

Abstract
We applied spFRET microscopy for direct observation of intranucleosomal DNA dynamics. Mononucleosomes, reconstituted with DNA containing a FRET pair at the dyad axis and exit of the nucleosome core particle, were immobilized through a 30 bp DNA tether on a polyethyleneglycol functionalized slide and visualized using Total Internal Reflection Fluorescence microscopy. FRET efficiency time-traces revealed two types of dynamics: acceptor blinking and intramolecular rearrangements. Both Cy5 and ATTO647N acceptor dyes showed severe blinking in a deoxygenated buffer in the presence of 2% βME. Replacing the triplet quencher βME with 1 mM Trolox eliminated most blinking effects. After suppression of blinking three subpopulations were observed: 90% appeared as dissociated complexes; the remaining 10% featured an average FRET efficiency in agreement with intact nucleosomes. In 97% of these intact nucleosomes no significant changes in FRET efficiency were observed in the experimentally accessible time window ranging from 10 ms to 10’s of seconds. However, 3% of the intact nucleosomes showed intervals with reduced FRET efficiency, clearly distinct from blinking, with a lifetime of 120 ms. These fluctuations can unambiguously be attributed to DNA breathing. Our findings illustrate not only the merits but also typical caveats encountered in single-molecule FRET studies on complex biological systems.
Keywords: FRET, Nucleosome, Single-molecule, Blinking
Introduction
Fluorescence (or Förster) Resonance Energy Transfer (FRET) is a process in which the energy of an excited donor fluorophore is transferred non-radiatively to an acceptor molecule [1]. The efficiency of energy transfer E is given by:
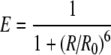 |
1 |
where R is the distance between donor and acceptor and R0 is the Förster radius, at which 50% energy transfer occurs (typically 5 nm for Cy3–Cy5, a commonly used FRET pair). FRET is a powerful tool to study the structure and function of biological molecules, such as DNA. When extended to the single-molecule level, single pair FRET (spFRET) can potentially be applied to determine the conformational distribution of an ensemble of molecules and the dynamics of individual molecules [2–4]. We exploited spFRET to study the structure and dynamics of single nucleosomes, the fundamental units of compaction and organization of eukaryotic DNA.
The nucleosome core particle consists of ∼ 50 nm DNA wrapped nearly twice around a histone-octamer protein-core [5]. Nucleosomal DNA has to unwrap from the nucleosome core to sterically allow processes such as transcription, replication and repair. Accessibility to nucleosomal DNA is facilitated by ATP-dependent remodeling enzymes in vivo [6]. However, it is known that spontaneous conformational changes of the nucleosome expose occluded sites in the DNA as well [7]. DNA breathing, the transient unwrapping and rewrapping of a stretch of DNA from the nucleosome core, has recently been studied in detail with a variety of fluorescence techniques. The equilibrium constant of this process was determined with bulk FRET measurements [8]. Unwrapping lifetimes of 10–50 ms were obtained with stopped-flow FRET measurements and Fluorescence Cross Correlation Spectroscopy (FCCS) [9]. Interestingly, based on their single pair FRET work, Tomschik et al. concluded that unwrapping of nucleosomal DNA occurs to a much larger extent than was previously anticipated [10]: they suggested that 30–60% of the nucleosomal DNA was unwrapped with a lifetime on the order of ∼150 ms before rewrapping.
Although the conceptual beauty of FRET studies is undisputed, there are a number of important caveats in single-molecule FRET studies of biomolecules, such as fluorophore blinking, photobleaching and sample immobilization. Here, we addressed these issues. spFRET microscopy on mononucleosomes revealed two dominant types of dynamics: acceptor blinking and intramolecular rearrangements that we attribute to DNA breathing, which only became apparent after suppression of blinking. Upon immobilization, we observed three different populations: 90% of the nucleosomes dissociated or represented donor-only species, and 10% remained intact. Of these fully wrapped nucleosomes, 97% showed stable FRET on timescales between 0.01–10 s, while 3% showed dynamics with a dwell time of 120 ms that we attribute to conformational changes in the nucleosome.
Material and methods
DNA preparation
A 177 base pair (bp) DNA was constructed by PCR using the 601 nucleosome positioning element [11] as template. PCR primers were as follows. Forward primer: Biotin-TTTGAATTCC CAGGGAATTG GGCGGCCGCC CTGGAGAATC CCGGTGCCGA GGCCGC (acceptor labeled nucleotide is underlined). Reverse primer: ACAGGATGTA TATATCTGAC ACGTGCCTGG AGACTAGGGA GTAATCCCCT TGGCGGTTAA AACGCGGGGG ACAGCGCGTA CG (donor labeled nucleotide is underlined). We used either Cy3–Cy5 or ATTO550-ATTO647N as donor-acceptor FRET pair. PCR products were purified with a GFX PCR DNA & Gel Band Purification Kit (GE Healthcare). The position of the labels was chosen such that after reconstitution the acceptor was located at the nucleosome exit, and the donor near the dyad axis, as illustrated in Fig. 1. Donor and acceptor were predicted to be ∼4 nm apart, as deduced from the nucleosome crystal structure [12], resulting in a FRET efficiency E of approximately 0.8 for the Cy3–Cy5 pair (R0 ∼5 nm), and of approximately 0.9 for the ATTO550-ATTO647N pair (R0 ∼6 nm).
