

Abstract
Pulmonary arterial hypertension (PAH), once considered a rare complication of sickle cell disease (SCD) and thalassemia, appears to be more common in adults with hemoglobinopathy than previously appreciated. On prospective screening of adults with SCD, approximately one-third of adults are found on echocardiography to have a tricuspid regurgitant jet velocity (TRV) of 2.5 m/s or higher, many of whom are asymptomatic. Dyspnea on exertion is the most common presenting symptom. This TRV abnormality is a marker for approximately 40% 3-year mortality in adults, and it is associated with laboratory values suggestive of more severe intravascular hemolysis. Release of hemoglobin and arginase from lysed red cells causes scavenging of nitric oxide (NO) and catabolism of L-arginine, the obligate substrate for NO synthase. The resulting impairment in NO bioavailability is associated with pulmonary vasoconstriction, endothelial dysfunction, thrombosis, and eventual development of plexogenic arterial lesions, the histological hallmark of all forms of PAH. Undoubtedly, additional pathophysiological mechanisms will also play a role in its multifactorial pathogenesis. Early data from children with SCD indicate a similar prevalence of elevated TRV, but the prognostic implications of this remain to be established. Individual patient diagnosis of PAH requires confirmation by right heart catheterization studies and individualized management. Hemolysis-associated PAH with impairments in NO bioavailability is being identified in thalassemia and other hemolytic disorders, and may be a general consequence of long-standing, severe intravascular hemolytic anemia.
Keywords: sickle cell, thalassemia, pulmonary hypertension, nitric oxide, arginase
Pulmonary hypertension is a complication of sickle cell disease (SCD) and thalassemia gaining considerable attention in the past 4 years. Cor pulmonale, the clinical syndrome of right ventricular failure, has been considered for decades to be a rare, but well-known, terminal complication in adults with both hemoglobinopathies [1, 2]. As the survival of patients with hemoglobinopathies has improved dramatically in the last forty years, the prevalence of such chronic complications has increased. Well-documented studies of pulmonary arterial hypertension (PAH) have drawn the attention of the hematology community to its frequent occurrence and poor prognosis [3–7]. This article will review the current understanding of hemoglobinopathy-associated pulmonary hypertension, with special attention to the most recently developing data in children.
PREVALENCE AND PROGNOSIS
In the last few years, echocardiographic screening studies have suggested that the prevalence of hemoglobinopathy-associated PAH is much higher than previously known. In SCD, approximately one-third of adult patients have an elevated tricuspid regurgitant jet velocity (TRV) of 2.5 m/s or higher, a threshold that correlates in right heart catheterization studies to a pulmonary artery systolic pressure of at least 30 mm Hg [8–10]. Even though this threshold represents quite mild pulmonary hypertension, SCD patients with TRV above this threshold have a 9- to 10-fold higher risk for early mortality than those with a lower TRV [8, 9]. Our own update to these mortality figures indicate that adults with SCD and TRV ≥2.5 m/s have 40% mortality at 40 months of follow-up. It is as yet unproven whether PAH is the actual cause of death in these cases, although PAH is a frequent cause of death in other studies [11–13]. If it is, then patients with SCD are dying of PAH before the pressures rise to the levels commonly seen in the terminal phase of primary pulmonary hypertension. It appears that the baseline compromised oxygen delivery and co-morbid organ dysfunction of SCD diminishes the physiological reserve to tolerate even modest pulmonary arterial pressures. Large-scale screening in thalassemia intermedia and major also has indicated a high prevalence and adverse prognosis for PAH as estimated by echocardiography [14–17].
PATHOLOGY
Autopsy studies in SCD have revealed histopathological findings common to all forms of PAH, including plexiform and concentric medial hyperplastic pulmonary vascular lesions, and in situ pulmonary arterial thrombosis, although interpretations of these findings has varied (Figure 1) [12, 13, 18, 19]. Similar autopsy findings also have been observed in thalassemia, particularly pulmonary thrombi in patients who had previously undergone splenectomy [20–24]. It remains undetermined in what proportion of these cases pulmonary thrombosis is a cause or effect of PAH. In summary, the histopathology of PAH in SCD and thalassemia is associated with vasoconstriction, proliferative vascular smooth muscle and irregular endothelium in pulmonary arteries with associated thrombosis, all combining to produce luminal narrowing, and eventual right ventricular failure (Figure 1).
FIGURE 1.
