

Abstract
Adhesion to the extracellular matrix regulates numerous changes in the actin cytoskeleton by regulating the activity of the Rho family of small GTPases. Here, we report that adhesion, and the associated changes in cell shape and cytoskeletal tension, are all required for GTP-bound RhoA to activate its downstream effector, ROCK. Using an in vitro kinase assay for endogenous ROCK, we found that cells in suspension, attached on substrates coated with low density fibronectin, or on spreading-restrictive micropatterned islands all exhibited low ROCK activity and correspondingly low myosin light chain phosphorylation, in the face of high levels of GTP-bound RhoA. In contrast, allowing cells to spread against substrates rescued ROCK and myosin activity. Interestingly, inhibition of tension with cytochalasin D or blebbistatin also inhibited ROCK activity within 20 minutes. The abrogation of ROCK activity by cell detachment or inhibition of tension could not be rescued by constitutively active RhoA-V14. These results suggest the existence of a feedback loop between cytoskeletal tension, adhesion maturation, and ROCK signaling that likely contributes to numerous mechanochemical processes.
Keywords: Rho; ROCK; kinase assay; myosin phosphorylation; cell shape; focal adhesions, FAK, hypertension
INTRODUCTION
Contractile tension generated within the actin cytoskeleton by myosin II is emerging as a key player in many cellular processes, from the stabilization of cell-matrix adhesions to the modulation of gene expression, cell proliferation, and differentiation [1-5]. A principal mediator of cytoskeletal tension is the small GTPase RhoA and its downstream effector Rho-associated kinase (ROCK). GTP binding and hydrolysis switches RhoA between a GTP-bound active and a GDP-bound inactive state [6]. The conformationally active RhoA propagates downstream signals in turn by binding to effector proteins such as ROCK. ROCK, through the phosphorylation and deactivation of the myosin binding subunit of myosin phosphatase (MYPT1), and direct phosphorylation of myosin light chain (MLC), leads to contractile force generation [7].
Integrin mediated adhesion to the extracellular matrix (ECM) has been shown to regulate RhoA signaling [8, 9]. It has been shown that placing cells in suspension leads to increased GTP-RhoA levels, [10] and replating cells on ECM leads to a transient down regulation of RhoA activity followed by a slow recovery [11]. While it has largely been thought that adhesion regulates the RhoA-ROCK-tension pathway through its effects on RhoA [8, 9], evidence suggests the possibility that the coupling of RhoA to ROCK activity also may be an important control point. For example, despite high levels of GTP-RhoA [11], suspended cells exhibit low MLC phosphorylation [2][10]. Similarly, while cytochalasin D treatment has long been known to decrease MLC phosphorylation [12], it has also been shown to increase cellular GTP-RhoA [11]. Thus while high RhoA activity is often used to indirectly implicate high contractility, RhoA activity may in certain circumstances actually be decoupled from myosin-based contractility. Decoupling between RhoA and another effector, mDia, has recently been reported [13]. Elimination of signaling by FAK, a principal mediator of integrin signaling, abrogates mDia-mediated stabilization of microtubules at the leading edge despite the presence of active RhoA [13]. Furthermore, the related Rho family GTPase, Rac, requires adhesion to couple to its effector PAK. By recruiting cholesterol rich lipid rafts to the plasma membrane, integrin ligation localizes active Rac to the membrane, permitting Rac to activate PAK [14]. Together, these findings suggest that RhoA-mediated ROCK activity may also be regulated by adhesion.
Interestingly, RhoA signaling not only is modulated by cell-ECM adhesion, but also acts as an important regulator of adhesion. Adhesion to the ECM involves numerous interrelated processes including integrin binding, cell spreading and flattening against the substrate, and condensation of integrins to form large structures known as focal adhesions. RhoA-ROCK mediated cytoskeletal tension appears to limit the degree to which cells spread and is critical for focal adhesion maturation and signaling [15-17], and ROCK-generated tension may feedback to regulate RhoA at least in cells on collagen gels [3]. These data highlight the possible existence of feedback and feed forward regulatory loops that, if present, would play a critical role in how cells adaptively alter adhesion, morphology, and mechanics in response to their ECM environment.
In this study, we investigated the possibility that both adhesion and tension are involved in regulating RhoA, ROCK and myosin activity. We examined how different aspects of adhesion (integrin ligation, cell spreading, focal adhesion formation and cytoskeletal tension) modulated RhoA-ROCK signaling. Our findings demonstrate that cell adhesion and spreading are required for RhoA to activate ROCK, and that this direct modulation of ROCK activity by adhesion requires traction forces mediated by cytoskeletal tension. Thus, full myosin activation by RhoA requires adhesion to establish a positive feedback loop that links RhoA-myosin signaling and force-mediated adhesion maturation. This control system provides an explanation for how adhesion, cell mechanics, and RhoA GTPase signaling are so closely intertwined in many mechanotransduction processes.