Fig. 1.

FRET system for the study of mononucleosome dynamics. a The 177 bp DNA construct, indicating the position of the labels 80 bp apart in a fragment containing the 601 nucleosome positioning sequence. A biotin label allowed for immobilization of the construct. b, c Illustrations of the mononucleosome structure, indicating the position of donor and acceptor upon reconstitution. The distance between the labels was ∼4 nm, at which efficient FRET takes place. Unwrapping of the DNA from the nucleosome core will be accompanied by a decrease in FRET due to increasing separation between donor and acceptor
Nucleosome reconstitution
Recombinant histone octamers were mixed with the DNA construct at a 1:1 ratio, in TE (1 mM EDTA, 10 mM TRIS pH 8.0) and 2 M NaCl. Mononucleosomes were reconstituted by salt dialysis against 0.85, 0.65, 0.5 and finally against 0.1 M NaCl, all buffered with TE.
Bulk fluorescence measurements
Bulk fluorescence experiments were carried out on a Luminescence Spectrometer (LS55, Perkins Elmer). All experiments were performed at room temperature (22°C). The nucleosome concentration was 10–50 nM. The donor dye was excited at 515 nm and the emission was recorded from 535 to 700 nm. The acceptor dye was excited at 615 nm and the emission was recorded from 635 to 700 nm, to obtain acceptor-only emission spectra. The FRET efficiency was determined from the enhanced fluorescence of the acceptor using the (ratio)A method [13]:
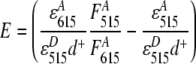 |
2 |
where 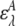 and
and 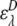 are the acceptor and donor extinction coefficient respectively at wavelength λ,
are the acceptor and donor extinction coefficient respectively at wavelength λ, 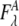 is the fluorescence intensity of the acceptor when excited at wavelength λ, and d+ is the fractional labeling coefficient of the donor. The fluorescence intensity of the acceptor was determined at its maximum value. d+ was determined from DNA and fluorophore absorption peaks in an absorption spectrum of the labeled DNA, measured from 230 to 700 nm with a spectrophotometer (Pharmaspec UV-1700, Shimadzu).
is the fluorescence intensity of the acceptor when excited at wavelength λ, and d+ is the fractional labeling coefficient of the donor. The fluorescence intensity of the acceptor was determined at its maximum value. d+ was determined from DNA and fluorophore absorption peaks in an absorption spectrum of the labeled DNA, measured from 230 to 700 nm with a spectrophotometer (Pharmaspec UV-1700, Shimadzu).
Single-molecule FRET measurements
Cleaned glass slides were amino functionalized with 10 μg/ml poly-d-lysine, and subsequently incubated for 4 h with an amine reactive polyethylene glycol (PEG) mixture: 20% mPEG-succinimidyl propionate 5,000 molecular weight (Nektar Therapeutics) and 0.2% biotin-PEG-n-hydroxysuccinimide 3,400 molecular weight (Nektar Therapeutics) in 0.1 M sodium carbonate buffer (pH 8.2). A flow cell was assembled by sealing a poly-dimethylsiloxane channel with a PEG functionalized slide. A 0.1 mg/ml streptavidin (Roche) solution was incubated for 5 min, and subsequently washed away. A sample, which typically consists of 10–50 pM of labeled mononucleosomes in 50–200 mM NaCl, 10 mM TRIS.HCl pH 8.0, 0.03% NP-40, and 10–50 nM unlabeled mononucleosomes, was injected in the channel and immobilized. An enzymatic oxygen scavenger system (1% glucose, 2% β-mercaptoethanol (βME) or 1 to 2 mM Trolox (Sigma), 0.2 mg/ml glucose oxidase, and 0.04 mg/ml catalase), was added to the buffer to extend the lifetime of the fluorophores before photobleaching. The buffer was degassed prior to use to further reduce the oxygen concentration. The flow cell was mounted on a microscope equipped with a 100X oil-immersion TIRF microscope objective (NA = 1.45, NIKON) and temperature-stabilized at 22°C using a water circulating bath connected to all parts of the setup in contact with the sample. The 514 nm line of an Ar+ laser (Coherent) was used to illuminate an area of ∼ 600 μm2 with a power of 0.9 mW. In the case of alternating excitation, a 636 nm diode laser (Power Technology) was used to illuminate an area of ∼900 μm2 with a power of 0.3 mW. Both beams were circularly polarized and were displaced parallel to the optical axis of the objective, so that an evanescent excitation field was generated by total internal reflection of the light at the glass-water interface. The excitation intensity at the interface in the evanescent field is ∼4 times higher than the incident beam intensity at the critical angle [14]. We therefore estimated that the resulting excitation intensities at the interface were ∼0.6 kW/cm2 for the 514 nm excitation and ∼0.13 kW/cm2 for the 636 nm excitation respectively.
