3 E, increased levels of LC3-II were detected in cells depleted of Tsg101 or Vps24. This could in principle be caused either by elevated levels of autophagy or decreased degradation. However, the endogenous LC3-II level was similar in control and ESCRT-depleted cells treated with the proton ATPase inhibitor Bafilomycin A, known to inhibit lysosomal degradation (Fig. 3 F). If ESCRT knock-down lead to increased levels of autophagy, we would expect the LC3-II levels to be higher in ESCRT-depleted than control cells treated with Bafilomycin A. Moreover, the fact that LC3-B mRNA levels were unaffected in ESCRT-depleted cells compared with control cells (Fig. S2 B) also indicates, although indirectly, that LC3 synthesis is unchanged.
To further investigate whether autophagic degradation is inhibited in cells depleted for ESCRTs we took advantage of a recently developed double-tagged mCherry-GFP-LC3 construct (dtLC3) (Pankiv et al., 2007), which is detected as yellow fluorescent (green merged with red) in nonacidic structures (autophagosomes and amphisomes) and as red only in autolysosomes due to quenching of GFP in these acidic structures (see Fig. 4 A, cartoon). Whereas 50% of the total dtLC3 signal was red in control cells (Fig. 4, B and E), only 20% red was detected in Tsg101- (Fig. 4, C and E) and Vps24-depleted cells (Fig. 4, D and E). This indicates that transport of mCherry-GFP-LC3 to acidic lysosomes, i.e., formation of autolysosomes, is inhibited in ESCRT-depleted cells. Depletion of Tsg101 and Vps24 also inhibited starvation-induced degradation of long-lived proteins in HeLa cells (Fig. 4 F). Compared with control cells, there was an average 60 and 43% reduction of the level of starvation-induced degradation in Tsg101- and Vps24-depleted cells, respectively. Collectively, our data strongly indicate a general requirement for functional MVBs in autophagy.
Figure 4.

Formation of autolysosomes is inhibited in cells depleted of Tsg101 or Vps24. (A) The double-tagged LC3 protein (mCherry-GFP-LC3) will emit yellow (green merged with red) fluorescence in non-acidic structures and appear as red only in the autolysosomes due to quenching of GFP in these acidic structures. (B–F) HeLa cells were transfected with control (B), Tsg101 (C), or Vps24 (D) siRNA for 4 d and then with mCherry-GFP-LC3 for another 24 h before confocal microscopy analysis. Bar, 5 μm. (E) The mCherry-LC3 (red) signal was quantified in ∼30 cells with similar expression levels from three independent experiments using the Zeiss LSM 510 Meta software and the data are presented as % of total mCherry-GFP-LC3. Error bar = SEM. (F) Degradation of [14C]-valine- labeled proteins in cells incubated in complete medium (control), serum- and amino acid-deficient medium (starve), or starvation media in the presence of 3-methyl adenine (3-MA) to inhibit autophagic degradation. Average degradation per hour from four experiments done in duplicates ±1 SEM are shown.
Formation of autolysosomes is inhibited in ESCRT-depleted cells
Our data indicate that ubiquitin-positive, p62-positive aggregates accumulate in ESCRT-depleted cells due to defective autophagic degradation, but at what step is this pathway impeded? Although the classical view is that autophagosomes fuse directly with lysosomes, autophagosomes have also been found to undergo fusions with earlier parts of the endocytic pathway (Tooze et al., 1990; Punnonen et al., 1993; Liou et al., 1997; Berg et al., 1998) and the term amphisome is used to describe pre-autolysosomal compartments containing both autophagic and endocytic material (Gordon and Seglen, 1988). Because the ESCRT-complexes are required for proper formation of MVBs, it is likely that loss of ESCRT subunits inhibits either the fusion of autophagosomes with MVBs or fusion of amphisomes with lysosomes.
To address this issue we used immunoelectron microscopy (EM). To perform double-labeling experiments with autophagic and endosomal markers, we used HeLa cells stably expressing GFP-LC3 (Bampton et al., 2005). Cryosections were incubated with antibodies against GFP and the MVB/late endosome marker lyso-bisphosphatidic acid (LBPA), and the presence of amphisomes could then be scored based on both LC3-LBPA colocalization and morphology (Fig. 5). GFP-LC3 and LBPA positive amphisomes were detected in control cells (Fig. 5 A), but more frequently in cells lacking Tsg101 (42% increase) (Fig. 5, B, D, and F) and Vps24 (29% increase) (Fig. 5, C and F). In addition, clusters of double-membrane structures, consisting of autophagosomes and tubular structures which might represent phagophores, all labeling strongly for GFP-LC3, are typically found in Tsg101-depleted cells (seen in ∼25% of the cells) (Fig. 5, B and E), but were not so prominent in Vps24-depleted cells and never seen in control cells. Increased levels of amphisomes in ESCRT-depleted cells, visualized as colocalization between Alfy and LBPA, was also found using confocal immunofluorescence (IF) microscopy (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200702115/DC1). In some cells Alfy positive structures seemed to be surrounded by LBPA-positive membranes (Fig. S4 D), suggesting that protein aggregates may accumulate in amphisomes when the MVB pathway is impeded. However, due to the lower resolution of the confocal microscope we cannot distinguish fused (amphisomes) from docked autophagosome–endosome vesicles, and EM analysis is thus a better way of addressing this issue.
Figure 5.

Autophagosomes and amphisomes do form in ESCRT-depleted cells. HeLa cells stably expressing GFP-LC3 were transfected with control (A), Tsg101 (B, D, and E), or Vps24 (C) siRNA and processed for immuno-EM analysis. Cryosections were incubated with antibodies against GFP (15-nm gold) and LBPA (10-nm gold). Amphisomes (Am), characterized by their content of electron-dense LC3-positive areas and intraluminal vesicles, were observed in both control (A) and ESCRT-depleted cells (B and C). (D) In the Tsg101-depleted cells we found large clusters of more typical autophagosomes (Au) and (E) clusters of double-membrane structures, consisting of autophagosomes and tubular structures which might represent phagophores, all labeling strongly for GFP-LC3. We never observed similar clusters in control cells and very rarely in Vps24-depleted cells. Bars, 200 nm. (F) Quantification of amphisomes in control cells and cells depleted for Tsg101 or Vps24. Approximately 20 cells were included per siRNA depletion per experiment (n = 3). Error bars = SEM.
To determine whether p62-positive structures could be found within autophagic vesicles, cryosections of HeLa cells treated with control, Tsg101, or Vps24 siRNA were incubated with antibodies against p62, followed by colloidal gold-labeled secondary antibodies. p62-positive structures were detected within amphisomes both in control cells and in cells depleted of Tsg101 or Vps24 (Fig. 6, E and F). In addition, clusters of small vesicular–tubular elements and larger vesicles of typical endosomal morphology were often found within p62-labeled areas in Tsg101- and Vps24-depleted cells (Fig. 6, A and B). Membrane-free dense p62-positive cytosolic aggregates were also frequently found in these cells (Fig. 6, C and D; and Table I), but never in control cells (Table I).
