

Abstract
We present an example of expression and purification of a biologically active G protein coupled receptor (GPCR) from yeast. An expression vector was constructed to encode the Saccharomyces cerevisiae GPCR α-factor receptor (Ste2p, the STE2 gene product) containing a 9-amino acid sequence of rhodopsin that served as an epitope/affinity tag. In the construct, two glycosylation sites and two cysteine residues were removed to aid future structural and functional studies. The receptor was expressed in yeast cells and was detected as a single band in a western blot indicating the absence of glycosylation. Ligand binding and signaling assays of the epitope-tagged, mutated receptor showed it maintained the full wild-type biological activity. For extraction of Ste2p, yeast membranes were solubilized with 0.5 % n-dodecyl maltoside (DM). Approximately 120 μg of purified α-factor receptor was obtained per liter of culture by single-step affinity chromatography using a monoclonal antibody to the rhodopsin epitope. The binding affinity (Kd) of the purified α-factor receptor in DM micelles was 28 nM as compared to Kd = 12.7 nM for Ste2p in cell membranes, and approximately 40 % of the purified receptor was correctly folded as judged by ligand saturation binding. About 50 % of the receptor sequence was retrieved from MALDI-TOF and nanospray mass spectrometry after CNBr digestion of the purified receptor. The methods described will enable structural studies of the α-factor receptor and may provide an efficient technique to purify other GPCRs that have been functionally expressed in yeast.
Introduction
G-protein coupled receptors (GPCRs) are integral membrane proteins characterized by seven transmembrane helices and comprise one of the largest known superfamilies of receptors with in excess of 2000 genes identified across taxa [1]. They are attractive and proven drug targets in a wide range of therapeutic areas due to their involvement in signaling and response to diverse external stimuli in nearly all human cell types [2].
Because of their central role in biological systems, a detailed understanding of the interaction of GPCRs with their ligands and associated G proteins is essential. Structural characterization of these receptors has been limited, however, because of the difficulty in producing significant quantities of GPCRs in functionally active form. Furthermore, once sufficiently purified, high resolution structural analysis of GPCRs are not straightforward owing to their high degree of hydrophobicity and conformational flexibility [3]. Thus far, bovine rhodopsin is the only GPCR whose crystal structure has been reported [4]. This structure resulted from high-level purification from bovine retina in which over 50% of the membrane protein is rhodopsin so that extending this purification approach to other receptors is not applicable. For the purification of GPCRs various expression hosts and purification strategies have been developed and tested. These include E. coli, yeasts, insect and mammalian cells for the expression system, and adopting ligand-, metal-, or immuno-affinity tags for the purification purpose. It is widely perceived that no specific expression or purification strategy can be considered as a standard method due to the intrinsic diversity of the behavior of each GPCR during expression and purification processes (for a review, see [5]).
GPCRs for peptide ligands exist in yeast cells. Haploid Saccharomyces cerevisiae exists in one of two mating types, (MATa and MATα). Mating occurs between cells of opposite type and is controlled by the exchange of mating peptides acting as pheromones: MATa-cells secrete a-factor and express the α-factor receptor (Ste2p) whereas MATα-cells secrete α-factor and express the a-factor receptor (Ste3p). Both receptors couple to the same heterotrimeric G-protein composed of Gα (Gpa1p), Gβ (Ste4p) and Gγ (Ste18p), and stimulation results in the release of the Gβγ dimer, which then interacts with effectors to bring about conjugation. α-Factor receptor (Ste2p) is one of the prototypical GPCRs that has been widely used as a model system for studying structure-function relationships of ligand-GPCR interaction, the mechanism of G-protein activation upon ligand binding and the regulation of downstream signaling processes via heterotrimeric G-protein [6].
Versatility of the S. cerevisiae system in studying GPCRs has been also manifested by many successful functional expressions of mammalian GPCRs in this yeast [7]. In addition, the S. cerevisiae system offers many benefits, such as robust and fast growth, a completely sequenced genome, easy genetic manipulation, inexpensive media and simple nutritional requirement.