The fluorescence was collected by the objective and filtered through a custom-made dual color band pass filter (Chroma), that rejects scattered laser light, and a long pass filter (OG530, Schott). The fluorescence was further split into a donor and an acceptor channel by a custom-made dichroic wedge mirror (0.5° angle, center wavelength of 630 nm, Chroma) placed in the infinity path of the microscope [15]. A +150 mm achromatic lens (Thorlabs) projected the separate images on a multiplication gain CCD camera (Cascade 512B, Roper Scientific) operating at a frame rate of 20 to 100 Hz.
Data analysis
The simultaneously acquired donor and acceptor images (typically 80 by 80 pixels) were aligned with respect to one another through their cross correlation. The first 50 donor and acceptor frames were overlaid, and their intensities averaged. Low frequency background signal was filtered out with a high-pass FFT filter. The location of the fluorophores was then determined by applying a threshold of 2 times the background noise level. A time-trace of donor and acceptor intensities was then calculated by integrating the pixel intensities 1.5 pixel around the fluorophore center for each frame and each image. In the case of alternating excitation, the acceptor intensity upon direct excitation was retrieved by deinterleaving the acceptor time-trace. The FRET efficiency was calculated from [2]:
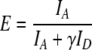 |
3 |
where IA and ID are acceptor and donor intensity respectively, and 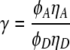 is a parameter to correct for photophysical properties of the dyes. ΦA and ΦD are acceptor and donor quantum yield, and ηA and ηD are acceptor and donor detector efficiency respectively. As a first approximation γ was set to unity.
is a parameter to correct for photophysical properties of the dyes. ΦA and ΦD are acceptor and donor quantum yield, and ηA and ηD are acceptor and donor detector efficiency respectively. As a first approximation γ was set to unity.
A more accurate estimate for γ was obtained from experimental intensity time traces where donor bleaching took place after acceptor bleaching. In these cases the FRET efficiency could also be calculated from donor quenching:
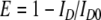 |
4 |
where ID0 is the donor intensity after bleaching of the acceptor. Combining Eqs. 3 and 4 results in:
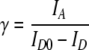 |
5 |
Experimental results
Bulk fluorescence spectra reveal proper reconstitution of mononucleosomes
The results of bulk fluorescence and absorption experiments on reconstituted mononucleosomes are shown in Fig. 2. The reconstituted sample showed efficient FRET, indicated by a distinct peak of fluorescence at the acceptor maximum emission wavelength (670 nm for ATTO647N, see Fig. 2a). This peak was not present in the labeled DNA-only sample, confirming that the donor and the acceptor were in close proximity due to mononucleosome reconstitution. As a control, we diluted the mononucleosome sample in 2 M NaCl, as this high ionic strength disrupts nucleosome structure [16]. As predicted, over 90% of the energy transfer signal was lost.
Fig. 2.
Bulk fluorescence emission and absorption spectra revealed proper reconstitution of mononucleosomes. a Bulk fluorescence emission spectra. A distinct peak of fluorescence at the acceptor emission wavelength was seen after reconstitution, which was not present for the labeled DNA only. The peak disappeared upon dilution of the reconstituted material in 2M NaCl, an ionic strength at which nucleosome structure is disrupted. b Bulk absorption spectrum of the fluorescently labeled DNA construct. The stoichiometry of the labels was obtained by comparison with the absorption spectra of ATTO550 and ATTO647N (as provided by the manufacturer), which are plotted with dotted lines
The observed average FRET efficiency in the reconstituted mononucleosomes was 0.75 ± 0.1, which was in good agreement with FRET values predicted by the position of the FRET pair in the nucleosome. From the bulk FRET experiments, and the predicted maximum FRET efficiency of ∼0.9 for a mononucleosome with the ATTO550-ATTO647N FRET-pair, we estimated the reconstitution yield to be at least 85%. Some residual donor emission can be accounted for by incomplete acceptor labeling. With absorption measurements on the DNA construct (Fig. 2b) we determined that the acceptor:donor:DNA stoichiometry was ∼0.7:0.9:1. Together, these bulk data show that the labeled DNA construct and the histone proteins properly formed mononucleosomes upon reconstitution (see Fig. 1).
spFRET microscopy reveals individual nucleosomes together with a large population of dissociated nucleosomes
To investigate mononucleosome subpopulations and dynamics, spFRET measurements were performed in a wide field microscope. Figure 3 shows an example of typical single-molecule fluorescence images of immobilized mononucleosomes. In Fig. 3a, an acceptor channel image was superimposed on a donor channel image. 10% of the immobilized fluorophores showed efficient FRET, as indicated by colocalized fluorescent spots in the acceptor channel upon donor excitation, and thus represent fully reconstituted mononucleosomes.
Fig. 3.

Single molecule fluorescence image of immobilized mononucleosomes. a False color representation of averaged donor and acceptor channel images, excited at 514 nm. The arrows point at molecules that featured efficient FRET from donor to acceptor. The majority of the molecules however did not show FRET and appears in red. b The same field of view excited at 636 nm, allowing for unambiguous identification of acceptor fluorophores
In contrast, 90% of the fluorophores did not show FRET at all. This conflicts with the bulk experiments, where after correction for incomplete labeling an average FRET efficiency of 0.75 was found. As mentioned before, there was a fraction of donor only labeled species (∼30%), but this alone could not explain the observed discrepancy between bulk and single-molecule measurement. When we directly excited the acceptor fluorophores (see Fig. 3b), we found that most of the donors were colocalized with an acceptor. Therefore we conclude that FRET signal was lost during the single-molecule measurement, due to disassembly of a large fraction of the nucleosomes.