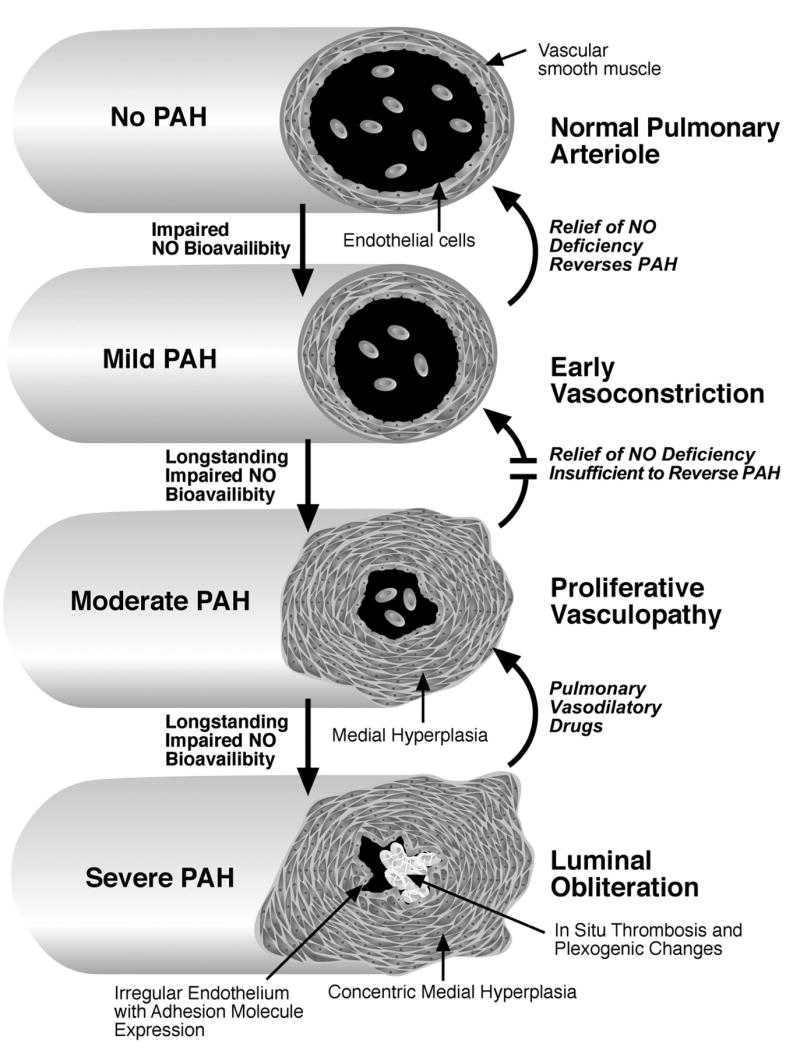
Hypothetical model of the progression of pulmonary arterial hypertension during childhood and adulthood in patients with SCD. In this model, impaired NO bioavailability related to chronic hemolysis results in chronic pulmonary vasoconstriction, mildly elevating pulmonary vascular resistance and pulmonary arterial pressures. As this becomes more long-standing, vascular smooth muscle hyperplasia begins to create a relatively fixed lesion, compounded in later stages by irregular, activated endothelium with expression of adhesion molecules. In situ thrombosis further occludes the vessel lumen, and results in plexogenic changes, further accelerating the progression of the pulmonary arterial hypertension.
The pathophysiology of PAH in hemoglobinopathies is undoubtedly multifactorial, but epidemiological and biochemical data support a prominent role for intravascular hemolysis inducing a state of vascular dysfunction. Clinical variables independently associated with PAH in adults with SCD include the following: low hemoglobin levels, suggestive of more severe hemolytic anemia; high steady-state serum lactate dehydrogenase (LDH) levels, largely reflecting intravascular hemolysis [25]; high serum creatinine levels, indicative of renal insufficiency; high serum direct bilirubin and alkaline phosphatase levels, suggesting cholestatic hepatic dysfunction; low serum transferrin or high serum ferritin levels, indicative of iron overload (Table 1) [8]. In addition, the prevalence of an elevated TRV appears to rise with age during adulthood, exceeding 65% after 50 years of age [8, 9] One study has conflicting findings regarding the relationship of high TRV to systolic blood pressure [9]. Another has found an additional correlation of high TRV to proteinuria [10]. Overall, there is a relationship of elevated TRV to the degree of intravascular hemolysis, and the degrees of renal and hepatic dysfunction, and to iron overload. Except for the contribution of hemolysis, there are no studies to indicate whether the other factors contribute to the development of PAH, or whether they reflect other organs concurrently affected by vasculopathy.
TABLE 1.