Figure 6.

Immuno-EM of p62-positive structures in Vps24- and Tsg101-depleted cells. Cryosections of HeLa cells transfected with Vps24 (A, C, and E) or Tsg101 (B, D, and F) siRNA were incubated with antibodies against p62 (15-nm protein A gold, 10-nm in D). Membrane-free p62-positive aggregates (C and D) or p62-positive structures contained within dense clusters of vesicular-tubular elements (A and B) were detected. Vesicles with early endosomal morphology (EE) or multivesicular appearance (MVB) associated with these clusters. We also observed p62 labeling in electron-dense structures sequestered within amphisomes in cell depleted of Vps24 (E) or Tsg101 (F). The amphisomes typically consist of an electron-dense p62 positive body (E and F, arrowhead), and a multivesicular endosomal structure (E and F, arrow). Bars, 200 nm.
Table I.
Membrane-free p62-positive aggregates accumulate in ESCRT depleted cells
| Aggregates per cell ± SD | |
|---|---|
| Control | 0 ± 0 |
| Tsg101 siRNA | 0.095 ± 0.037* |
| Vps 24 siRNA | 0.100 ± 0.050* |
The number of p62-positive aggregates in control, Tsg101, and Vps24 siRNA treated cells was quantified by counting approximately 200 randomly chosen cell profiles per group from 3 different experiments. *, P ≤ 0.05 according to t test.
Collectively, our EM and IF analyses indicate that autophagosomes and amphisomes accumulate in ESCRT-depleted cells, but also clusters of endosomal and autophagic vesicles and membrane-free aggregates are formed.
Ubiquitin- and p62-positive aggregates accumulate in CHMP2B mutant cells
Mutations in the ESCRT-III subunit CHPM2B were recently linked to FTD (Skibinski et al., 2005) and ALS (Parkinson et al., 2006). Sequencing of CHMP2B in a Danish pedigree with autosomal-dominant FTD identified a G-to-C transition in the acceptor splice site of exon 6 in CHMP2B in affected individuals, generating two aberrant transcripts. One transcript contained the 201-bp intronic sequence in between exon 5 and 6 (CHMP2BIntron5), resulting in a premature stop codon, and thus a 36-amino acid C-terminal truncation. The other had a 10-bp deletion due to use of a cryptic splice site located 10 bp from the 5′ end of exon 6 (CHMP2BΔ10), leading to the final 36 amino acids of CHMP2B being replaced with an abnormal 29-amino acid C terminus (Skibinski et al., 2005).
The patient brains from this family contain ubiquitin and p62-positive inclusions (Holm et al., 2007) and therefore, one hypothesis that could explain the neurodegenerative phenotype seen in these patients is that mutations in CHMP2B result in repressed autophagic degradation. To investigate this hypothesis, HeLa cells were transfected with myc-tagged wild-type or mutant CHMP2B, stained with antibodies against myc, ubiquitin, and p62, and analyzed by confocal microscopy. As can be seen in Fig. 7 A, untransfected cells and cells expressing wild-type CHMP2B showed weak staining for ubiquitin and p62 on small cytoplasmic structures. In contrast, the levels of ubiquitin and p62 were strongly increased (20–40 times) in cells expressing CHMP2BIntron5 (Fig. 7, B and D) and CHMP2BΔ10 (Fig. 7, C and D). Increased p62 levels were also detected by immunoblotting in cells expressing CHMP2BIntron5 compared with mock-transfected cells or cells transfected with wild-type CHMP2B (Fig. 7 E). Although the transfection efficiency was below 30%, the total expression levels of CHMP2Bwt and CHMP2BIntron5 were equal (Fig. 7 E). p62 was also found to accumulate in the Triton-X-100 insoluble fraction in CHMP2BIntron5-expressing cells, as determined by differential detergent extraction (unpublished data). As in ESCRT-depleted cells, increased levels of GFP-LC3 were seen in HeLa GFP-LC3 cells expressing CHMP2B mutants (Fig. S5, B and C, available at http://www.jcb.org/cgi/content/full/jcb.200702115/DC1) compared with untransfected cells and cells expressing wild-type CHMP2B (Fig. S5 A).
Figure 7.

Ubiquitin and p62 positive structures accumulate in cells expressing CHMP2 mutants. HeLa cells transfected with myc-tagged wild-type CHMP2B (A), CHMP2BIntron5 (B), or CHMP2BΔ10 (C) were labeled with antibodies against c-Myc (red), ubiquitin (green), and p62 (blue) and analyzed by confocal microscopy. Single channel images in black and white are shown. Bars,10 μm. (D) Total levels of p62 in these cells were quantified in 20 cells from three independent experiments using the Zeiss LSM 510 Meta software and the average normalized to the average p62 level in control (mock transfected) cells. Error bars = SEM. (E) Western blot analysis showing accumulation of p62 in cells transfected with myc-CHMP2BIntron5 compared with mock-transfected cells (control) and cells transfected with CHMP2Bwt. (F) Immuno-EM showing p62-positive aggregates containing dense clusters of vesicular–tubular structures in cells transfected with myc-CHMP2BIntron5. Bar, 200 nm.
We then used immuno-EM to further characterize the morphology of p62-positive structures formed in cells expressing CHMP2B mutants. In cells depleted of Tsg101 and Vps24, p62-positive autophagosomes, amphisomes, clusters of small vesicular–tubular elements, and membrane-free dense p62-positive cytosolic aggregates were found (Fig. 6). Similar p62-positive structures were detected in cells expressing the CHMP2BIntron5 mutant and a p62-positive cluster of small vesicular–tubular elements can be seen in Fig. 7 F. Collectively, our results indicate that expression of CHMP2B mutants inhibit autophagic degradation, leading to accumulation of ubiquitin, p62, and GFP-LC3.
TDP-43 accumulates in cytoplasmic inclusions in ESCRT-depleted cells
FTLD-U and ALS are characterized by abnormal accumulation of p62- and ubiquitin-positive, tau- and α-synuclein-negative neuronal cytoplasmic inclusions, and TDP-43 was recently identified as the major ubiquitinated protein of these diseases (Arai et al., 2006; Neumann et al., 2006). We therefore asked whether TDP-43 accumulates in cytoplasmic aggregates in HeLa cells depleted of Tsg101 or Vps24. In control cells, TDP-43 was mainly detected in the nucleus and in a few cells also in small cytoplasmic structures positive for p62 (Fig. 8 A). In contrast, in cells depleted of Tsg101 (Fig. 8 B) or Vps24 (Fig. 8 C), TDP-43 accumulated in aggregates that also stained positive for p62 and ubiquitin. Thus, our data show, for the first time, a link between TDP-43–positive inclusions and depletion of proteins required for MVB formation and autophagic degradation.
Figure 8.