It is known that nucleosomes become unstable and dissociate when they are diluted to low concentrations [17, 18]. For our wide field spFRET measurements we diluted to pM fluorophore concentrations to resolve individual fluorophores. We ensured that the nucleosome concentration was always above 10–50 nM. By adding an excess of unlabeled mononucleosomes and 0.03% non-ionic detergent (NP-40) to our buffer. Thastrom et al. [19] reported that under these conditions nucleosomes do not dissociate in bulk solutions. We found that even 50 nM of unlabeled nucleosomes, far above the dilution-driven dissociation threshold, did not retain proper nucleosome folding, excluding dilution effects to be the cause.
It is known that H2A–H2B histone dimers can spontaneously be exchanged from the protein core [20], which in our case would result in a transient loss of FRET. However, we found the same amount of disassembled nucleosomes upon immobilization when the octamer protein core was crosslinked by dialysis against 0.05% glutaraldehyde in 1 mM EDTA. We confirmed that the crosslinking itself did not dissociate nucleosomes with bulk fluorescence experiments. This suggests that not the histone protein core dissociates, but rather that the wrapped DNA loosens or significantly rearranges itself around the protein core. We confirmed that mononucleosomes in free solution (in the same buffer used for single-molecule experiments) remain stable for hours at room temperature using bulk fluorescence measurements. Therefore we consider the dissociation of the nucleosomes described here to be associated with their immobilization to the functionalized cover glass. As an alternative immobilization strategy we performed experiments with biotinylated BSA-functionalized cover glasses instead of PEGs. Biotinylated BSA is often used for single-molecule studies involving nucleic acids, whereas PEGs are often used for studies involving DNA-protein complexes [21]. Biotinylated BSA yielded even less intact nucleosomes. The exact nature of the interactions of the nucleosomes with the modified cover slides remains unclear, but the destabilizing effect of the surface forms a hurdle for obtaining large datasets of spFRET measurements.
On the 10% immobilized mononucleosomes showing FRET, irreversible loss of FRET was only found after photobleaching, implying that their nucleosomal structure remained intact after immobilization.
Single-molecule fluorescence footprint of individual nucleosomes
Example intensity time-traces of intact single nucleosomes are shown in Fig. 4a and b. Donor and acceptor intensity were clearly anti-correlated, indicative of their FRET interaction. The intensity of a single donor (Cy3) was 1.4 ± 0.3 × 103 counts/10 ms at a signal to noise ratio (SNR) of 5. When the donor was quenched by FRET, both the intensity and SNR decreased. The intensity of an acceptor (Cy5) excited via FRET was 0.9 ± 0.2 × 103 counts/10 ms at a SNR of 3. After ∼4 s of continuous illumination at ∼0.6 kW/cm2 either donor or acceptor photobleached, limiting the observational window to a few seconds. The total number of emitted photons until bleaching from a FRET pair was ∼105 (calculated with gain G = 33 counts/photon, detection efficiency ηD and ηA ∼15%). The observed average FRET efficiency of the high FRET level was ∼0.5 ± 0.13, slightly lower than the values measured in the bulk. From the traces where donor bleaching takes place after acceptor bleaching, we estimated the correction factor ã for photophysical parameters of Cy3/Cy5 to be ∼0.7 ± 0.3 (Eq. 4). The corrected FRET efficiency was then ∼0.6 ± 0.3, in good agreement with values obtained from bulk measurements.
Fig. 4.
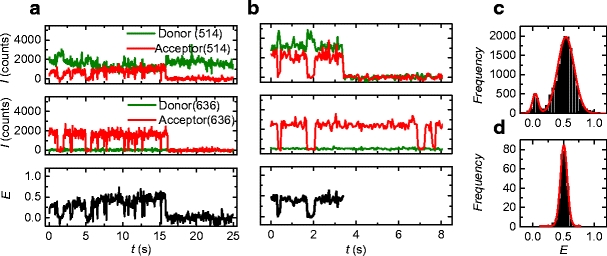
Single molecule FRET traces from individual mononucleosomes. The top panels in a and b show the intensity time traces of donor and acceptor for green excitation; the middle panel shows the intensity time traces of donor and acceptor for red excitation, which were acquired in alternation with the green excitations. The bottom panels show the calculated FRET efficiency. The fluctuations between high and low FRET states featured perfect correlation with the corresponding acceptor intensity traces excited at 636 nm. c Histogram of the FRET efficiencies of multiple single molecule traces. d Histogram of the FRET efficiencies of a single trace. The spread in FRET efficiency was larger between different traces than within a single trace
We found that the standard deviation in FRET efficiency of the high FRET state of the entire population (0.13) was larger than the standard deviation within individual traces (0.06), as shown in the histograms in Fig. 4c and d. This observation can be accounted for either by different nucleosome populations with slight variations in FRET efficiency, or by local differences in rotational freedom of the dyes due to immobilization.