Purification of α-factor receptor in two different systems has been reported. In a study using a native S. cerevisiae expression system [8], 1 mg quantities of His-epitope tagged α-factor receptor was purified on a Ni-NTA column and reconstituted in lipid vesicles. The binding activity of the reconstituted receptor indicated that only 6 % of the receptor was capable of ligand binding, but the addition of solubilized membranes from S. cerevisiae to the artificial membrane restored most of expected ligand binding activity (at least 80 %). Nevertheless, the co-factors for this effect were not identified despite extensive efforts. In another study, α-factor receptor was purified using a transient expression system in HEK293 EBNA cell line [9]. About 1 mg of Ste2p was purified per liter of culture with relatively high affinity to a fluorescently-labeled α-factor. However, for large scale purification a stable expression system is required, and the heterogeneous glycosylation of the recombinant protein in a mammalian system may interfere with the formation of diffractable crystals.
In this work we describe methods for efficient affinity purification of Ste2p expressed in native yeast cells. Non-glycosylated, cysteine-less α-factor receptor containing a rhodopsin affinity tag was successfully purified yielding mg quantities of Ste2p with high ligand affinity. Liquid chromatography tandem mass spectrometry analysis revealed that the purified preparation was highly enriched; with strict filtering criteria no extraneous proteins was identified in the purified Ste2p preparation. The quantity and purity obtained should facilitate biophysical and structural studies of Ste2p, and the methods used should guide future efforts to purify GPCRs expressed in yeast.
Materials and Methods
Yeast strains, plasmids, and media
S. cerevisiae strains used were LM102 [10] and BJS21 [11]. The relevant genotype of LM102 is MATa, bar1, leu2, ura3, FUS1-lacZ::URA3, ste2-dl (the α-factor receptor coding region deletion) and that of BJS21 is MATa, prc1-407 prb1-1122 pep4-3 leu2 trp1 ura3-52 ste2::KanR. LM102 was used to measure α-factor-induced growth arrest (halo assay) and to determine α-factor binding using whole cells. The strain carried the bar1 mutant allele inactivating the Bar1p protease that is responsible for degradation of α-factor. Protease deficient BJS21 strain was used for receptor purification due to its high level of receptor expression (see Results). The wild-type STE2 gene, with a native promoter was cloned into a yeast/bacterial shuttle vector, pGA314.WT [12] and used as a template for the generation of an expression construct in this study. The pGA314 plasmid carries the TRP1 gene as a selectable marker and is a low copy CEN-based plasmid.
Site-directed Mutagenesis
Single-stranded phagemid DNA of pGA314.WT was prepared by infecting Escherichia coli strain CJ236 (ung−, dut−) carrying pGA314.WT with the helper phage M13KO7. Oligonucleotide-directed mutagenesis of single-stranded phagemid DNA was constructed as described by Kunkel et al. [13]. The product of the mutagenesis reaction mixture was transformed into E. coli strain DH5 (Life Technologies, Inc.), and transformants were selected on ampicillin-containing plates. Plasmids were then isolated from transformants using the Wizard Miniprep kit (Promega). Two cysteines at residue 59 and 252 and two known glycosylation sites (Asn 25 and Asn32) were replaced by Serine and Glutamine, respectively. An epitope tag comprising codons encoding a 9 amino acid sequence of rhodopsin (rho-tag) was introduced at the C-terminal end of the receptor sequence for the affinity purification of the receptor. The rho epitope was first used successfully in a purification scheme for the parathyroid hormone receptor [14] and subsequently has been used for purification of other GPCRs [5]. After sequence confirmation, the resulting plasmid was digested with Bam HI and EcoRI, ligated into p424GPD overexpression vector [15], and digested with the same restriction enzymes generating pBKY1. Throughout this report, α-factor receptor expressed from pBKY1 is denoted as Ste2p-rho while the receptor with no cysteine residues is denoted as wild type. Plasmid pBKY1 was transformed into the yeast strains LM102 or BJS21, and transformants were selected by their growth in medium lacking tryptophan. All primers were purchased from Sigma/Genosys (The Woodlands, TX). DNA sequencing was carried out in the DNA sequencing facility located on the campus of the University of Tennessee.