TDP-43 accumulates in cytoplasmic ubiquitin-positive structures in Tsg101- and Vps24-depleted cells. HeLa cells transfected with control (A), Tsg101 (B), or Vps24 (C) siRNA were fixed, permeabilized, and stained with antibodies against TDP-43 (green), ubiquitin (red), and p62 (blue). Single channel images in black and white are shown. Colocalization of all proteins is indicated in white in the merged picture. Bars, 10 μm.
We did not detect TDP-43 in the cytoplasmic ubiquitin-, p62-positive structures found in cells expressing CHMP2BIntron5 and CHMP2BΔ10 (Fig. S5, E and F). This is in line with recent data showing that ubiquitin-positive inclusions in patients from the Danish family with the CHMP2B mutation are TDP-43 negative (Holm et al., 2007). This is in contrast to other cases of FTLD-U, and might suggest that two distinct effects of MVBs may be occurring in TDP-43–positive and–negative FTLD-U.
Vps24 is required for clearance of Huntingtin polyQ inclusions
Our data indicate that functional MVBs play an important role in preventing formation of ubiquitin-positive inclusions, and we next asked whether clearance of expanded polyQ inclusions associated with HD also requires functional MVBs. Conditional expression of exon 1 of Huntingtin with a pathogenic polyglutamine stretch of 65 or 103 repeats fused to monomeric CFP (Htt 65Q- or 103Q-mCFP) in HeLa and Neuro2a (N2a) cell lines was recently shown to lead to formation of inclusions that were readily cleared by autophagy within 5 d after shutting off protein expression (Yamamoto et al., 2006). The time required for aggregate clearance was comparable to the clearance observed in primary neurons generated from an inducible mouse model of HD (Yamamoto et al., 2000; Martin-Aparicio et al., 2001). We therefore used these cells to determine whether inclusion clearance still occurred in cells depleted of Vps24. This was analyzed by filter-trap experiments (amount of SDS-insoluble material) (Fig. 9, A and C) or confocal analysis (number of cells having aggregates) (Fig. 9 B). Although 25–30% of HeLa Htt103Q-mCFP transfected with control siRNA had inclusions in the absence of doxycycline (control-dox), only 5% of the control cells had visible inclusions after 3 d of dox treatment (control+dox) (Fig. 9 B). Aggregate clearance was severely reduced in cells depleted of Vps24, as 15–20% of the cells had visible Htt103Q-mCFP inclusions after 3 d of dox treatment (Vps24+dox) (Fig. 9 B). Similar results were obtained using HeLa Htt65Q-mCFP cells, as analyzed by filter-trap experiments (Fig. 9 A). Moreover, depletion of Vps24 in mouse neuronal cells (N2a Htt103Q-mCFP) also inhibited inclusion clearance compared with control cells after 3 d of dox treatment (Fig. 9 C).
Figure 9.
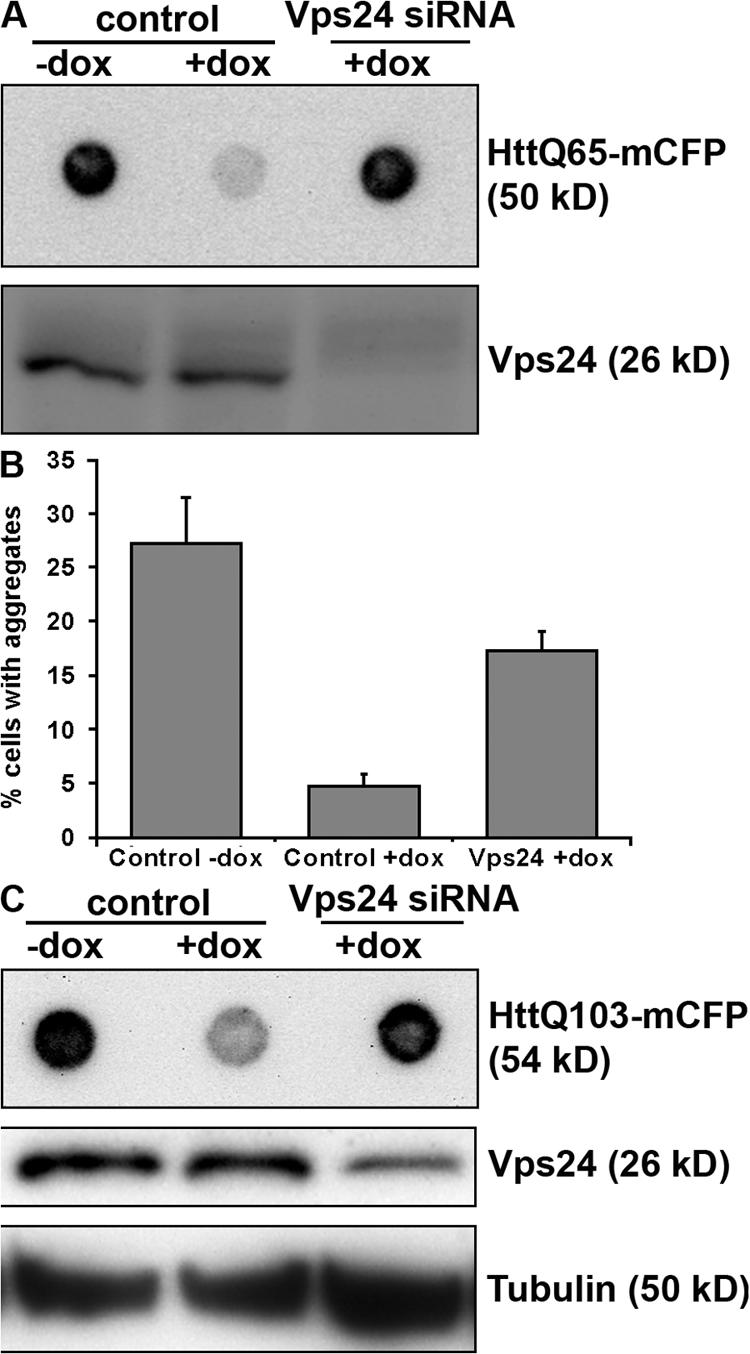
Vps24 is required for efficient clearance of Htt inclusions. HeLa cells expressing HttQ65-mCFP (A) or HttQ103-mCFP (B) and N2a cells expressing HttQ103-mCFP (C) were transfected with control or Vps24 siRNA for 2 d and then incubated in the absence or presence of doxycycline for 3 d to turn off expression of HttQ65/Q103-mCFP. Vps24 depletion was determined by Western blot analysis. (A and C) Clearance of SDS-insoluble Htt inclusions was analyzed by filter-trap assays. (B) Alternatively, aggregate clearance was analyzed by confocal quantification of the number of HeLa HttQ103 cells having visible inclusions after 3 d in the absence or presence of dox. 300 cells from three independent experiments were counted for each condition. Error bars = SD.
Together, our data indicate that clearance of Htt-positive inclusions depends on functional MVBs. This adds to previous reports showing that pathogenic Htt proteins are cleared by autophagy (Ravikumar et al., 2004; Iwata et al., 2005; Ravikumar and Rubinsztein, 2006; Yamamoto et al., 2006) and reveals an important role of ESCRT complexes in autophagic degradation of protein aggregates.