Acceptor blinking is the dominant source of spFRET dynamics
The FRET traces shown in Fig. 4a and b are highly dynamic and fluctuate between a high FRET state (E ∼0.6, lifetime 2.5 s) and a low FRET state (E ∼0.1, lifetime 0.13 s). Interestingly, the characteristics of these fluctuations, i.e. both on and off time, and the low FRET level, are remarkably similar to those observed by Tomschik et al. [10] who performed analogous experiments. This similarity however, is remarkable in view of the completely different FRET-label location. Tomschik et al. labeled the nucleosome opposite to the dyad axis and probed the DNA at the most internal position of the nucleosome, whereas our substrate has labels at the most exterior position. Widom et al. have previously shown that the enzymatic accessibility of the DNA inside a nucleosome strongly reduces as the DNA is more internal in the nucleosome [7], suggesting a higher frequency of unwrapping events in our experiments.
Because of the nearly complete absence of acceptor emission, we investigated the nature of these fluctuations in order to exclude reversible transitions of the acceptor to an inactive state (acceptor blinking, resulting in a Förster radius of effectively zero [22]) as the origin of these events. By alternating donor excitation with direct acceptor excitation we could directly monitor the acceptor condition as shown in Fig. 3b. After deinterleaving the data into two time-traces, one for green excitation and one for red excitation, it became obvious that the fluorescence intensity of the acceptor upon direct excitation correlated perfectly with the enhanced emission of the acceptor due to FRET. Thus, the low FRET state must be attributed to blinking, due to a dark-state level of the acceptor. Further evidence that these fluctuations were caused by acceptor blinking was provided by experiments with alternative acceptor dye (ATTO647N, emission spectrum similar to Cy5). Alternating excitation of the acceptor dye revealed a strong positive correlation between sensitized emission of the acceptor and direct excitation of the acceptor. In this case the low FRET state was also present, but with a much shorter lifetime of 0.046 s. In conclusion, our data confirm that the fluctuations between a high and a low FRET state reflect photophysical processes in the acceptor dye rather than nucleosome conformational changes.
We further analyzed the single-molecule FRET traces for dynamics other than blinking. Therefore we filtered out blinking events by the application of a threshold on low FRET efficiencies (≤ ∼0.1–0.2, dependent on the noise in the measurement). Although careful inspection did occasionally reveal anticorrelated features of donor and acceptor channel, these features had a lifetime below the time resolution of our measurements. To confirm that we did not overlook any dynamics, we analyzed the fluorophore intensity noise in the high FRET state, which in the absence of dynamics should be limited by shot noise. The theoretical noise σtot in the measurement was estimated by [23]:
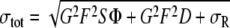 |
6 |
where G is the multiplication gain factor, F is the excess noise factor due to the multiplication gain register, S is the number of photons that reach the camera, Φ is the camera quantum yield, D is the dark count, and σR is the readout noise. The first contribution represents photon shot noise after multiplication, the second contribution represents the camera dark noise after multiplication, and the third the ADC converter electronic noise. Readout noise and dark noise were calculated from the standard deviation of an area of the chip that was not illuminated by fluorescence to be 130 counts/10 ms. The actual noise σ in the single-molecule fluorescence traces was estimated by the standard deviation of the measured fluorophore intensity. The measured and calculated noise were tested for equality with an F test: 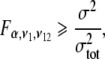 where α is the significance level at which the test was performed (0.05), and ν1, ν2 are the degrees of freedom used to calculate σ and σtot respectively. We found that the total measured noise was significantly (typically 1.5 times) higher than that predicted by photon statistics and camera noise only. This implied that the traces contained dynamic events that cannot be fully resolved, originating from either photophysical processes (short blinking events, or intersystem crossing), or fast nucleosome dynamics. Hence, to accurately capture these events, blinking had to be further suppressed, and the sampling frequency had to be increased.
where α is the significance level at which the test was performed (0.05), and ν1, ν2 are the degrees of freedom used to calculate σ and σtot respectively. We found that the total measured noise was significantly (typically 1.5 times) higher than that predicted by photon statistics and camera noise only. This implied that the traces contained dynamic events that cannot be fully resolved, originating from either photophysical processes (short blinking events, or intersystem crossing), or fast nucleosome dynamics. Hence, to accurately capture these events, blinking had to be further suppressed, and the sampling frequency had to be increased.
Suppression of blinking
In order to suppress blinking, we first tested a different acceptor dye (ATTO647N), which was reported to have superior photochemical stability compared to Cy5 [24]. As mentioned before, this acceptor dye showed blinking as well, as seen in the example traces and histograms of Fig. 5a and b. Although a small fraction of molecules did not show any dynamics in FRET, the majority significantly blinked. In the case of Cy5 93% of all acceptors excited via FRET showed significant blinking, with a lifetime of ∼0.13 ± 0.05 s, and lifetime of the high state of ∼0.8 ± 0.1 s. In the case of ATTO647N, 94% of all acceptors excited via FRET showed blinking, with a lifetime of ∼0.046 ± 0.02 s, and lifetime of the high state of ∼1.2 ± 0.2 s. In conclusion, the use of a different dye did not suppress blinking to the required level, but just yielded different blinking statistics.
Fig. 5.