Factors Linked Epidemiologically to PAH in Adults with SCD
| Low hemoglobin |
| High LDH |
| High creatinine levels |
| High direct bilirubin |
| High alkaline phosphatase |
| Low serum transferrin |
| High ferritin levels |
| Low arginine:ornithine ratio |
| High systolic blood pressure |
| Increasing age |
| Proteinuria |
Our group has accumulated additional biochemical and physiological evidence implicating intravascular hemolysis in the development of impaired nitric oxide bioavailability and chronic vascular dysfunction [26, 27]. Nitric oxide (NO) is a master regulator of vascular health, producing vasodilation and increased blood flow, in part by promoting vasodilation, and by inhibiting endothelial adhesion molecule expression, vascular smooth muscle proliferation, platelet aggregation and blood coagulation [28]. Pathological processes in SCD accelerate the destruction of NO, and limit the compensatory increase in NO production. Hemoglobin decompartmentalized from the red cell into blood plasma by intravascular hemolysis reaches steady-state levels of 5–10 μM, and at times exceeding 50 μM [29–31]. This hemoglobin reacts with NO in a rapid, nearly diffusion-limited reaction to produce methemoglobin and inert nitrate [29]. This plasma hemoglobin is associated with a relative resistance of vasodilation to NO donors in patients with SCD [32]. It is also clear that from canine hemolysis studies that plasma hemoglobin scavenges NO and produces pulmonary vasoconstriction, hypertension, and renal dysfunction, all partly reversible by inhaled NO [27]. In addition, NO is also consumed by reaction with reactive oxygen species produced as a by-product of the highly expressed enzymatic activities of xanthine oxidase and NADPH oxidase (Table 2) [33, 34]. Lastly, more recent studies suggest that hemolysis leads to uncoupling of endothelial NO synthase activity, likely secondary to heme-mediated oxidative damage to the enzyme, also producing reactive oxygen species [35]. These mechanisms may combine additively to markedly accelerate NO destruction.
TABLE 2.
Mechanisms Implicated in the Vascular Dysfunction of SCD
| NO scavenging by cell-free plasma hemoglobin |
| Plasma L-arginine depletion by cell-free plasma arginase |
| Oxidation of NO by reactive oxygen species produced by xanthine oxidase and NADPH oxidase |
| Endothelial adhesion to red cells, white cells and platelets |
Intravascular hemolysis also limits the expected compensatory increase in NO synthase activity. Arginase, concurrently released from red cells into blood plasma during intravascular hemolysis, converts plasma L-arginine to ornithine, depleting plasma levels of L-arginine, the obligate substrate for NO synthase [36]. The arginine:ornithine ratio, a convenient marker of plasma arginase activity, correlates with pulmonary hypertension and risk of death in patients with sickle cell disease and thalassemia [36, 37]. In this manner, intravascular hemolysis contributes to both decreased production and increased destruction of NO, compounded by reactive oxygen species produced as a side product of xanthine oxidase and NADPH oxidase activity, which are both expressed abundantly in SCD. It is also conceivable that oxidant stress due to chemistry generated by iron and heme released from intravascular hemolysis may also deplete NO [38, 39]. Multiple mechanisms directly and indirectly attributable to hemolysis reduce NO bioavailability in SCD and thalassemia. A similar mechanism also may pertain in malaria and paroxysmal nocturnal hemoglobinuria [40, 41].
CLINICAL PRESENTATION AND COURSE
The most common presenting symptom for PAH in SCD is worsening dyspnea on exertion. This is often attributed by the clinician to anemia, and cardiopulmonary evaluation may be delayed. Symptomatic patients may also have systolic systemic hypertension and peripheral edema. Clubbing of the digits is commonly observed [42]. Laboratory tests in adults with SCD frequently display markers of hemolysis, renal insufficiency, iron overload, and a subtle cholestatic abnormality, out of proportion to other patients with SCD. Such laboratory markers include lower hemoglobin and serum transferrin, higher reticulocyte count, and higher levels of serum lactate dehydrogenase, aspartate aminotransferase, alkaline phosphatase, creatinine and ferritin. The plasma arginine:ornithine ratio is often <0.7. Proteinuria may also be found. Symptomatic patients often have a TRV ≥ 3 m/s. Chest x-ray and computed tomography of the chest may show prominent vascularity, and enlargement of the pulmonary artery. In more advanced cases, computed tomography scans may demonstrate bibasilar interstitial fibrosis and a mosaic perfusion pattern. The diagnosis of PAH must be confirmed with right heart catheterization studies, with expected findings summarized elsewhere [3, 42]. Besides the frequent contribution of hemolysis-associated pulmonary vasoconstriction, in a minority of patients, the pulmonary hypertension may be compounded by mitral valvular insufficiency or left ventricular diastolic dysfunction [43].
As might be expected, in mild PAH, the patients may report no symptoms, but screening echocardiography reveals a TRV of 2.5–2.9 m/s. The same laboratory abnormalities as above may be seen, but typically to a milder degree than in patients with more severe PAH. This mild degree of elevation in the TRV frequently goes unreported on echocardiography reports, with the inappropriate interpretation that it is too small to have clinical consequences. However, adults with SCD have severe anemia with very high cardiac output, less cardiopulmonary reserve, and frequent co-morbid organ dysfunction, and even this small degree of TRV elevation epidemiologically is associated with early mortality.
Adults with SCD during vaso-occlusive pain crisis (VOC) may sometimes develop peripheral or periorbital edema. Although this may be related to overly vigorous hydration, many of these patients have acutely elevated TRV during VOC, with the edema and TRV both returning to normal afterward [44]. These findings suggest that NO bioavailability falls further during hyperhemolysis associated with VOC [30, 31, 45], leading to acute worsening of pulmonary vasoconstriction and consequent right ventricular failure [44]. This may contribute to deaths observed during VOC [12, 44, 46].