Discussion
The ESCRT complexes are involved in sorting of endocytosed ubiquitinated integral membrane proteins into the ILVs of MVBs. Their depletion in mammalian cells has been found to inhibit degradation of proteins like the EGF-R and results in MVBs with abnormal morphology (Slagsvold et al., 2006). However, little is known about the role of MVBs, and the consequence of depleting ESCRT subunits, for autophagic degradation. In this study we show that siRNA-mediated depletion of ESCRT subunits inhibits autophagic degradation, leading to accumulation of large ubiquitin-positive protein aggregates, also containing Alfy and p62, proteins known to closely associate with cytoplasmic ubiquitin-positive structures and autophagic membranes (Simonsen et al., 2004; Bjorkoy et al., 2005). We show that degradation of the autophagic membrane protein Atg8/LC3-II and the autophagic substrate p62 is inhibited and that there is a shift toward p62 insolubility, indicative of a more aggregated conformation, in cells depleted of Tsg101 and Vps24. Autophagosomes and amphisomes are formed in ESCRT-depleted cells, but the formation of autolysosomes is inhibited. This is in line with previous studies, showing that mutations in Vps class C proteins (Rieder and Emr, 1997; Lindmo et al., 2006) and the class E Vps proteins Vps4p/SKD1 (Shirahama et al., 1997; Nara et al., 2002; Besteiro et al., 2006) and CeVPS-27 (Roudier et al., 2005) impede the formation of autolysosomes. In addition, we observed clusters of small vesicular–tubular elements and membrane-free dense p62-positive cytosolic aggregates in ESCRT-depleted cells. Although we cannot exclude the possibility that ESCRTs also have non-MVB related functions in autophagy, our data strongly indicate that dysfunctional MVBs inhibit autophagic degradation, leading to formation of large aggregates that eventually may cause neurodegenerative disease.
The ESCRT-III subunit CHMP2B, forming a complex with Vps24 (Babst et al., 2002; von Schwedler et al., 2003), was recently found to be mutated in a large Danish family with familial FTD (Skibinski et al., 2005) and in patients with ALS (Parkinson et al., 2006). CHMP2B mutations are not a common cause of FTD, as several studies have failed to identify CHMP2B mutations in FTD patients (Cannon et al., 2006; Rizzu et al., 2006). We show here that cells expressing CHMP2B mutants corresponding to the mutation found in the Danish FTD patients are characterized by accumulation of ubiquitin, p62, and LC3, indicating that autophagic degradation is impeded in these cells. Our data thus suggest a possible explanation to the observed neurodegenerative phenotypes seen in CHMP2B mutant patients. Ubiquitin and p62 are common components of protein inclusions associated with neurodegenerative disease (Talbot and Ansorge, 2006) and are found in the brains of the Danish FTD patients (Holm et al., 2007). However, the ubiquitin- and p62-positive inclusions observed in the Danish FTD patient brains with the CHMP2B mutation occur at low frequency as compared with other cases of FTLD-U and are generally observed mostly in the hippocampus, which is not a site of the neurodegenerative pathology (Holm et al., 2007). This suggests that although autophagy may be impaired globally, leading to cell death, the formation of ubiquitin- and p62-positive inclusions occurs only in a subset of cells in vivo. This could be because the majority of cells with such inclusions have degenerated by the end stage of the disease that is observed in post mortem material, or that the inclusions are a protective mechanism against impaired autophagy.
Ubiquitin-positive tau-negative neuronal cytoplasmic inclusions are common pathological features in FTLD-U and ALS, and TDP-43 was recently identified as the major ubiquitinated protein in these disorders (Arai et al., 2006; Neumann et al., 2006). We here show that TDP-43 accumulates in cells depleted of Tsg101 and Vps24, suggesting that impaired MVB function could have a role in TDP-43 aggregate formation in FTLD-U and ALS. It is not clear from our experiments if TDP-43 itself is degraded via autophagy, although this could be one explanation for its accumulation. In contrast to other cases of FTLD-U, the ubiquitin-positive inclusions found in patients from the Danish CHMP2B mutant family are TDP-43 negative (Holm et al., 2007). We also failed to detect TDP-43 in the cytoplasmic ubiquitin-, p62-positive structures that accumulate in cells expressing mutant CMHP2B, suggesting that the molecular mechanisms responsible for accumulation of p62 and TDP-43 differ.
Using a cell-based system for HD, we show that ESCRTs (Vps24) are required also for efficient clearance of the Htt polyQ aggregates, both in human HeLa and mouse neuronal cells. It has previously been shown that the autophagic pathway is responsible for clearance of Htt polyQ aggregates (Ravikumar et al., 2004; Iwata et al., 2005; Ravikumar and Rubinsztein, 2006; Yamamoto et al., 2006), but our results show, for the first time, that functional MVBs and a Vps class E protein are required for efficient clearance of Htt inclusions.
Recently, it was found that mice with neuronal-specific deficiencies for Atg5 or Atg7, proteins known to be essential for autophagy, are characterized by accumulation of cytoplasmic inclusion bodies and a neurodegenerative phenotype (Hara et al., 2006; Komatsu et al., 2006). Our results indicate that depletion of ESCRT proteins likely will result in a similar phenotype, but do not allow us to conclude about when these proteins become ubiquitinated. There is little or no evidence to date showing that polyubiquitinated proteins are normally degraded by autophagy and it is probably more likely that proteins that are normally turned over by autophagy become polyubiquitinated when autophagic degradation is inhibited. However, it was recently found that induction of autophagy leads to enhanced delivery of ubiquitin to lysosomes and that this correlates with enhanced lysosomal bactericidal capacity (Alonso et al., 2007), which might support the idea that polyubiquitinated proteins are autophagic substrates. p62, containing a ubiquitin-binding UBA domain (Vadlamudi et al., 1996), would be an excellent candidate for recognition and targeting of polyubiquitin linked proteins to the autophagic pathway (Fig. 10).
Figure 10.

Model for autophagic degradation in control and ESCRT-depleted cells. In control cells, cytoplasmic cargo (proteins and organelles) is sequestered by an isolation membrane/ phagophore, forming double-membrane autophagosomes that can fuse with MVBs, forming amphisomes. Amphisomes, containing both endocytic and autophagic cargo, then fuse with lysosomes, forming autolysosomes, where the content is degraded. Autophagosomes may also fuse directly with lysosomes, although the amphisome pathway seems to be the major pathway in HeLa cells. The ESCRT complexes are required for proper sorting and degradation of ubiquitinated integral membrane proteins (e.g., EGFR) and for proper MVB morphology, and depletion of ESCRT subunits results in the formation of aberrant MVBs (morphology depending of which ESCRT subunit is depleted) (red box A). Degradation of autophagic cargo is also inhibited in ESCRT-depleted cells, proposedly because of inhibited formation of autolysosomes (red box B), although autophagosomes and amphisomes are still formed. In addition, large p62- and ubiquitin-positive membrane-free aggregates accumulate in ESCRT- depleted cells (red box C), indicating that continuous autophagic clearance of cytoplasmic proteins is important to avoid accumulation of ubiquitin-positive aggregates that may cause neurodegeneration.