Fluorophore blinking in spFRET traces obtained from mononucleosomes. In the presence of βME, nucleosomes labeled with Cy5 a or ATTO647N b both show severe blinking in 95% of the traces. Example traces (top) and blinking lifetime histograms (bottom) are shown. c,d In the presence of an alternative triplet quencher, Trolox, blinking of both dyes is significantly suppressed. The example traces (top) show the absence of blinking in 90% of the traces, while the blinking lifetime histograms (bottom) show a small but finite amount of fast blinking still present in ∼10% of the traces
Recently a different approach to reduce blinking was described by Rasnik et al. [25]. They replaced the triplet quencher βME in the oxygen scavenger system by a water-soluble analog of vitamin E, Trolox. Using this approach, Cy5 blinking in single-molecule FRET measurements on DNA constructs was eliminated. We tested the effect of Trolox in the imaging buffer on blinking of FRET pair labeled mononucleosomes. Results are shown in example traces and histograms in Fig. 5c and d. Both for Cy5 and ATTO647N blinking was dramatically suppressed in the presence of 1 to 2 mM Trolox: over 90% of the traces showed no observable blinking. Noise analyses of the intensity fluctuations in most of these traces were fully accounted for by camera noise and photon statistics (shot noise) only. Thus, within our time resolution (10 ms), no effect of short time scale blinking, or inter system crossing, was detected.
Surprisingly, less than 10% of the observed FRET pairs still showed some extent of blinking indicated by fast excursions into a FRET state below 0.2, with a typical off time of 14 ± 1 and 13 ± 1 ms respectively, as shown in the histograms in Fig. 5c and d. Because the lifetime of these blinking events was on the order of the smallest sampling time used, blinking events were not identified by alternating excitation of the acceptor dye, but only by FRET efficiencies below the noise threshold. Direct excitation of the acceptor did only reveal some occasional blinking in the acceptor traces, with the same lifetime of 13–14 ms. To confirm that a small but finite amount of fast blinking still occurred in the presence of Trolox, we performed spFRET measurements on a FRET pair that was separated by 11 basepair duplex DNA. This construct does not exhibit structural changes that affect the FRET intensity. In this case we also observed a small, but finite amount of blinking in a number of traces (data not shown), with a lifetime similar to that measured on mononucleosomes.
A fraction of the immobilized nucleosomes shows dynamics clearly distinct from blinking
The suppression of blinking finally allowed us to unambiguously identify non-blinking dynamic events in the FRET traces. From a sample of 236 mononucleosomes that showed FRET, we found that over 95% of the traces essentially show stable FRET efficiency, as illustrated in Fig. 6a and b; all anti-correlated features in the FRET efficiency were short-lived and fall within the noise of the measurement. Thus, the upper limit for dynamic events that could have been missed in this population was 10 ms (the sampling time used). Interestingly, 3% of the traces showed dynamic events clearly distinct from blinking (examples shown in Fig. 6c and d), as judged by the following criteria: (1) the acceptor signal of a low FRET event was significantly higher than zero. (2) No correlated change in acceptor intensity was detected using alternating excitation. (3) Events persisted for at least two data-points. We found 14 events with an average FRET change ΔE of −0.23 and an average dwell time of 120 ± 5 ms, as summarized in the histogram and cumulative distribution function in Fig. 6e and f respectively. The lifetime was determined by fitting a cumulative exponential distribution to the data, independent of binning and therefore a more accurate way of determining the lifetime when using small datasets (Fig. 6f) than fitting a distribution to binned data. The lifetime of the high FRET state could not be determined accurately, due to the short time window that was available due to photobleaching. Since we explicitly checked the vitality of both fluorophores, we ruled out photodynamics and we could unambiguously attribute the observed features to DNA breathing dynamics.
Fig. 6.

A fraction of the immobilized nucleosomes showed dynamics clearly distinct from blinking. a,b After suppression of blinking with Trolox, over 95% of the FRET traces do not show FRET dynamics. The theoretical photon and instrument noise is approximately indicated by the grey bars. c,d ∼3% of the intensity traces (top panels) showed FRET fluctuations (bottom panels) clearly distinct from blinking: the acceptor intensity was significantly higher than zero, and events persisted multiple data points (see insets). These fluctuations clearly exceeded the noise. e Histogram and cumulative distribution plot f of the lifetime of the dynamic events. An exponential fit to the data gave an average lifetime of 120 ms
Discussion and conclusion
Time-traces of spFRET microscopy on single reconstituted mononucleosomes revealed two types of dynamics: acceptor blinking and intramolecular rearrangements. Intramolecular rearrangements became only apparent after suppression of blinking. Both Cy5 and ATTO647N showed severe blinking in a deoxygenated buffer in the presence of 2% βME. Replacing the triplet quencher βME with Trolox effectively eliminated most blinking effects. The lifetime of DNA unwrapping that we obtained after rigorous elimination of blinking events (∼120 ms) was comparable to the 150–180 ms obtained by Tomschik et al. [10], despite the very different location of the labels in the nucleosome, and probably less important, the different DNA sequence and origin of the histones. However, we observed a very similar lifetime (∼130 ms) for Cy5 blinking under comparable buffer conditions (2% βME). The FRET efficiency of the open states in our experiments was significantly above the detection threshold, so we can explicitly exclude photophysics as the origin of the observed changes in FRET efficiency.