Hemoglobin SC disease has a lower prevalence of PAH than homozygous SCD or S-β0-thalassemia, as might be expected based on the lower hemolytic rate [8;10] Fetal hemoglobin expression, while protective for other complications of SCD, has been variably associated with lower rates of PAH [9]. The effect of coinheritance of α-thalassemia and SCD on PAH prevalence has not been yet reported.
TREATMENT
There are currently several FDA-approved drugs for the treatment of pulmonary hypertension, but there are few studies of the efficacy or toxicities of these drugs in patients with hemoglobinopathies. In published pilot experience by our group and others, the phosphodiesterase-5 inhibitor sildenafil reduces pulmonary pressures and improves cardiopulmonary performance in patients with SCD and thalassemia with pulmonary hypertension [47, 48]. The oral endothelin receptor blocker bosentan is being tested in patients with SCD and PAH, but no results are available as yet. There are minimal published experiences as yet in SCD with FDA-approved intravenous, subcutaneous, or inhaled prostacyclins [49], although our group has found anecdotal success using intravenous epoprostenol to stabilize two SCD patients with life-threatening right ventricular failure due to PAH. Additional clinical trials are needed to help establish the safety and efficacy of these agents in hemoglobinopathy patients.
HIGH TRV IN PEDIATRIC PATIENTS WITH SCD
Much less is known regarding the prevalence and natural history of PAH in pediatric patients with hemoglobinopathies. However, the results of 11 small screening studies have been published or presented in abstract form, and all the authors have consented to have their data summarized here (Table 3). Remarkably, aggregated screening results from over 600 children with SCD indicate that 30% have with TRV ≥ 2.5 m/s, nearly identical to 32% in the summarized screening experience in adults with SCD. Six studies have reported a combined prevalence of 8% for TRV ≥ 3 m/s, suggestive of moderate to severe PAH, slightly less than the combined prevalence of 14% in adults. Many of these pediatric studies indicate several correlations of high TRV to the same variables seen in adults: low hemoglobin levels, high reticulocyte counts, bilirubin, and LDH levels (personal communication, Stephen C. Nelson, Farzana D. Pashankar, and Margaret T. Lee) [50–54]. As in adults, these markers implicate hemolysis-associated mechanisms in pediatric PAH. A correlation of high TRV to systolic blood pressure has also been observed in children, confirming a relationship seen in one study in adults (personal communication, Farzana D. Pashkar) [55].
TABLE 3.
Summary of Reports of Echocardiographic Screening in Children and Adults with Sickle Cell Disease
| TRV ≥ 2.5
|
TRV ≥ 3
|
Reported significant correlations | ||||||
|---|---|---|---|---|---|---|---|---|
| Author | Location | Ages (years) | Screened n | n | Percentage | n | Percentage | |
| Morris et al. [58] | CA | 7–17 | 60 | 15 | 25 | n.r. | ||
| Young et al. [50] | MI | 0–18 | 36 | 24a | 66 | n.r. | Low Hb, Plt; high retic, ACS | |
| Nelson et al.b | MN | 4–18 | 53 | 15 | 28 | 3 | 5 | High absolute retic |
| Onyekwere et al. [51] | DC, MI | 8–24 | 52 | 24 | 46 | 6 | 12 | Low Hb; high LDH, bilirubin |
| Pashankar et al.c | CT | 0–18 | 25 | 6 | 24 | 1 | 4 | Low Hb; high SBP |
| Lee at al.d | NY | 7–19 | 55 | 12 | 22 | n.r. | High retic, LDH, AST | |
| Suell et al. [59] | TX | 12–22 | 80 | 21 | 26 | 4 | 5 | None |
| Ambrusko et al. [52] | PA | 0–21 | 44 | 13 | 30 | 6 | 14 | Low Hb; high retic, CVD, chronic Tx |
| Qureshi et al. [60] | CA | 0–21 | 31 | 5a | 16 | n.r. | n.r. | |
| Liem et al. [53] | IL | 10–20 | 43 | 13 | 30 | n.r. | Low Hb; high LDH, retic, WBC | |
| Sedrak et al. [54] | NY | 5–21 | 48 | 4 | 8 | n.r. | High bilirubin | |
| Joyce et al. [57] | DC | 0–21 | 76 | 29 | 38 | 6 | 8 | n.r. |
| Gladwin et al. [8] | MD, DC | ≥ 18 | 195 | 63 | 32 | 17 | 9 | Low Hb, Tf; high LDH, alk phos, SBP |
| Ataga et al. [9] | NC | ≥18 | 60 | 18 | 30 | 7 | 12 | Low Hb, SBP |
| De Castro et al. [10] | NC | ≥18 | 125 | 40 | 32 | 28 | 22 | Low Hb |
| Pediatric totals | 603 | 181 | 30 | 26 | 8 | |||
| Adult totals | 380 | 121 | 32 | 52 | 14 | |||
Note. TRV, tricuspid regurgitant jet velocity; n.r., not reported; Hb, hemoglobin; Plt, platelet count; retic, reticulocyte count; ACS, acute chest syndrome incidence; LDH, serum lactate dehydrogenase; SBP, systolic blood pressure; AST, serum aspartate aminotransferase; CVD, history of cerebrovascular disease; Tx, transfusion therapy; Tf, serum transferrin; alk phos, serum alkaline phosphatase; WBC, white blood cell count.