MATERIALS AND METHODS
Cell culture and reagents
Primary bovine pulmonary artery endothelial cells (VEC Technologies, Rensselaer, NY) were grown in DMEM supplemented with 5% bovine serum, 100U/ml penicillin, and 100μg/ml streptomycin (all from Invitrogen). Cells were used between passages 5 to 8. Human umbilical vein endothelial cells (gift from Dr. Guillermo Garcia-Cardena, Harvard Medical School) were grown in M199 medium (Cambrex) supplemented with 20% FBS (Invitrogen), 50μg/ml endothelial mitogen (Biomedical Technologies), 100μg/ml heparin (Sigma), penicillin and streptomycin. Cells were used between passages 5 to 7. Other reagents used were: fibronectin (BD Biosciences); BSA (Serologicals Corporation); anti-phospho-myosin light chain 2 (Thr18/Ser19) (Cell Signaling Technologies), anti-nonmuscle myosin II heavy chain (Biomedical Technologies); recombinant MYPT1 fragment, anti-ROCK-2, anti-phospho MYPT1 Thr 696, Rhotekin agarose beads (all from Upstate); anti-RhoA (Santa Cruz), anti-vinculin, protease and phosphatase inhibitor cocktails (all from Sigma), protein-G sepharose beads (Amersham), ATP (Sigma), Blebbistatin (25μM; Tocris), cytochalasin D (2 μg/ml; Sigma); TRITC-phalloidin (Molecular Probes); NaCl, EDTA, MgCl2, glycerol (all from JT Baker); HEPES (Gibco); NP-40 (Igepal CA-630), leupeptin, pepstatin, aprotinin (all from Sigma). The RhoA-V14 adenovirus was prepared and used as previously described [4].
Manipulation of cell adhesion
For experiments using cells in suspension, cells were plated on petri dishes coated with 25μg/ml of fibronectin (FN) and allowed to spread for 3h at which time they were trypsinized and maintained in suspension with periodic gentle shaking in polystyrene tubes in the incubator for 1h. ECM density was varied for some experiments by varying the concentration of the FN coating solution, as indicated in the text. Micropatterned substrates were generated on PDMS (polydimethylsiloxane)-coated petri dishes by using a protein stamping technique described before [18].
Measurement of cytoskeletal tension
Traction forces were measured by using microfabricated substrates previously described [19]. Briefly, cells were plated on microfabricated elastomeric posts on which they attach and spread, applying tractions forces to the posts. The resulting bending of the posts was measured and converted to a traction force. Fifteen cells were quantified for each condition, averaged over 3 different samples.
Immunofluorescence microscopy
Cells were washed twice with PBS and fixed with 3.7% paraformaldehyde for 30 minutes at room temperature. F-actin was visualized by staining with TRITC-phalloidin and epifluorescence images were captured on a Nikon TE2000 microscope using an Orca digital camera (Hamamatsu). Focal adhesions were quantified by vinculin staining and digital image analysis as previously described, including only adhesions larger than 0.25 μm2 [17].
ROCK activity assay
Endogenous ROCK kinase activity in immunoprecipitated ROCK was measured by an in vitro kinase assay using a recombinant fragment (aa 654-880) of human MYPT1 (rMYPT1) as substrate. Cells were washed twice with ice cold PBS and lysed into IP buffer: 50 mM HEPES pH 7.4, 150 mM NaCl, 1 mM MgCl2, 10 mM NaF, 1 mM Na3VO4, 5% glycerol, 1% Nonidet P-40, 1 mM dithiothreitol, 1 mM EGTA, 5μl/ml each of protease inhibitor and phosphatase inhibitor cocktails. All manipulations after cell lysis were done at ice-cold temperature. Cell lysates were centrifuged at 14000 rpm for 4 minutes to remove insoluble debris. Supernatants were transferred to fresh tubes, precleared with protein-G sepharose beads for 15 minutes, and incubated with anti-ROCK antibody for 40 minutes. The antibody-ROCK complexes were captured by incubating with protein G sepharose beads for 20 minutes. Beads were washed 3X in IP buffer and used for the kinase reaction. The lysis buffer quantitatively solubilized cellular ROCK and the immunopecipitation process depleted greater than 85% of ROCK from the lysates (data not shown). We also verified that cellular ROCK levels did not change by the experimental treatments prior to lysis (data not shown). The kinase reaction mixture included, in a final volume of 100 μl, 0.5μg rMYPT1, 50 μl of beads, and 100 μM ATP. rMYPT1 phosphorylation was measured by Western blotting using rabbit anti-phospho MYPT1 Thr 696. Results were normalized for total protein measured by BCA assay.