Our single-molecule measurements revealed at least three subpopulations in the reconstituted and immobilized nucleosome sample: 90% of the fluorophores represented dissociated nucleosomes or donor only species, 10% represented intact nucleosomes. Of these, 97% remained stable on time-scales ranging from 10 ms to 10 s of seconds, while 3% showed intervals with reduced FRET efficiency and a lifetime of 120 ms clearly distinct from blinking.
Why most nucleosomes dissociate upon immobilization to the cover slip remains unknown. Immobilization of the molecules is necessary for extension of the available observation time. The time limit is given by photobleaching, one of the key advantages of this method with respect to, for example, Fluorescence Correlation Spectroscopy. However, the close proximity to the surface provides ample opportunity for interactions with it. Surface induced nucleosome dissociation has been reported before in Atomic Force Microscopy (AFM) studies. Using AFM in liquid, Nikova et al. observed an unwrapping of ∼25 nm of DNA from nucleosomes absorbed to a mica surface [26]. This unwrapping was attributed to a depletion of H2A–H2B histone dimers induced by the high surface charge of the mica, resulting in unwrapping of DNA. Although PEGs are neutral polymers that are commonly used to reduce non-specific surface binding of proteins, they may affect nucleosomes in different ways: PEG molecules have been reported to interact strongly with unfolded proteins [21], and could therefore possibly interact with histone tails. Furthermore, histone proteins are known to be adhesive to glass or plastic [27].
The large fraction of dissociated nucleosomes we report here was not observed by Tomschik et al. [10]. Because of the internal position of the labels they used, at least 50 bp of DNA had to be detached from the histone core before FRET was completely lost. We labeled the DNA at the very end of the histone bound part, and accordingly a detachment of 10–20 bp of DNA would already result in complete loss of FRET. Furthermore, the exterior part of the DNA is largely constrained by the mobile H2A–H2B dimer, whereas the labeled part of the DNA in the nucleosomes used by Tomschik et al. is mostly constrained by the more stable H3–H4 tetramer.
A labeling strategy by Li et al. [8, 9], who end-labeled a 601 nucleosome positioning element together with either histone H3 or H2A, provides a more comparable construct. Based on stopped-flow FRET and FCCS experiments they deduced an unwrapping rate of 4 s−1 an unwrapping lifetime of 10–50 ms. The 3% of our traces that showed dynamics typically featured multiple unwrapping events before photobleaching. Though photobleaching obstructs quantification of the unwrapping rate, it is of the same order of magnitude as observed by Li et al. The lifetime of the unwrapped state we observed is five to ten times larger. This discrepancy may in part be explained by differences in experimental conditions and nucleosome constructs; we can however not exclude the possibility that we overlook short-lived unwrapped states, biasing our data to a longer lifetime.
The absence of observations of DNA unwrapping in the majority of the intact nucleosomes reported in this study is in strong contrast with the extent of DNA breathing dynamics found by Li et al. [8, 9]. Two possible explanations could account for this difference: (1) The most frequently occurring DNA unwrapping occurs at a rate that exceeds the time resolution of our experiment. The rare dynamics (3%) that we observe would reflect the release of multiple histone-DNA contacts, a process that would occur less often and on longer time scales than unwrapping of only the first DNA-octamer. However, unwrapping of 10–20 bp of DNA would induce a more dramatic reduction in FRET efficiency than the reduction we observed, which is consistent with unwrapping of 10 bp or less. (2) The immobilized nucleosomes did not undergo breathing dynamics. It should be kept in mind that because of the disruption of 90% of the nucleosome upon immobilization, we only probed a subset of nucleosomes that do not dissociate upon immobilization. These nucleosomes could either be resistant to unwrapping of the DNA, or immobilized in such a way that DNA dynamics are inhibited due to interactions with the surface, while still retaining proper folding. In either case, immobilization is expected to have major impact on nucleosome dynamics, emphasizing the need for a more inert immobilization than point attachment to a PEG coated surface.
Our findings demonstrate that experimental conditions can have a profound impact on the data obtained when probing nucleosome structure and conformational dynamics. Immobilization effects and blinking dynamics have to be accounted for, and where possible suppressed in order to extract biologically relevant data from spFRET experiments. We have shown that DNA breathing kinetics obtained from carefully optimized spFRET experiments approaches values obtained from bulk experiments, opening the way to more complex single-molecule studies of chromatin dynamics.
Acknowledgement
We would like to thank Jon Widom for providing a plasmid containing the 601 sequence; Jesper Donsmark, John van Egmond and Ineke de Boer for technical assistance, and Paige Shaklee for help with the manuscript. AB, CL, and SJTvN acknowledge support from the EuroDYNA eurocores program. This work is part of the research programme of the ‘Stichting voor Fundamenteel Onderzoek der Materie (FOM)’, which is financially supported by the ‘Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO)’.