Reported number of patients with estimated right ventricular systolic pressure >30 mm Hg instead of TRV ≥ 2.5, which is approximately equivalent.
Personal communication, Stephen Nelson.
Personal communication, Farzana Pashankar.
Personal communication, Margaret T. Lee.
Overall, preliminary studies suggest a provocatively high prevalence of elevated TRV in children with SCD. There are several caveats in these data. These pediatric studies are a mixture of retrospective and prospective designs with variable entry criteria and most have yet to undergo peer review. Many of them likely reflect a bias toward increased screening in more symptomatic patients. Prospective screening studies are needed to indicate the true prevalence and prognosis of elevate TRV in the pediatric SCD population.
DOES A HIGH TRV MEAN PAH?
The TRV has been a highly useful tool in adults with SCD, in whom there is a good statistical correlation with right heart catheterization, the gold standard for the diagnosis of PAH. The TRV is very useful in epidemiological studies and for screening purposes. However, its specificity is not sufficient to diagnose PAH in individual patients. In our own anecdotal observations, young adults with particularly high cardiac output occasionally have a mildly elevated TRV without evidence on right heart catheterization of increased pulmonary pressures or vascular resistance. In adults, a high TRV is a marker that predicts early mortality in SCD, and might be a possible indication to consider general measures such as institution of hydroxyurea or chronic transfusion therapy. Pulmonary vasodilator therapy should be considered only in those patients with PAH confirmed by right heart catheterization. It is not yet proven whether such interventions prolong survival in SCD patients with elevated TRV. In children, the sensitivity and specificity of the TRV for identifying PAH requires more study.
IS IT BETTER TO DIAGNOSE PAH AT A YOUNG AGE?
Lessons learned in PAH due to other causes make a case for screening for PAH in children with SCD. In an example familiar to pediatricians, diagnosis of an atrial septal defect in childhood generally leads to surgical repair and reversal of PAH with no significant sequelae. However, if diagnosis of the defect is delayed to early adulthood, PAH may become more fixed with potentially irreversible histological and functional changes, leading to early death or need for lung transplantation.
We propose that a similar sequence of events may occur in patients with SCD (Figure 1). In this model, hemolysis-associated impairment of NO bioavailability over decades causes chronic vasoconstriction and mild pulmonary hypertension. At this point, decreasing hemolysis by chronic transfusion might ameliorate the NO consumption and deficiency, and thereby reverse the pulmonary vasoconstriction and pulmonary hypertension. There are some data available to support this model. Lezcano et al. have reported that chronic transfusion lowers plasma hemoglobin in children with sickle cell disease [56]. Furthermore, one group has indicated that children with SCD on chronic transfusion have lower average TRV and estimated RV pressure than those not chronically transfused [57]. In an informative case, a 5-year old child with a TRV of 3.1 m/s was treated with chronic transfusion therapy, on which the TRV declined to 0.7 m/s after 19 months, supporting a potential for reversal of high TRV in young children (personal communication, Margaret T. Lee).
By age 20–50 years, we suggest that more extensive vascular smooth muscle hyperplasia will have occurred, with luminal narrowing that may not respond to therapeutic maneuvers to reduce hemolysis (Figure 1, bottom two stages). In even more advanced cases, irregular, chronically activated endothelium may have accrued in situ thrombosis and plexogenic changes that dramatically increase pulmonary vascular resistance. In such advanced stages, reduction of hemolysis by transfusion may be ineffective, and only pharmacological treatment with pulmonary vasodilator drugs may produce improvement. These drugs are expected to improve pulmonary pressures and cardiopulmonary performance, but gradual progression of the disease is still likely, as in other forms of PAH.
This is a model that merits additional testing, and several sickle cell centers are accumulating more TRV data in their pediatric patients with SCD. Such prospectively gathered data, supplemented by long-term follow-up data, will provide valuable information regarding the prognostic significance of an elevated TRV in childhood. This will guide future clinical decision making regarding the appropriateness of hydroxyurea, transfusions, or pulmonary vasodilator therapy.
Acknowledgments
G.J.K. and M.T.G. are supported by intramural funding from the National Heart, Lung and Blood Institute and the Clinical Center of the National Institutes of Health. M.T.G. also receives research support through a cooperative research and development agreement with INO Therapeutics. O.C.O. receives support from Novartis Pharmaceuticals and INO Therapeutics. We thank Stephen C. Nelson, Farzana D. Pashankar, and Margaret T. Lee for sharing unpublished data.