In conclusion, we have shown that depletion of ESCRT subunits or overexpression of CHMP2B mutant proteins inhibit autophagic degradation, leading to accumulation of ubiquitin-positive aggregates that contain proteins associated with neurodegenerative disease. Our data indicate that functional MVBs are required to prevent accumulation of abnormal proteins that can disrupt neural function and ultimately lead to neurodegeneration.
Materials and methods
Cell culture
HeLa cell cultures were maintained as recommended by American Type Culture Collection (Manassas, VA). HeLa and N2a cells stably expressing a Tet-off inducible exon1 of Htt carrying a polyQ expansion of 65 or 103 residues, fused to monomeric enhanced CFP at the C termini (Htt 65Q- or 103Q-mCFP), were cultured as described (Yamamoto et al., 2006) and 100 ng/ml doxycycline (dox) was used to shut off production of new protein. HeLa cells stably expressing GFP-LC3 were a gift from Aviva Tolkovsky (University of Cambridge, Cambridge, UK; Bampton et al., 2005).
Antibodies
Rabbit antibodies against Hrs (Raiborg et al., 2001), Vps24 (Bache et al., 2006), and Alfy (Simonsen et al., 2004) have been described before. An antibody recognizing human Vps22 was made by injecting rabbits with recombinant Vps22 as a fusion with maltose binding protein (MBP) (Eurogenetec). The antiserum was affinity purified on Vps22-MBP Affi-Gel beads (Bio-Rad Laboratories). A mouse monoclonal antibody against Tsg101 was obtained from GeneTex. Rabbit anti-LC3 antibody was a gift from Tamotsu Yoshimori (Osaka University, Osaka, Japan). Human anti-early endosomal antigen (EEA)1 antiserum was a gift from Ban-Hock Toh (Monash University, Melbourne, Australia). Rabbit anti-lysosomal-associated membrane protein (LAMP)2 was a gift from Gillian Griffiths (University of Oxford, Oxford, UK). Mouse monoclonal anti-lyso-bisphosphatidic acid (LBPA) was provided by Jean Gruenberg (University of Geneva, Geneva, Switzerland). Guinea pig anti-p62 C-terminal antibody was from PROGEN Biotechnik GmbH. Mouse monoclonal antibodies against α-tubulin and against conjugated mono- and polyubiquitin (FK2) were from Sigma-Aldrich and Affiniti Research Products, respectively. Anti-GFP antibody was from AbCam. Rabbit anti-TDP-43 antibody was from ProteinTech Group. Rabbit anti- c-Myc antibody was from Santa Cruz Biotechnology, Inc. Cy2-, Cy3-, and Cy5-labeled secondary antibodies were from Jackson ImmunoResearch Laboratories.
Transfection of siRNA oligonucleotides and plasmids
The following previously described siRNA oligonucleotides were used: Hrs (Bache et al., 2003), Tgs101 (Bishop et al., 2002), Vps24 (Bache et al., 2006), and control (Cabezas et al., 2005). Vps22 was depleted by the siRNA duplex: sense 5′-CUUGCAGAGGCCAAGUAUA-3′ and antisense 5′-UAUACUUGGCCUCUGCAAG-3′ (MWG-Biotech). Results were confirmed by the use of ON-TARGETplus SMARTpool siRNA (Dharmacon) against human Hrs, Tsg101, and Vps22. ON-TARGETplus SMARTpool siRNA against human Atg5 and against mouse Vps24 were also used. Transfection of HeLa cells with siRNA oligonucleotides was performed as described previously (Bache et al., 2003). In brief, the cells were transfected with 40–100 nM siRNA using Oligofectamine (Invitrogen) for 3 d; the cells were then replated and left for another 2 d before experiments were performed. Specific protein knockdown was demonstrated by running equal amounts of cell lysate on SDS-PAGE, followed by Western blotting using antibodies against Hrs, Tsg101, Vps22, or Vps24.
HeLa cells were transfected with mCherry-GFP-LC3 after 4 d of siRNA transfection and incubated for another 24 h before analysis by confocal microscopy. Transfection of HeLa cells with cDNA encoding myc-tagged wild-type CHMP2B, CHMP2BIntron5, or CHMP2BΔ10 in pLNCX2 (CLONTECH Laboratories, Inc.), or mCherry-GFP-LC3 was performed using FuGene6 (Roche), according to the manufacturer's instruction.
Differential detergent extraction and Western blot analysis
To analyze the cellular levels of different proteins and their solubility, cells were first extracted in ice-cold lysis buffer (50 mM NaCl, 10 mM Tris, 5 mM EDTA, 0.1% SDS, and 1% Triton X-100 + protease and phosphatase inhibitor cocktails), centrifuged (14,000 rpm) for 10 min, and the supernatants (soluble fraction) collected. The remaining protein pellets were washed with phosphate-buffered saline (PBS) before extraction with 2% SDS-containing sample buffer (insoluble fraction). Protein concentrations in the soluble fractions were determined and ∼20 μg of protein per sample was loaded and resolved on 15% or 4–20% gradient gels (Pierce Chemical Co.) followed by electro-blotting to Immobilon-P membranes (Millipore). The blots were probed with specific antibodies, which were detected using standard ECL reagents. The intensities of the of the different bands obtained were quantified using the software provided by the ChemiGenius imaging system (Syngene) and relative amounts quantified using tubulin as a loading control. 200 nM Bafilomycin A was added for 8 h to inhibit lysosomal degradation where indicated.
Confocal immunofluorescence microscopy
HeLa cells grown on coverslips, transfected or not with the indicated siRNA or plasmid, were fixed in 3% paraformaldehyde, permeabilized with 0.05% saponin, and stained for fluorescence microscopy as described previously (Simonsen et al., 1998). Coverslips were examined using a microscope (LSM 510 META; Carl Zeiss MicroImaging, Inc.) equipped with a Neo-Fluar 100×/1.45 oil immersion objective. Image processing and analysis were done with Zeiss LSM 510 software version 3.2, ImageJ (National Institutes of Health, Bethesda, MD; http://rsb.info.nih.gov/ij/, 1997–2007), and Adobe Photoshop 7.0.