References
- 1.Förster T (1965) In: Sinanoglu O (ed.), Modern quantum chemistry. Academic, New York, pp 93–137
- 2.Ha T (2001) Single-molecule fluorescence resonance energy transfer. Methods 25(1):78–86 [DOI] [PubMed]
- 3.Ha T, Rasnik I, Cheng W, Babcock HP, Gauss GH, Lohman TM, Chu S (2002) Initiation and re-initiation of DNA unwinding by the Escherichia coli Rep helicase. Nature 419(6907):638–641 [DOI] [PubMed]
- 4.Mckinney SA, Freeman ADJ, Lilley DMJ, Ha TJ (2005) Observing spontaneous branch migration of Holliday junctions one step at a time. Proc Natl Acad Sci USA 102(16):5715–5720 [DOI] [PMC free article] [PubMed]
- 5.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 angstrom resolution. Nature 389(6648):251–260 [DOI] [PubMed]
- 6.Saha A, Wittmeyer J, Cairns BR (2006) Chromatin remodelling: the industrial revolution of DNA around histones. Nat Rev Mol Cell Biol 7(6):437–447 [DOI] [PubMed]
- 7.Polach KJ, Widom J (1995) Mechanism of protein access to specific Dna-sequences in chromatin-a dynamic equilibrium-model for gene-regulation. J Mol Biol 254(2):130–149 [DOI] [PubMed]
- 8.Li G, Widom J (2004) Nucleosomes facilitate their own invasion. Nat Struct Mol Biol 11(8):763–769 [DOI] [PubMed]
- 9.Li G, Levitus M, Bustamante C, Widom J (2005) Rapid spontaneous accesibility of nucleosomal DNA. Nat Struct Mol Biol 12(1):46–53 [DOI] [PubMed]
- 10.Tomschik M, Zheng HC, van Holde K, Zlatanova J, Leuba SH (2005) Fast, long-range, reversible conformational fluctuations in nucleosomes revealed by single-pair fluorescence resonance energy transfer. Proc Natl Acad Sci USA 102(9):3278–3283 [DOI] [PMC free article] [PubMed]
- 11.Lowary PT, Widom J (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol 276(1):19–42 [DOI] [PubMed]
- 12.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ (2002) Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 angstrom resolution. J Mol Biol 319(5):1097–1113 [DOI] [PubMed]
- 13.Clegg RM (1992) Fluorescence resonance energy-transfer and nucleic-acids. Methods Enzymol 211:353–388 [DOI] [PubMed]
- 14.Thompson NL, Pearce KH, Hsieh HV (1993) Total internal-reflection fluorescence microscopy-application to substrate-supported planar membranes. Eur Biophys J Biophys Lett 22(5): 367–378 [DOI] [PubMed]
- 15.Cognet L, Harms GS, Blab GA, Lommerse PHM, Schmidt T (2000) Simultaneous dual-color and dual-polarization imaging of single molecules. Appl Phys Lett 77(24):4052–4054 [DOI]
- 16.Yager TD, Mcmurray CT, Vanholde KE (1989) Salt-induced release of Dna from nucleosome core particles. Biochemistry 28(5):2271–2281 [DOI] [PubMed]
- 17.Claudet C, Angelov D, Bouvet P, Dimitrov S, Bednar J (2005) Histone octamer instability under single molecule experiment conditions. J Biol Chem 280(20):19958–19965 [DOI] [PubMed]
- 18.Godde JS, Wolffe AP (1995) Disruption of reconstituted nucleosomes—the effect of particle concentration, MgCl2 and KCl concentration, the histone tails, and temperature. J Biol Chem 270(46):27399–27402 [DOI] [PubMed]
- 19.Thastrom A, Gottesfeld JM, Luger K, Widom J (2004) Histone—DNA binding free energy cannot be measured in dilution-driven dissociation experiments. Biochemistry 43(3):736–741 [DOI] [PubMed]
- 20.Eickbush TH, Moudrianakis EN (1978) Histone core complex-octamer assembled by 2 sets of protein–protein interactions. Biochemistry 17(23):4955–4964 [DOI] [PubMed]
- 21.Rasnik I, Mckinney SA, Ha T (2005) Surfaces and orientations: Much to FRET about? Acc Chem Res 38(7):542–548 [DOI] [PubMed]
- 22.Sabanayagam CR, Eid JS, Meller A (2005) Long time scale blinking kinetics of cyanine fluorophores conjugated to DNA and its effect on Forster resonance energy transfer. J Chem Phys 123(22) [DOI] [PubMed]
- 23.Roper Scientific (2003) On-chip multiplication gain. Technical Note #14
- 24.Eggeling C, Widengren J, Brand L, Schaffer J, Felekyan S, Seidel CAM (2006) Analysis of photobleaching in single-molecule multicolor excitation and forster resonance energy transfer measurement. J Phys Chem A 110(9):2979–2995 [DOI] [PubMed]
- 25.Rasnik I, Mckinney SA, Ha T (2006) Nonblinking and longlasting single-molecule fluorescence imaging. Nature Methods 3(11):891–893 [DOI] [PubMed]
- 26.Nikova DN, Pope LH, Bennink ML, van Leijenhorst-Groener KA, van der Werf K, Greve J (2004) Unexpected binding motifs for subnucleosomal particles revealed by atomic force microscopy. Biophys J 87(6):4135–4145 [DOI] [PMC free article] [PubMed]
- 27.Lusser A, Kadonaga JT (2004) Strategies for the reconstitution of chromatin. Nature Methods 1(1):19–26 [DOI] [PubMed]