Contributor Information
Gregory J. Kato, Vascular Medicine Branch, National Heart, Lung and Blood Institute, and Critical Care Medicine Department, Clinical Center, National Institutes of Health, Bethesda, Maryland, USA
Onyinye C. Onyekwere, Center for Sickle Cell Disease, Howard University College of Medicine, Washington, DC, USA
Mark T. Gladwin, Vascular Medicine Branch, National Heart, Lung and Blood Institute, and Critical Care Medicine Department, Clinical Center, National Institutes of Health, Bethesda, Maryland, USA
References
- 1.Moser KM, Shea JG. The relationship between pulmonary infarction, cor pulmonale and the sickle states. Am J Med. 1957;22:561–579. doi: 10.1016/0002-9343(57)90110-9. [DOI] [PubMed] [Google Scholar]
- 2.Koren A, Garty I, Antonelli D, et al. Right ventricular cardiac dysfunction in beta-thalassemia major. Am J Dis Child. 1987;141:93–96. [PubMed] [Google Scholar]
- 3.Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood. 2003;101:1257–1261. doi: 10.1182/blood-2002-03-0948. [DOI] [PubMed] [Google Scholar]
- 4.Collins FS, Orringer EP. Pulmonary hypertension and cor pulmonale in the sickle hemoglobinopathies. Am J Med. 1982;73:814–821. doi: 10.1016/0002-9343(82)90763-x. [DOI] [PubMed] [Google Scholar]
- 5.Sutton LL, Castro O, Cross DJ, et al. Pulmonary hypertension in sickle cell disease. Am J Cardiol. 1994;74:626–628. doi: 10.1016/0002-9149(94)90760-9. [DOI] [PubMed] [Google Scholar]
- 6.Aessopos A, Stamatelos G, Skoumas V, et al. Pulmonary hypertension and right heart failure in patients with beta-thalassemia intermedia. Chest. 1995;107:50–53. doi: 10.1378/chest.107.1.50. [DOI] [PubMed] [Google Scholar]
- 7.Atichartakarn V, Likittanasombat K, Chuncharunee S, et al. Pulmonary arterial hypertension in previously splenectomized patients with beta-thalassemic disorders. Int J Hematol. 2003;78:139–145. doi: 10.1007/BF02983382. [DOI] [PubMed] [Google Scholar]
- 8.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350:886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 9.Ataga KI, Sood N, De GG, et al. Pulmonary hypertension in sickle cell disease. Am J Med. 2004;117:665–669. doi: 10.1016/j.amjmed.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 10.De Castro LM, Jonassaint JC, Graham FL, et al. Pulmonary hypertension in SS, SC and Sβ thalassemia: prevalence, associated clinical syndromes, and mortality. Blood. 2004;104:462a. [Google Scholar]
- 11.Powars DR, Chan LS, Hiti A, et al. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore) 2005;84:363–376. doi: 10.1097/01.md.0000189089.45003.52. [DOI] [PubMed] [Google Scholar]
- 12.Manci EA, Culberson DE, Yang YM, et al. Causes of death in sickle cell disease: an autopsy study. Br J Haematol. 2003;123:359–365. doi: 10.1046/j.1365-2141.2003.04594.x. [DOI] [PubMed] [Google Scholar]
- 13.Adedeji MO, Cespedes J, Allen K, et al. Pulmonary thrombotic arteriopathy in patients with sickle cell disease. Arch Pathol Lab Med. 2001;125:1436–1441. doi: 10.5858/2001-125-1436-PTAIPW. [DOI] [PubMed] [Google Scholar]
- 14.Aessopos A, Farmakis D, Karagiorga M, et al. Cardiac involvement in thalassemia intermedia: a multicenter study. Blood. 2001;97:3411–3416. doi: 10.1182/blood.v97.11.3411. [DOI] [PubMed] [Google Scholar]
- 15.Aessopos A, Farmakis D, Deftereos S, et al. Thalassemia heart disease: a comparative evaluation of thalassemia major and thalassemia intermedia. Chest. 2005;127:1523–1530. doi: 10.1378/chest.127.5.1523. [DOI] [PubMed] [Google Scholar]
- 16.Du ZD, Roguin N, Milgram E, et al. Pulmonary hypertension in patients with thalassemia major. Am Heart J. 1997;134:532–537. doi: 10.1016/s0002-8703(97)70091-7. [DOI] [PubMed] [Google Scholar]
- 17.Wu KH, Chang JS, Su BH, et al. Tricuspid regurgitation in patients with beta-thalassemia major. Ann Hematol. 2004;83:779–783. doi: 10.1007/s00277-004-0954-8. [DOI] [PubMed] [Google Scholar]
- 18.Gerry JL, Bulkley BH, Hutchins GM. Clinicopathologic analysis of cardiac dysfunction in 52 patients with sickle cell anemia. Am J Cardiol. 1978;42:211–216. doi: 10.1016/0002-9149(78)90902-5. [DOI] [PubMed] [Google Scholar]