Electron microscopy
HeLa cells, transfected or not with the indicated siRNA or plasmid, were fixed in 4% formaldehyde/0.2% gluteraldehyde in 0.1 M phosphate buffer at room temperature for 40 min, washed, scraped, and pelleted in 12% gelatin at 10,000 rpm. Specimens were infiltrated with 2.3 M sucrose, mounted on silver pins, and frozen in liquid nitrogen. Ultrathin cryosections were cut at −110°C (EM FCS ultramicrotome; Leica) and collected with a 1:1 mixture of 2% methyl cellulose and 2.3 M sucrose. Sections were transferred to formvar/carbon-coated grids and labeled with antibodies against p62 or GFP and LBPA, followed by Protein A conjugates essentially as described (Slot et al., 1991). Sections were observed at 60–80 kV using a Philips CM10 and a JEOL JEM-1230 electron microscope, equipped with a SIS Megaview 3 or Morada camera, respectively. Quantification of labeling was performed by counting gold particles on randomly chosen cell profiles. The number of p62-positive aggregates was counted in ∼200 cells per group (control, Tsg101-, and Vps24-siRNA) from three different experiments.
Degradation of long-lived proteins in response to amino acid deprivation
HeLa cells treated with control or siRNA against Tsg101 or hVps24 were incubated for 24 h with 0.25 μCi/ml l-[14C] valine–supplemented media. Cells were rinsed three times with PBS to remove unincorporated radioisotopes and then chased in fresh complete media containing 10 mM cold valine for 2 h to allow degradation of short-lived proteins. Cells were rinsed in HBSS + 10 mM Hepes and incubated for 4 h with either complete media or HBSS + 10 mM Hepes + 10 mM Valine ± 10 mM 3-methyl adenine (3-MA) (Sigma-Aldrich). Cells were then scraped and, using TCA, protein was precipitated from both the incubation media and the cells. Proteolysis was assessed as the acid-soluble radioactivity divided by the radioactivity maintained in the precipitate.
Promoter shutdown experiments and filter-trap assay
48 h after siRNA transfection, HeLa Htt65Q- or 103Q-mCFP cells or N2a Htt103Q-mCFP cells were exposed to 100 ng/ml dox for another 3 d to shut down production of new Htt65Q/103Q-mCFP protein and permit more than 50% of clearance to occur (Yamamoto et al., 2006). The effect of siRNA treatment on polyQ aggregate clearance was analyzed by using the membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates, according to the published protocol (Scherzinger et al., 1997; Wanker et al., 1999). An anti-GFP antibody was used to detect 65Q/103QmCFP. The total protein load was normalized to the volume of the soluble fraction. Alternatively, clearance of Htt103QmCFP aggregates was analyzed by confocal immunofluorescence microscopy. The percentage of cell having aggregates was quantified by counting 300 cells for each condition from three independent experiments.
Protein synthesis assay
Cells were incubated for 2 h in Hepes medium lacking leucine. The cells were then incubated in Hepes medium containing 2 μCi ml−1 [3H]-leucine for 20 min at 37°C. Cells were extracted with 5% trichloroacetic acid (TCA) for 10 min, followed by a wash (5 min) in 5% TCA and subsequently dissolved in 0.1 M KOH. The cell-associated radioactivity was measured.
Proteasome assay
Proteasome activity was analyzed using the Proteasome-Glo Cell-based assay (Promega), which measures the chymotrypsin-like protease activity associated with the proteasome complex, according to the manufacturer's instruction. The proteasome inhibitor PSI (50 μM) was added 5 h before the analysis.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from 5 × 105 HeLa cells using the Aurum Total RNA mini kit (Bio-Rad Laboratories), according to the manufacturer's instructions. Purity and quantity were measured by optical density. 1 μg total RNA was used for cDNA synthesis using the iScript cDNA Synthesis kit (Bio-Rad Laboratories). Real-time PCR was performed in parallel 20-μl reactions containing 10 μl 2× QuantiTect SYBR Green PCR master mix (QIAGEN), 2 μl 10× QuantiTect Primer Assay (QIAGEN), and 20 ng cDNA (2 ng were used for actin) in 96-well optical plates. The cycling conditions for the LightCycler480 (Roche) were 95°C for 15 min, 40 cycles of 94°C 15 s, 55°C 20 s, and 72°C 20 s The following prevalidated QuantiTect Primer Assays were used: Hs_SQSTM1_1_SG, Hs_MAP1LC3B_1_SG, Hs_ACTB_1_SG, Hs_TBP_1_SG. Real-time efficiencies were calculated from the slopes of the standard dilution curves. RNA transcription levels were determined by the method of direct comparison of C T values (C T > 35 rejected) and relative quantities calculated by the ΔΔC T equation or transformed into linear form by 2−ΔΔCt. Transcripts were normalized to the quantity of actin and TBP for each condition.
Online supplemental material
Fig. S1 shows single channel images of the insets in Fig. 1. Fig. S2 shows that neither protein synthesis, transcription, nor proteasome activity is drastically affected by depletion of ESCRT subunits. Fig. S3 shows that both the number and size of p62-positive structures increase strongly in cells depleted of Atg5. Fig. S4 shows increased colocalization of Alfy and LBPA in HeLa cells depleted of ESCRT subunits. Fig. S5 shows that p62 and GFP-LC3, but not TDP-43 accumulate in cells expressing CHMP2B mutants. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200702115/DC1.
Acknowledgments
We are very grateful to Aviva Tolkovsky for the gift of HeLa GFP-LC3 cells and to Terje Johansen for providing the mCherry-GFP-LC3 construct before publication.
This work was supported by grants from the Research Council of Norway, the Norwegian Cancer Society, and the Hartmann Family Foundation.
S. Stuffers and C. Raiborg contributed equally to this paper.
Abbreviations used in this paper: Alfy, autophagy-linked FYVE protein; ALS, amyotrophic lateral sclerosis; Atg, autophagy defective; CHMP2B, charged multivesicular body protein 2B/chromatin-modifying protein 2B; dox, doxycycline; EEA1, early endosome antigen 1; ESCRT, endosomal sorting complex required for transport; FTD, frontotemporal dementia; FTLD-U, frontotemporal lobar degeneration with ubiquitin deposits; HD, Huntington's disease; Hrs, hepatocyte growth factor–regulated tyrosine kinase substrate; Htt, Huntingtin; ILV, intraluminal vesicles; LBPA, lyso-bisphosphatidic acid; LC3, light chain 3; MVB, multivesicular body; TDP-43, TAR DNA-binding protein 43; Tsg101, tumor susceptibility gene 101; Vps, vacuolar protein sorting.