RhoA activity assay
RhoA activity was measured using a previously described method [11]. Briefly, cells were lysed in Rho buffer (25mM HEPES, pH 7.5, 150mM NaCl, 1% NP-40 (Igepal CA-630), 10mM MgCl2, 1mM EDTA and 10% glycerol, 10μg/ml leupeptin, 10μg/ml pepstatin, and 10 μg/ml aprotinin) and GTP-Rho was affinity precipitated using Rhotekin-agarose beads. Affinity precipitated RhoA was quantified in parallel with total cellular RhoA from cell lysates by western blot analysis with an antibody against RhoA.
Western blot analysis
Samples were resolved on 4-20% Ready Gels by SDS-PAGE, transferred to PVDF membranes, blocked using 5% nonfat milk powder, and incubated in primary antibody overnight, washed twice with 0.2% Tween in Tris-buffered saline (TBST), incubated with the secondary antibody for 1h at room temperature, and washed thrice with TBST. Bands were detected using Pierce West Dura chemiluminescence system and quantified using unmodified, raw data obtained from images taken on a Versadoc imager and Quantity One software (Biorad). For measuring MLC phosphorylation, samples were collected by washing cells 2X in ice-cold PBS, lysed into 1X Laemmli sample buffer, boiled and analyzed by western blotting. Diphospho-MLC (Ser19/Thr18) signal was normalized to that of nonmuscle myosin II heavy chain.
RESULTS
Adhesion is required for the functional coupling of RhoA activity and ROCK activity
For the first set of experiments, we examined how complete loss of adhesion to fibronectin (FN) affected myosin phosphorylation, RhoA activity and ROCK activity. Endothelial cells were plated on a high density coating (25 μg/ml) of fibronectin, allowed to attach and spread, and then were either released from the substrate or left attached for one additional hour. As an additional comparison to the suspension condition, some of the suspended cells were incubated with fibronectin-coated polystyrene beads to promote integrin engagement in the absence of cell spreading. Cells were lysed and assayed for myosin light chain (MLC) dual phosphorylation at Ser19/Thr18. MLC phosphorylation was low in suspended compared to attached cells even in the presence of integrin engagement with fibronectin-coated beads (Fig. 1A). Interestingly, loss of adhesion did not the change GTP-loading of RhoA in suspended cells as compared to adherent cells, though, integrin engagement through fibronectin-coated beads decreased RhoA activity (Fig. 1B). To determine whether the disconnect between RhoA and myosin signaling occurred upstream or downstream of ROCK, the major regulator of myosin in these cells (Suppl. Fig 1), we examined the activity of endogenous ROCK by kinase assay. Cells in suspension exhibited dramatically lower ROCK activity compared to adherent cells (Fig. 1C), mimicking the loss of myosin activity. These initial findings suggested that in the absence of adhesion, RhoA fails to activate ROCK, thereby disconnecting RhoA activity from myosin phosphorylation.
Fig. 1.

Placing cells in suspension decouples Rho activity from ROCK. A) Western blots (top) and normalized graphs (bottom) of phosphorylated myosin light chain (Ser19/Thr18) and myosin heavy chain in adherent, suspended, and suspended cells incubated with fibronectin-coated beads; n=2. B) Western blots (top) and normalized graphs (bottom) of GTP-Rho and total Rho in adherent, suspended, and suspended cells incubated with fibronectin-coated beads; n=2. C) Western blot (top) and normalized graphs (bottom) of ROCK kinase assay product, phosphoMYPT1 Thr696 adherent, suspended, and suspended cells incubated with fibronectin-coated beads; n=3. Error bars indicate standard errors of mean; ** (p<0.005).
Graded ECM ligand density and cell spreading modulate RhoA-ROCK coupling
It has previously been shown that Rac-PAK coupling depends only on the presence or absence of adhesion; integrin ligation through fibronectin-coated beads in suspended cells is sufficient for Rac to activate PAK [14]. We examined whether Rho-ROCK signaling was also unaffected by varying the degree of substrate adhesion. Endothelial cells were plated on either high (25 μg/ml) or low (0.1 μg/ml) density of FN coated on polystyrene dishes and allowed to attach and spread (Fig. 2A). Cells on low FN exhibited 3-fold lower MLC phosphorylation relative to those on high FN (Fig. 2B) while RhoA activity was not different (Fig. 2C). ROCK activity again correlated with myosin phosphorylation, being substantially lower in the cells on low FN (Fig. 2D). Thus, ROCK activity was uncoupled from RhoA even under adherent conditions, albeit of low integrin ligation.
Fig. 2.