- 19.Haque AK, Gokhale S, Rampy BA, et al. Pulmonary hypertension in sickle cell hemoglobinopathy: a clinicopathologic study of 20 cases. Hum Pathol. 2002;33:1037–1043. doi: 10.1053/hupa.2002.128059. [DOI] [PubMed] [Google Scholar]
- 20.Grisaru D, Rachmilewitz EA, Mosseri M, et al. Cardiopulmonary assessment in beta-thalassemia major. Chest. 1990;98:1138–1142. doi: 10.1378/chest.98.5.1138. [DOI] [PubMed] [Google Scholar]
- 21.Sonakul D, Pacharee P, Laohapand T, et al. Pulmonary artery obstruction in thalassaemia. Southeast Asian J Trop Med Public Health. 1980;11:516–523. [PubMed] [Google Scholar]
- 22.Sonakul D, Thakerngpol K, Pacharee P. Cardiac pathology in 76 thalassemic patients. Birth Defects Orig Artic Ser. 1988;23:177–191. [PubMed] [Google Scholar]
- 23.Sonakul D, Fucharoen S. Pulmonary thromboembolism in thalassemic patients. Southeast Asian J Trop Med Public Health. 1992;23(Suppl 2):25–28. [PubMed] [Google Scholar]
- 24.Hahalis G, Manolis AS, Apostolopoulos D, et al. Right ventricular cardiomyopathy in beta-thalassaemia major. Eur Heart J. 2002;23:147–156. doi: 10.1053/euhj.2001.2709. [DOI] [PubMed] [Google Scholar]
- 25.Kato GJ, McGowan VR, Machado RF, et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension and death in patients with sickle cell disease. Blood. 2006;107:2279–2285. doi: 10.1182/blood-2005-06-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rother RP, Bell L, Hillmen P, et al. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293:1653–1662. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- 27.Minneci PC, Deans KJ, Zhi H, et al. Hemolysis-associated endothelial dysfunction mediated by accelerated NO inactivation by decompartmentalized oxyhemoglobin. J Clin Invest. 2005;115:3409–3417. doi: 10.1172/JCI25040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walford G, Loscalzo J. Nitric oxide in vascular biology. J Thromb Haemost. 2003;1:2112–2118. doi: 10.1046/j.1538-7836.2003.00345.x. [DOI] [PubMed] [Google Scholar]
- 29.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 30.Neely CL, Wajima T, Kraus AP, et al. Lactic acid dehydrogenase activity and plasma hemoglobin elevations in sickle cell disease. Am J Clin Pathol. 1969;52:167–169. doi: 10.1093/ajcp/52.2.167. [DOI] [PubMed] [Google Scholar]
- 31.Naumann HN, Diggs LW, Barreras L, et al. Plasma hemoglobin and hemoglobin fractions in sickle cell crisis. Am J Clin Pathol. 1971;56:137–147. doi: 10.1093/ajcp/56.2.137. [DOI] [PubMed] [Google Scholar]
- 32.Gladwin MT, Schechter AN, Ognibene FP, et al. Divergent nitric oxide bioavailability in men and women with sickle cell disease. Circulation. 2003;107:271–278. doi: 10.1161/01.cir.0000044943.12533.a8. [DOI] [PubMed] [Google Scholar]
- 33.Aslan M, Freeman BA. Oxidant-mediated impairment of nitric oxide signaling in sickle cell disease—mechanisms and consequences. Cell Mol Biol (Noisy-le-grand) 2004;50:95–105. [PubMed] [Google Scholar]
- 34.Wood KC, Hebbel RP, Granger DN. Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J. 2005;19:989–991. doi: 10.1096/fj.04-3218fje. [DOI] [PubMed] [Google Scholar]
- 35.Hsu LL, Champion HC, Campbell-Lee SA, et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood. doi: 10.1182/blood-2006-08-039438. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris CR, Kato GJ, Poljakovic M, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension and mortality in sickle cell disease. JAMA. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris CR, Kuypers FA, Kato GJ, et al. Hemolysis-associated pulmonary hypertension in thalassemia. Ann N Y Acad Sci. 2005;1054:481–5. 481–485. doi: 10.1196/annals.1345.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hebbel RP. Auto-oxidation and a membrane-associated ‘Fenton reagent’: a possible explanation for development of membrane lesions in sickle erythrocytes. Clin Haematol. 1985;14:129–140. [PubMed] [Google Scholar]
- 39.Sadrzadeh SM, Graf E, Panter SS, et al. Hemoglobin: a biologic Fenton reagent. J Biol Chem. 1984;259:14354–14356. [PubMed] [Google Scholar]
- 40.Gramaglia I, Sobolewski P, Meays D, et al. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat Med. 2006;12:1417–1422. doi: 10.1038/nm1499. [DOI] [PubMed] [Google Scholar]