References
- Alonso, S., K. Pethe, D.G. Russell, and G.E. Purdy. 2007. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc. Natl. Acad. Sci. USA. 104:6031–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai, T., M. Hasegawa, H. Akiyama, K. Ikeda, T. Nonaka, H. Mori, D. Mann, K. Tsuchiya, M. Yoshida, Y. Hashizume, and T. Oda. 2006. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351:602–611. [DOI] [PubMed] [Google Scholar]
- Babst, M., D.J. Katzmann, E.J. Estepa-Sabal, T. Meerloo, and S.D. Emr. 2002. Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev. Cell. 3:271–282. [DOI] [PubMed] [Google Scholar]
- Bache, K.G., C. Raiborg, A. Mehlum, and H. Stenmark. 2003. STAM and Hrs are subunits of a multivalent ubiquitin-binding complex on early endosomes. J. Biol. Chem. 278:12513–12521. [DOI] [PubMed] [Google Scholar]
- Bache, K.G., S. Stuffers, L. Malerod, T. Slagsvold, C. Raiborg, D. Lechardeur, S. Walchli, G.L. Lukacs, A. Brech, and H. Stenmark. 2006. The ESCRT-III subunit hVps24 is required for degradation but not silencing of the epidermal growth factor receptor. Mol. Biol. Cell. 17:2513–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bampton, E.T., C.G. Goemans, D. Niranjan, N. Mizushima, and A.M. Tolkovsky. 2005. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 1:23–36. [DOI] [PubMed] [Google Scholar]
- Bence, N.F., R.M. Sampat, and R.R. Kopito. 2001. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 292:1552–1555. [DOI] [PubMed] [Google Scholar]
- Berg, T.O., M. Fengsrud, P.E. Stromhaug, T. Berg, and P.O. Seglen. 1998. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J. Biol. Chem. 273:21883–21892. [DOI] [PubMed] [Google Scholar]
- Besteiro, S., R.A. Williams, L.S. Morrison, G.H. Coombs, and J.C. Mottram. 2006. Endosome sorting and autophagy are essential for differentiation and virulence of Leishmania major. J. Biol. Chem. 281:11384–11396. [DOI] [PubMed] [Google Scholar]
- Bishop, N., A. Horman, and P. Woodman. 2002. Mammalian class E vps proteins recognize ubiquitin and act in the removal of endosomal protein-ubiquitin conjugates. J. Cell Biol. 157:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy, G., T. Lamark, A. Brech, H. Outzen, M. Perander, A. Overvatn, H. Stenmark, and T. Johansen. 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas, A., K.G. Bache, A. Brech, and H. Stenmark. 2005. Alix regulates cortical actin and the spatial distribution of endosomes. J. Cell Sci. 118:2625–2635. [DOI] [PubMed] [Google Scholar]
- Cannon, A., M. Baker, B. Boeve, K. Josephs, D. Knopman, R. Petersen, J. Parisi, D. Dickison, J. Adamson, J. Snowden, et al. 2006. CHMP2B mutations are not a common cause of frontotemporal lobar degeneration. Neurosci. Lett. 398:83–84. [DOI] [PubMed] [Google Scholar]
- Gordon, P.B., and P.O. Seglen. 1988. Prelysosomal convergence of autophagic and endocytic pathways. Biochem. Biophys. Res. Commun. 151:40–47. [DOI] [PubMed] [Google Scholar]
- Hara, T., K. Nakamura, M. Matsui, A. Yamamoto, Y. Nakahara, R. Suzuki-Migishima, M. Yokoyama, K. Mishima, I. Saito, H. Okano, and N. Mizushima. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 441:885–889. [DOI] [PubMed] [Google Scholar]
- Harvey, R.J., M. Skelton-Robinson, and M.N. Rossor. 2003. The prevalence and causes of dementia in people under the age of 65 years. J. Neurol. Neurosurg. Psychiatry. 74:1206–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm,I.E., E.Englund, I.R.A.Mackenzie, P.Johannsen, and A.M.Isaacs. 2007. A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3 (FTD-3). J. Neuropathol. Exp. Neurol. 66:884–891. [DOI] [PubMed] [Google Scholar]
- Hurley, J.H., and S.D. Emr. 2006. The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu. Rev. Biophys. Biomol. Struct. 35:277–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata, A., J.C. Christianson, M. Bucci, L.M. Ellerby, N. Nukina, L.S. Forno, and R.R. Kopito. 2005. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc. Natl. Acad. Sci. USA. 102:13135–13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana, N.R., E.A. Zemskov, G. Wang, and N. Nukina. 2001. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 10:1049–1059. [DOI] [PubMed] [Google Scholar]
- Kabeya, Y., N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi, and T. Yoshimori. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya, Y., N. Mizushima, A. Yamamoto, S. Oshitani-Okamoto, Y. Ohsumi, and T. Yoshimori. 2004. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 117:2805–2812. [DOI] [PubMed] [Google Scholar]
- Klionsky, D.J., and Y. Ohsumi. 1999. Vacuolar import of proteins and organelles from the cytoplasm. Annu. Rev. Cell Dev. Biol. 15:1–32. [DOI] [PubMed] [Google Scholar]
- Klionsky, D.J., and S.D. Emr. 2000. Autophagy as a regulated pathway of cellular degradation. Science. 290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky, D.J., J.M. Cregg, W.A. Dunn Jr., S.D. Emr, Y. Sakai, I.V. Sandoval, A. Sibirny, S. Subramani, M. Thumm, M. Veenhuis, and Y. Ohsumi. 2003. A unified nomenclature for yeast autophagy-related genes. Dev. Cell. 5:539–545. [DOI] [PubMed] [Google Scholar]
- Komatsu, M., S. Waguri, T. Chiba, S. Murata, J. Iwata, I. Tanida, T. Ueno, M. Koike, Y. Uchiyama, E. Kominami, and K. Tanaka. 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 441:880–884. [DOI] [PubMed] [Google Scholar]
- Kuma, A., M. Matsui, and N. Mizushima. 2007. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 3:323–328. [DOI] [PubMed] [Google Scholar]
- Lindmo, K., A. Simonsen, A. Brech, K. Finley, T.E. Rusten, and H. Stenmark. 2006. A dual function for Deep orange in programmed autophagy in the Drosophila melanogaster fat body. Exp. Cell Res. 312:2018–2027. [DOI] [PubMed] [Google Scholar]
- Liou, W., H.J. Geuze, M.J. Geelen, and J.W. Slot. 1997. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J. Cell Biol. 136:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Aparicio, E., A. Yamamoto, F. Hernandez, R. Hen, J. Avila, and J.J. Lucas. 2001. Proteasomal-dependent aggregate reversal and absence of cell death in a conditional mouse model of Huntington's disease. J. Neurosci. 21:8772–8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima, N., A. Yamamoto, M. Hatano, Y. Kobayashi, Y. Kabeya, K. Suzuki, T. Tokuhisa, Y. Ohsumi, and T. Yoshimori. 2001. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nara, A., N. Mizushima, A. Yamamoto, Y. Kabeya, Y. Ohsumi, and T. Yoshimori. 2002. SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Struct. Funct. 27:29–37. [DOI] [PubMed] [Google Scholar]