ECM density and cell shape regulate Rho-ROCK coupling. A and E: Phase contrast micrograph of cells on high fibronectin or ‘spread’ condition (A,E top, respectively) or low fibronectin (A,E bottom, respectively) substrates. B and F: Western blots (top) and normalized graphs (bottom) of phosphorylated myosin light chain (Ser19/Thr18) and myosin heavy chain in cells on hiFN and loFN (B; n=3) or ‘spread’ or ‘unspread’ condition (F; n=3). C and G: Western blots (top) and normalized graphs (bottom) of GTP-Rho and total Rho in cells on hiFN and loFN (C; n=8) or in ‘spread’ or ‘unspread’ condition (G; n=2 experiments). D and H: Western blot (top) and normalized graphs (bottom) of ROCK kinase assay product, phosphoMYPT1 Thr696 in cells on hiFN and loFN (D; n=6) and ‘spread’ and ‘unspread’ condition (H; n=4). Error bars indicate standard errors of mean; * (p<0.05), ** (p<0.005).
Cell adhesion involves not only changes in integrin ligation and clustering, but also changes in cell spreading and flattening on the substrate, both of which have distinct effects on cell signaling and function. To directly modulate the degree of cell spreading against a substrate without altering ECM density, we used a micropatterning approach to generate patches of ECM the size of individual cells. Cells were plated on 25 × 25 μm size patterns of FN that restricted them from spreading (‘unspread’) or allowed to freely spread on unpatterned FN (‘spread’) (Fig. 2E) and assayed for RhoA, ROCK and myosin activity. We found that myosin light chain phosphorylation was lower in the spreading-restricted cells on patterns (Fig. 2F) much like the low vs. high density FN condition (Fig. 2B). Interestingly, we found that RhoA activity was higher in the spreading-restricted cells (Fig. 2G). Despite the increase in RhoA signaling, ROCK activity was lower in unspread cells compared to spread cells (Fig. 2H). Together, these data suggest that cell spreading regulates both RhoA activity and the degree of coupling between RhoA and ROCK activity. That is, in unspread cells, RhoA activity is high but it fails to couple and activate ROCK and myosin. Cell spreading moderately decreases RhoA activity, but perhaps by allowing RhoA to act on it effector(s), ROCK and myosin activity dramatically increase.
Tension is required for the coupling of RhoA and ROCK activity
Cell shape could mediate RhoA-ROCK coupling through several potential mechanisms. Previous work from our lab and others has shown that unspread cells exert lower cytoskeletal tension than spread cells [19, 20], and that this tension in turn alters the number of focal adhesions that form in cells that are spread to different extents [21]. Hence, we examined the possibility that cytoskeletal tension may mediate the coupling of RhoA activity to ROCK. We used two mechanistically different drugs to disrupt cytoskeletal tension in cells - blebbistatin, which is a direct inhibitor of myosin II ATPase activity, and cytochalasin D, which disrupts the actin cytoskeleton. To validate the effects of these inhibitors, endothelial cells were allowed to attach and spread on FN for 4h and then treated with each inhibitor. Within 20 minutes, both of the treatments disrupted actin stress fibers, visualized by TRITC-phalloidin staining (Fig. 3A), and decreased the size and number of focal adhesions measured by vinculin staining (Fig. 3B). While these observations suggested that these inhibitors disrupted cytoskeletal tension, they were indirect measures of tension. Using a previously developed microfabricated system, we confirmed directly that cytoskeletal tension was inhibited by drug treatment within 20 minutes (Fig. 3 C). Using these inhibitors, we examined the activity of RhoA and ROCK following the 20-minute exposure. RhoA activity was unchanged upon blebbistatin treatment and increased upon cytochalasin D treatment (Fig. 4A). In contrast, ROCK activity was inhibited upon the disruption of tension in both cases (Fig. 4B). Thus, disrupting cytoskeletal tension decouples RhoA activity from ROCK.
Fig. 3.
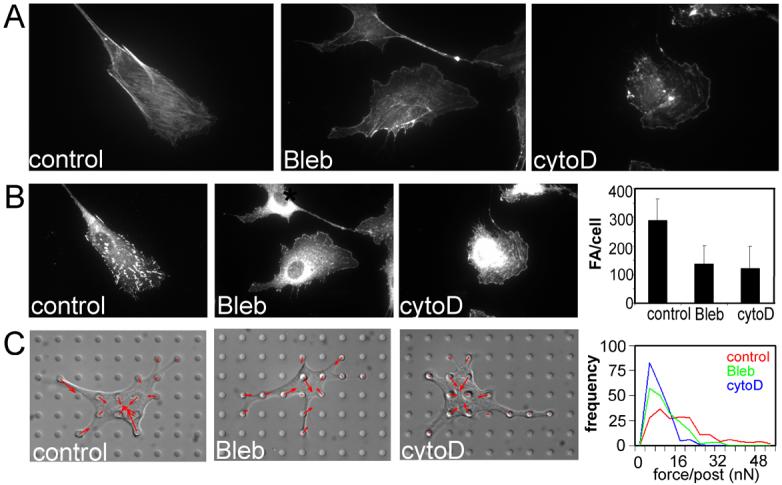
The effect of tension inhibiting drugs on stress fibers, focal adhesions and contractility. A) Immunofluorescence micrographs of stress fibers labeled with TRITC-phalloidin in control (left), blebbistatin-treated (25μM, middle), and cytochalasin D-treated (2μg/ml, right) cells. B) Immunofluorescence micrographs of focal adhesions labeled with anti-vinculin antibody in control (left), blebbistatin-treated (middle), and cytochalasin D-treated (right) cells. C) Graphs of quantified number of focal adhesions per cell in drug treated cells compared to controls. D) Differential interference contrast micrographs of control (left), blebbistatin-treated (middle), and cytochalasin D-treated (right) cells on the microfabricated force-measuring device, overlaid with contractile force vectors. The top graph to the right the average number of focal adhesions per cell in control, blebbistatin-treated and cytochalasin D-treated cells. The bottom graphs shows histograms of contractile forces, each histograms representing pooled data from 15 cells under the respective conditions from 3 experiments: red - control cells, green - blebbistatin-treated cells, blue - cytochalasin D treated cells. Error bars indicate standard deviations of measurement of about 50 cells from a representative experiment.