- 41.Heller PG, Grinberg AR, Lencioni M, et al. Pulmonary hypertension in paroxysmal nocturnal hemoglobinuria. Chest. 1992;102:642–643. doi: 10.1378/chest.102.2.642. [DOI] [PubMed] [Google Scholar]
- 42.Machado RF, Gladwin MT. Chronic sickle cell lung disease: new insights into the diagnosis, pathogenesis and treatment of pulmonary hypertension. Br J Haematol. 2005;129:449–464. doi: 10.1111/j.1365-2141.2005.05432.x. [DOI] [PubMed] [Google Scholar]
- 43.Sachdev V, Machado RF, Shizukuda Y, et al. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. 2007;49:472–479. doi: 10.1016/j.jacc.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Machado RF, Mack AK, Martyr S, et al. Severity of pulmonary hypertension during vaso-occlusive pain crisis and exercise in patients with sickle cell disease. Br J Haematol. 2007;136:319–325. doi: 10.1111/j.1365-2141.2006.06417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ballas SK, Marcolina MJ. Hyperhemolysis during the evolution of uncomplicated acute painful episodes in patients with sickle cell anemia. Transfusion (Paris) 2006;46:105–110. doi: 10.1111/j.1537-2995.2006.00679.x. [DOI] [PubMed] [Google Scholar]
- 46.Parfrey NA, Moore W, Hutchins GM. Is pain crisis a cause of death in sickle cell disease? Am J Clin Pathol. 1985;84:209–212. doi: 10.1093/ajcp/84.2.209. [DOI] [PubMed] [Google Scholar]
- 47.Machado RF, Martyr S, Kato GJ, et al. Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. Br J Haematol. 2005;130:445–453. doi: 10.1111/j.1365-2141.2005.05625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Derchi G, Forni GL, Formisano F, et al. Efficacy and safety of sildenafil in the treatment of severe pulmonary hypertension in patients with hemoglobinopathies. Haematologica. 2005;90:452–458. [PubMed] [Google Scholar]
- 49.Kaur K, Brown B, Lombardo F. Prostacyclin for secondary pulmonary hypertension. Ann Intern Med. 2000;132:165. doi: 10.7326/0003-4819-132-2-200001180-00019. [DOI] [PubMed] [Google Scholar]
- 50.Young EM, Zilberman MV, Du W, et al. Pulmonary hypertension in pediatric patients with sickle cell disease: a retrospective study. Blood. 2004;104:23b. [Google Scholar]
- 51.Onyekwere OC, Campbell AD, Teshome M, et al. Pulmonary hypertension in sickle cell disease children and adolescents. Pediatr Cardiol. doi: 10.1007/s00246-007-9018-x. in press. [DOI] [PubMed] [Google Scholar]
- 52.Ambrusko SJ, Gunawardena S, Sakara A, et al. Elevation of tricuspid regurgitant jet velocity, a marker for pulmonary hypertension in children with sickle cell disease. Pediatr Blood Cancer. 2006;47:907–913. doi: 10.1002/pbc.20791. [DOI] [PubMed] [Google Scholar]
- 53.Liem RI, Willingham NM, Young LT, et al. Tricuspid regurgitant jet velocity is signficantly associated with hemolysis in the evaluation of pulmonary hypertension in children and young adults with sickle cell disease. Blood. 2006;108:356a. [Google Scholar]
- 54.Sedrak A, Rao SP, Miller ST, et al. Pulmonary hypertension in children and adolescents with sickle cell disease. Blood. 2006;108:21b–22b. [Google Scholar]
- 55.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350:886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 56.Lezcano NE, Odo N, Kutlar A, et al. Regular transfusion lowers plasma free hemoglobin in children with sickle-cell disease at risk for stroke. Stroke. 2006;37:1424–1426. doi: 10.1161/01.STR.0000221173.97108.01. [DOI] [PubMed] [Google Scholar]
- 57.Joyce K, Sable C, Martin B, et al. Pulmonary artery hypertension in children with sickle cell disease: is chronic transfusion protective? Blood. 2006;108:356a. [Google Scholar]
- 58.Morris CR, Gardner J, Hagar W, et al. Pulmonary hypertension in sickle cell disease: a common complication for both adults and children. Blood. 2004;104:463a. [Google Scholar]
- 59.Suell MN, Bezold LI, Okcu MF, et al. Increased pulmonary artery pressures among adolescents with sickle cell disease. J Pediatr Hematol Oncol. 2005;27:654–658. doi: 10.1097/01.mph.0000194022.17968.bf. [DOI] [PubMed] [Google Scholar]
- 60.Qureshi N, Joyce JJ, Qi N, et al. Right ventricular abnormalities in sickle cell anemia: evidence of a progressive increase in pulmonary vascular resistance. J Pediatr. 2006;149:23–27. doi: 10.1016/j.jpeds.2005.12.055. [DOI] [PubMed] [Google Scholar]