- Neary, D., J. Snowden, and D. Mann. 2005. Frontotemporal dementia. Lancet Neurol. 4:771–780. [DOI] [PubMed] [Google Scholar]
- Neumann, M., D.M. Sampathu, L.K. Kwong, A.C. Truax, M.C. Micsenyi, T.T. Chou, J. Bruce, T. Schuck, M. Grossman, C.M. Clark, et al. 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 314:130–133. [DOI] [PubMed] [Google Scholar]
- Pankiv, S., T.H. Clausen, T. Lamark, A. Brech, J.A. Bruun, H. Outzen, A. Overvatn, G. Bjorkoy, and T. Johansen. 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282:24131–24145. [DOI] [PubMed] [Google Scholar]
- Parkinson, N., P.G. Ince, M.O. Smith, R. Highley, G. Skibinski, P.M. Andersen, K.E. Morrison, H.S. Pall, O. Hardiman, J. Collinge, et al. 2006. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology. 67:1074–1077. [DOI] [PubMed] [Google Scholar]
- Punnonen, E.L., S. Autio, H. Kaija, and H. Reunanen. 1993. Autophagic vacuoles fuse with the prelysosomal compartment in cultured rat fibroblasts. Eur. J. Cell Biol. 61:54–66. [PubMed] [Google Scholar]
- Raiborg, C., B. Bremnes, A. Mehlum, D.J. Gillooly, A. D'Arrigo, E. Stang, and H. Stenmark. 2001. FYVE and coiled-coil domains determine the specific localisation of Hrs to early endosomes. J. Cell Sci. 114:2255–2263. [DOI] [PubMed] [Google Scholar]
- Ratnavalli, E., C. Brayne, K. Dawson, and J.R. Hodges. 2002. The prevalence of frontotemporal dementia. Neurology. 58:1615–1621. [DOI] [PubMed] [Google Scholar]
- Ravikumar, B., and D.C. Rubinsztein. 2004. Can autophagy protect against neurodegeneration caused by aggregate-prone proteins? Neuroreport. 15:2443–2445. [DOI] [PubMed] [Google Scholar]
- Ravikumar, B., and D.C. Rubinsztein. 2006. Role of autophagy in the clearance of mutant huntingtin: a step towards therapy? Mol. Aspects Med. 27:520–527. [DOI] [PubMed] [Google Scholar]
- Ravikumar, B., C. Vacher, Z. Berger, J.E. Davies, S. Luo, L.G. Oroz, F. Scaravilli, D.F. Easton, R. Duden, C.J. O'Kane, and D.C. Rubinsztein. 2004. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36:585–595. [DOI] [PubMed] [Google Scholar]
- Rieder, S.E., and S.D. Emr. 1997. A novel RING finger protein complex essential for a late step in protein transport to the yeast vacuole. Mol. Biol. Cell. 8:2307–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzu, P., S.E. van Mil, B. Anar, S.M. Rosso, L.D. Kaat, P. Heutink, and J.C. van Swieten. 2006. CHMP2B mutations are not a cause of dementia in Dutch patients with familial and sporadic frontotemporal dementia. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 141:944–946. [DOI] [PubMed] [Google Scholar]
- Roudier, N., C. Lefebvre, and R. Legouis. 2005. CeVPS-27 is an endosomal protein required for the molting and the endocytic trafficking of the low-density lipoprotein receptor-related protein 1 in Caenorhabditis elegans. Traffic. 6:695–705. [DOI] [PubMed] [Google Scholar]
- Scherzinger, E., R. Lurz, M. Turmaine, L. Mangiarini, B. Hollenbach, R. Hasenbank, G.P. Bates, S.W. Davies, H. Lehrach, and E.E. Wanker. 1997. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 90:549–558. [DOI] [PubMed] [Google Scholar]
- Shirahama, K., T. Noda, and Y. Ohsumi. 1997. Mutational analysis of Csc1/Vps4p: involvement of endosome in regulation of autophagy in yeast. Cell Struct. Funct. 22:501–509. [DOI] [PubMed] [Google Scholar]
- Simonsen, A., R. Lippe, S. Christoforidis, J.M. Gaullier, A. Brech, J. Callaghan, B.H. Toh, C. Murphy, M. Zerial, and H. Stenmark. 1998. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 394:494–498. [DOI] [PubMed] [Google Scholar]
- Simonsen, A., H.C. Birkeland, D.J. Gillooly, N. Mizushima, A. Kuma, T. Yoshimori, T. Slagsvold, A. Brech, and H. Stenmark. 2004. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J. Cell Sci. 117:4239–4251. [DOI] [PubMed] [Google Scholar]
- Skibinski, G., N.J. Parkinson, J.M. Brown, L. Chakrabarti, S.L. Lloyd, H. Hummerich, J.E. Nielsen, J.R. Hodges, M.G. Spillantini, T. Thusgaard, et al. 2005. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37:806–808. [DOI] [PubMed] [Google Scholar]
- Slagsvold, T., K. Pattni, L. Malerod, and H. Stenmark. 2006. Endosomal and non-endosomal functions of ESCRT proteins. Trends Cell Biol. 16:317–326. [DOI] [PubMed] [Google Scholar]
- Slot, J.W., H.J. Geuze, S. Gigengack, G.E. Lienhard, and D.E. James. 1991. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J. Cell Biol. 113:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder, H., K. Mensah, C. Theisler, J. Lee, A. Matouschek, and B. Wolozin. 2003. Aggregated and monomeric alpha-synuclein bind to the S6' proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 278:11753–11759. [DOI] [PubMed] [Google Scholar]
- Talbot, K., and O.Ansorge. 2006. Recent advances in the genetics of amyotrophic lateral sclerosis and frontotemporal dementia: common pathways in neurodegenerative disease. Hum. Mol. Genet. 15:R182–R187. [DOI] [PubMed] [Google Scholar]
- Tooze, J., M. Hollinshead, T. Ludwig, K. Howell, B. Hoflack, and H. Kern. 1990. In exocrine pancreas, the basolateral endocytic pathway converges with the autophagic pathway immediately after the early endosome. J. Cell Biol. 111:329–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urushitani, M., J. Kurisu, K. Tsukita, and R. Takahashi. 2002. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J. Neurochem. 83:1030–1042. [DOI] [PubMed] [Google Scholar]
- Vadlamudi, R.K., I. Joung, J.L. Strominger, and J. Shin. 1996. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J. Biol. Chem. 271:20235–20237. [DOI] [PubMed] [Google Scholar]
- von Schwedler, U.K., M. Stuchell, B. Muller, D.M. Ward, H.Y. Chung, E. Morita, H.E. Wang, T. Davis, G.P. He, D.M. Cimbora, et al. 2003. The protein network of HIV budding. Cell. 114:701–713. [DOI] [PubMed] [Google Scholar]
- Wanker, E.E., E. Scherzinger, V. Heiser, A. Sittler, H. Eickhoff, and H. Lehrach. 1999. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol. 309:375–386. [DOI] [PubMed] [Google Scholar]
- Williams, R.L., and S. Urbe. 2007. The emerging shape of the ESCRT machinery. Nat. Rev. Mol. Cell Biol. 8:355–368. [DOI] [PubMed] [Google Scholar]
- Yamamoto, A., J.J. Lucas, and R. Hen. 2000. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 101:57–66. [DOI] [PubMed] [Google Scholar]
- Yamamoto, A., M.L. Cremona, and J.E. Rothman. 2006. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J. Cell Biol. 172:719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]