Fig. 4.
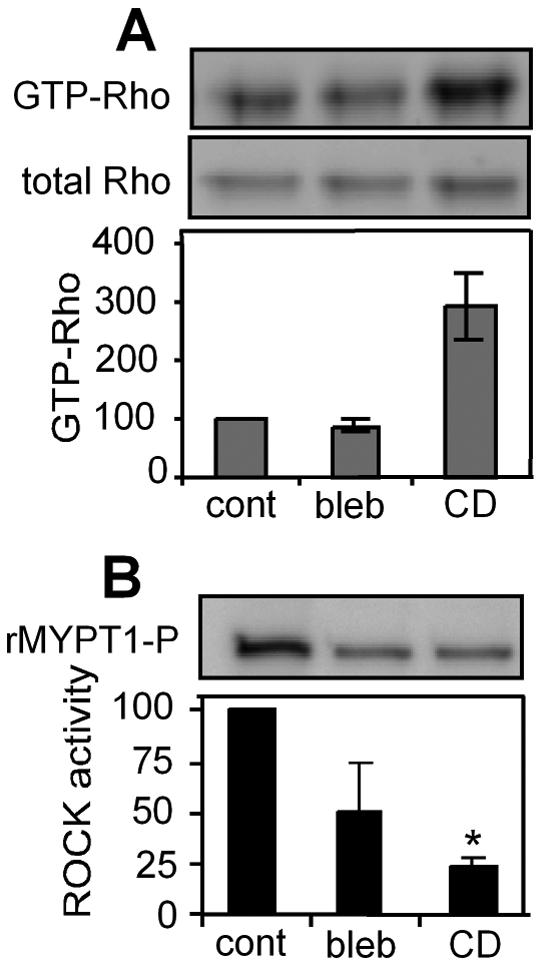
Blocking cytoskeletal tension blocks ROCK activity irrespective of GTP-Rho level. A) Western blot (top) and graph of normalized RhoA (bottom) of Rho pull down in control (cont), blebbistatin (bleb) and cytochalasin D (CD)-treated cells; n=2. B) Western blot (top) and graph of normalized ROCK activity (bottom) in control, blebbistatin and cytochalasin D-treated cells; n=3. Error bars indicate standard errors of mean; * (p<0.05)..
These findings together demonstrated that both the degree of adhesion and cytoskeletal tension regulated ROCK activity. While our studies demonstrated a loss of correlation between RhoA and ROCK activity when cell spreading or cytoskeletal tension was disrupted, they did not provide a direct demonstration of the functional decoupling between these signals. That is, the effects of these manipulations on RhoA and on ROCK could be unrelated, as ROCK activity can be modulated by several other signals [22-24]. To examine if either adhesion or cytoskeletal tension was required for ROCK activation by RhoA, we infected cells with an adenovirus that expressed the constitutively active RhoA-V14 (Fig. 5A) and measured ROCK activity under different conditions. As expected, RhoA-V14 expression increased myosin light chain phosphorylation as compared to GFP controls and maintained MLC phosphorylation even upon serum starvation (Fig. 5B). ROCK activity also increased in RhoA-V14 cells as compared to GFP controls (Fig. 5C). Importantly RhoA-mediated ROCK activity in RhoAV14 expressing cells was abrogated upon treatment with blebbistatin or cytochalasin D (Fig. 5C). Similarly, suspending RhoA-V14 expressing cells also eliminated RhoA-induced ROCK activity (Fig. 5D). Together, these data indicate that cell adhesion, spreading, and cytoskeletal tension are collectively necessary for RhoA to effectively activate ROCK.
Fig. 5.

Blocking tension blocks RhoA-activated ROCK activity. A) Phase contrast micrographs of control EGFP (top) and EGFP-RhoA-V14 (bottom) expressing cells. B) MLC phosphorylation of EGFP and RhoA-V14 expressing cells in serum-starved and serum-containing conditions; n=2. C) ROCK kinase activity in EGFP-expressing cells and in RhoA-V14-expressing cells without or with the inhibition of cytoskeletal tension; n=3. D) ROCK kinase activity in adherent or suspended RhoA-V14 expressing cells; n=3. Error bars indicate standard errors of mean; * (p<0.05).
DISCUSSION
Previous studies have suggested that RhoA-ROCK signaling is intricately linked to adhesion signaling. Here, by separating the effects of integrin ligation, cell spreading, and cytoskeletal tension, and directly measuring endogenous RhoA and ROCK activity, we have uncovered several important regulatory mechanisms. First, RhoA activity appears to be regulated by cell shape. Previous studies have shown that integrin-mediated adhesion can antagonize RhoA activity [11], and are consistent with our observation of decreased RhoA activity upon addition of fibronectin-coated beads to suspended cells. This same pathway may be responsible for the further suppression of RhoA activity as cells are allowed to spread on larger micropatterned islands of fibronectin. Although we have not examined the basis for this decrease, it is plausible that the mechanism involves the increased actin polymerization observed with cell adhesion and spreading [25]. It has been suggested that F-actin may sequester a Rho guanine-nucleotide exchange factor (Rho GEF) [26], such that increased actin polymerization would suppress RhoA activity. Consistent with this model, we observed increased RhoA-GTP loading upon cytochalasin D treatment, a phenomenon that has been previously noted by other researchers [11]. Additional studies will be required to further delineate this mechanism.
Second and perhaps most interestingly, the ability of RhoA to modulate ROCK kinase activity is dependent on cell adhesion, cell spreading and cytoskeletal tension. This decoupling of RhoA and ROCK is evident in suspended cells, which exhibited high levels of Rho-GTP but low MLC phosphorylation and ROCK activity. Remarkably, allowing cells to adhere on a high density of FN was not sufficient to rescue ROCK activity; instead, cell spreading and cytoskeletal tension were also required (Fig. 2, Fig. 4). Previous studies have shown for other small GTPase effectors PAK and mDia, that integrin ligation is sufficient to mediate their activation by Rac and Rho, respectively [13, 14]. This decoupling of Rho and ROCK by changes in cell shape may be one important specific mechanism by which cell shape modulates cell signaling, cytoskeletal tension, and adhesion maturation. The requirement of cytoskeletal tension to couple Rho-GTP to ROCK may be mediated through the effects of mechanical tension on the maturation of focal adhesions. That is, tension mediated changes in focal adhesion maturation could affect adhesion signaling and thereby the coupling of RhoA and ROCK activity. Nonetheless, these data importantly demonstrate that GTP-RhoA activity by itself is not an accurate measure of either ROCK or contractile activity in cells (Fig. 1, Fig. 4).
How does RhoA couple to ROCK in an adhesion dependent manner? Given that FAK is a major tyrosine kinase in focal adhesions and is important in transducing mechanical forces into biochemical signals, it may be involved in this control mechanism. FAK is required for microtubule stabilization by another RhoA effector, mDia [13]. Tyrosine kinase activity has been shown to be required for Rho induced stress fiber formation [27] and Rho can directly induce the phosphorylation of FAK in the absence of stress fiber formation [28], suggesting the possibility of FAK activation being upstream of ROCK. The colocalization of RhoA and ROCK may also play a role in the regulation of ROCK activity by adhesion and tension, perhaps mediated by membrane lipid rafts, as has been shown in two other cases. Adhesion-mediated activation of both PAK by GTP-Rac and mDia by GTP-Rho have been shown to be mediated by GM1-ganglioside containing lipid rafts [13, 14]. The dependence of lipid raft internalization or fusion to the plasma membrane by adhesion may represent a general mechanism by which Rho GTPases and their effectors are regulated. Similar mechanisms may underlie the adhesion and cytoskeletal regulation of ROCK that we observed in this study.
The bi-directional coupling between tension and ROCK activity has numerous implications. Once activated, this positive feedback loop may be an important mechanism to amplify and sustain ROCK activity, where ROCK activates tension, which in turn strengthens the RhoA-ROCK coupling. Conversely, this architecture also explains the observation that disruption of ROCK, tension, or adhesion, functionally disrupts the entire mechano-adhesive system and results often in similar phenotypes such as decreased proliferation [2, 5, 29]. It has recently been suggested that change in substrate stiffness may also affect adhesions and RhoA-ROCK coupling [3]. Here our findings would suggest that the decreased substrate stiffness alters the mechanical stress at adhesions generated by cytoskeletal tension, in turn decreasing ROCK activity, cytoskeletal tension and adhesion maturation, and, thus providing a general control mechanism whereby cell adhesion, substrate mechanics, cytoskeletal tension, and cell signaling are all intricately related (Fig. 6). Since ROCK is required for generating tension, this raises the question of what generates the tension required to activate ROCK in the first place. There are other well-characterized kinases for myosin such as MLCK and PAK, that may temporally precede ROCK activation [30]. There also exist as yet uncharacterized mechanisms for generating tension independent of MLC phosphorylation [31] that may precede and contribute to ROCK activation.
Fig. 6.
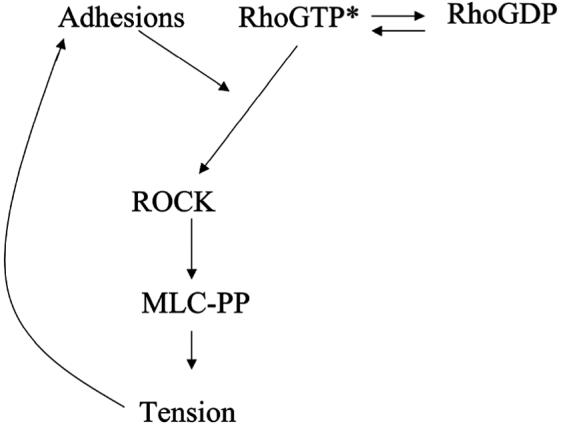
Schematic of the proposed model for how tension regulates ROCK activity. Cytoskeletal tension acts on cell-ECM adhesions to increase the signaling activity at focal adhesions. A molecular mediator at focal adhesions, allows the coupling of GTP-RhoA to ROCK and thereby increase ROCK kinase activity.
RhoA-ROCK signaling has long been known as an important regulator of cytoskeletal tension and adhesion maturation. The numerous feedback loops between these adhesive, cytoskeletal, and mechanical functions of the cell and this signaling pathway highlight the complex control system that cells have constructed, perhaps in order to navigate the complex relationship between ECM mechanics, structure, and composition. Understanding this control system ultimately will be central to our understanding of how cells coordinate the chemical and mechanical worlds.
Supplementary Material
Supplementary figure 1: Myosin light phosphorylation is regulated by ROCK. Western blot on the top shows the effect of inhibiting ROCK by adding Y27632 (50 μM) to spread cells on high density of fibronectin. The plot below is that of normalized MLC phosphorylation in control cells without and with Y27632 addition; n=2. Error bars indicate standard deviations.
Acknowledgements
We thank D. Cohen and N. Sniadecki for a critical reading of the manuscript, and Lixin Qi for expert technical assistance. This work was funded in part by grants from the NIH (HL073305, EB00262, GM74048, AR50850), and the Department of Defense Multidisciplinary University Research Initiative.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Chrzanowska WM, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 1996;133:1403–15. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang S, Chen CS, Ingber DE. Control of cyclin D1, p27(Kip1), and cell cycle progression in human capillary endothelial cells by cell shape and cytoskeletal tension. Mol. Biol. Cell. 1998;9:3179–93. doi: 10.1091/mbc.9.11.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J. Cell Biol. 2003;163:583–95. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mcbeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell Shape, Cytoskeletal Tension, and RhoA Regulate Stem Cell Lineage Commitment. Dev. Cell. 2004;6:483–95. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 5.Bhadriraju K, Hansen LK. Extracellular matrix-dependent myosin dynamics during G1-S phase cell cycle progression in hepatocytes. Exp. Cell. Res. 2004;300:259–71. doi: 10.1016/j.yexcr.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 6.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–35. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 7.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–8. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 8.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–80. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 9.Schiller MR. Coupling receptor tyrosine kinases to Rho GTPases--GEFs what’s the link. Cell Signal. 2006;18:1834–43. doi: 10.1016/j.cellsig.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 10.Ren XD, Wang R, Li Q, Kahek LAF, Kaibuchi K, Clark RAF. Disruption of Rho signal transduction upon cell detachment. J. Cell Sci. 2004;117:3511–3518. doi: 10.1242/jcs.01205. [DOI] [PubMed] [Google Scholar]
- 11.Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–85. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kreisberg JI, Venkatachalam MA, Radnik RA, Patel PY. Role of myosin light-chain phosphorylation and microtubules in stress fiber morphology in cultured mesangial cells. Am. J. Physiol. 1985;249:F227–35. doi: 10.1152/ajprenal.1985.249.2.F227. [DOI] [PubMed] [Google Scholar]
- 13.Palazzo AF, Eng CH, Schlaepfer DD, Marcantonio EE, Gundersen GG. Localized stabilization of microtubules by integrin- and FAK-facilitated Rho signaling. Science. 2004;303:836–9. doi: 10.1126/science.1091325. [DOI] [PubMed] [Google Scholar]
- 14.Del Pozo MA, Alderson NB, Kiosses WB, Chiang HH, Anderson RG, Schwartz MA. Integrins regulate Rac targeting by internalization of membrane domains. Science. 2004;303:839–42. doi: 10.1126/science.1092571. [DOI] [PubMed] [Google Scholar]
- 15.Galbraith CG, Yamada KM, Sheetz MP. The relationship between force and focal complex development. J. Cell Biol. 2002;159:695–705. doi: 10.1083/jcb.200204153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.A M, S.F H, K M, F OP, E D. Ingber, Role of RhoA, mDia, and ROCK in cell shape-dependent control of the Skp2-p27kip1 pathway and the G1/S transition. J. Biol. Chem. 2004;279:26323–30. doi: 10.1074/jbc.M402725200. [DOI] [PubMed] [Google Scholar]
- 17.Nelson CM, Pirone DM, Tan JL, Chen CS. Vascular endothelial-cadherin regulates cytoskeletal tension, cell spreading, and focal adhesions by stimulating RhoA. Mol. Biol. Cell. 2004;15:2943–53. doi: 10.1091/mbc.E03-10-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patan S. Vasculogenesis and angiogenesis. Cancer Treat. Res. 2004;117:3–32. doi: 10.1007/978-1-4419-8871-3_1. [DOI] [PubMed] [Google Scholar]
- 19.Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc. Natl. Acad. Sci. U. S. A. 2003;100:1484–9. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang N, Ostuni E, Whitesides GM, Ingber DE. Micropatterning tractional forces in living cells. Cell Motil. Cytoskeleton. 2002;52:97–106. doi: 10.1002/cm.10037. [DOI] [PubMed] [Google Scholar]
- 21.Chen CS, Alonso JL, Ostuni E, Whitesides GM, Ingber DE. Cell shape provides global control of focal adhesion assembly. Biochem. Biophys. Res. Commun. 2003;307:355–61. doi: 10.1016/s0006-291x(03)01165-3. [DOI] [PubMed] [Google Scholar]
- 22.Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, Watanabe N, Saito Y, Kakizuka A, Morii N, Narumiya S. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 1996;15:1885–93. [PMC free article] [PubMed] [Google Scholar]
- 23.Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 2001;3:346–52. doi: 10.1038/35070019. [DOI] [PubMed] [Google Scholar]
- 24.Feng J, Ito M, Kureishi Y, Ichikawa K, Amano M, Isaka N, Okawa K, Iwamatsu A, Kaibuchi K, Hartshorne DJ, Nakano T. Rho-associated kinase of chicken gizzard smooth muscle. J. Biol. Chem. 1999;274:3744–52. doi: 10.1074/jbc.274.6.3744. [DOI] [PubMed] [Google Scholar]
- 25.Mooney DJ, Langer R, Ingber DE. Cytoskeletal filament assembly and the control of cell spreading and function by extracellular matrix. J. Cell Sci. 1995;108:2311–2320. doi: 10.1242/jcs.108.6.2311. [DOI] [PubMed] [Google Scholar]
- 26.Banerjee J, Wedegaertner GB. Identification of a novel sequence in PDZ-RhoGEF that mediates interaction with the actin cytoskeleton. Mol. biol. Cell. 2004;15:1760–75. doi: 10.1091/mbc.E03-07-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ridley AJ, Hall A. Signal transduction pathways regulating Rho-mediated stress fibre formation: requirement for a tyrosine kinase. EMBO J. 1994;13:2600–10. doi: 10.1002/j.1460-2075.1994.tb06550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flinn HM, Ridley AJ. Rho stimulates tyrosine phosphorylation of focal adhesion kinase, p130 and paxillin. J. Cell Sci. 1996;109(Pt 5):1133–41. doi: 10.1242/jcs.109.5.1133. [DOI] [PubMed] [Google Scholar]
- 29.Nelson CM, Chen CS. VE-cadherin simultaneously stimulates and inhibits cell proliferation by altering cytoskeletal structure and tension. J. Cell. Sci. 2003;116:3571–81. doi: 10.1242/jcs.00680. [DOI] [PubMed] [Google Scholar]
- 30.Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne DJ, Sasaki Y, Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J. Cell. Biol. 2000;150:797–806. doi: 10.1083/jcb.150.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obara K, Nikcevic G, Pestic L, Nowak G, Lorimer DD, Guerriero VJ, Elson EL, Paul RJ, De LP. Fibroblast contractility without an increase in basal myosin light chain phosphorylation in wild type cells and cells expressing the catalytic domain of myosin light chain kinase. J. Biol. Chem. 1995;270:18734–7. doi: 10.1074/jbc.270.32.18734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1: Myosin light phosphorylation is regulated by ROCK. Western blot on the top shows the effect of inhibiting ROCK by adding Y27632 (50 μM) to spread cells on high density of fibronectin. The plot below is that of normalized MLC phosphorylation in control cells without and with Y27632 addition; n=2. Error bars indicate standard deviations.