Synthesis and Purification of Peptides
[Nle12]α-factor was synthesized as described previously [16]. This isosteric ]α-factor analog has identical biological and binding affinities as the native pheromone [17]. In this paper we refer to it as ]α-factor. The 9-amino acid peptide of the C terminus of rhodopsin (TETSQVAPA) was synthesized on an Applied Biosystems model 433A synthesizer using FastMoc chemistry. The purity and identity of peptide was determined by analytical HPLC followed by electrospray mass spectrometry (ES-MS). The rhodopsin C-terminal peptide calculated molecular mass was 945 Da and the experimental average molecular mass was 944.45.
Growth Arrest (Halo) Assay
Yeast nitrogen base medium (Difco) without amino acids (SD medium) supplemented with histidine (20 μg/ml), leucine (30 μg/ml), and methionine (20 μg/ml) was overlaid with 4 ml of cell suspension (2.5 × 105 cells/ml of Nobel agar). Filter disks (sterile blanks from Difco), 8 mm in diameter, were impregnated with 10-μl portions of α-factor at various concentrations and placed onto the overlay. The plates were incubated at 30 °C for 24–36 h and then observed for clear zones (halos) around the disks. The data were expressed as the diameter of the halo including the diameter of the disc. All assays were carried out at least three times with no more than a 2-mm variation in halo size at a particular amount applied for each peptide. The data were plotted as halo size versus the amount of peptide and linearized by regression analysis.
Binding Assays
Whole cell saturation binding assay was performed as described previously [18]. Briefly, cells were grown overnight at 30 °C and harvested at 1 × 107 cells/ml by centrifugation at 5000 g at 4 °C. The pelleted cells were washed two times in ice-cold YM-1 medium [18] and resuspended to 2 × 107 cells/ml. Various concentrations of [3H]α-factor were added to the cell suspension and incubated for 30 min. After incubation, triplicate samples of 200 μL were filtered and washed over glass fiber filtermats using the Standard Cell Harvester (Skatron Instruments, Sterling, VA) and placed in scintillation vials for counting. Specific binding was determined by subtracting counts associated with the LM102 (ste2 deletion) strain from counts bound to the strains harboring wild-type or Ste2p-rho receptors. Each experiment was carried out at least three times with similar results. Nonspecific binding of labeled α-factor to filters in the absence of cells was less than 20 cpm.
For the binding assay of the purified Ste2p-rho, the purified receptor (80 ng each) in Buffer B (50 mM HEPES, 0.15 M NaCl, 2 mM CaCl2, 5 mM KCl, 5 mM MgCl2, 4 mM EDTA, 15 % glycerol, pH 6.8) with 0.025% n-dodecyl maltoside (DM) was mixed with various concentration of either [3H]α-factor alone (for total binding) or [3H]α-factor plus 100-fold excess of non-labeled α-factor (for non-specific binding) in a final volume of 400 μl. The mixtures were swirled gently at RT for 30 min. Two-hundred μL of each reaction mixture was transferred into microcentrifuge tubes holding a binding membrane filter with 30 Kd MW cut-off size. The tubes were prewashed with Buffer B with 0.025% DM. After 1 min centrifugation at full speed in a microcentrifuge, the membrane filters were washed two times with 400 μL of Buffer B (0.025% DM). After the final wash, the filter units were transferred into scintillation vials containing 5 ml of scintillation fluid and read for radioactive counts. Data curves for binding were fitted from at least eight triplicate data points with GraphPad Prism software (nonlinear regression, one site competition or saturation).
Membrane preparation and Western blot
One to six liters of yeast culture, grown overnight, was harvested and suspended in Buffer A (10 mM HEPES and 4 mM EDTA, pH 7.0) with proteinase inhibitors [100X stock: pepstatin (1mg/mL), leupeptin (1mg/mL), and PMSF (10 mg/mL)]. Then the cells were homogenized using glass beads with a tissue grinder. The homogenates were centrifuged at 500 g for 5 min at 4 °C to remove debris and unbroken cells. Supernatants were centrifuged at 40,000 g for 30 min at 4 °C, the supernatants were discarded, and the membrane pellets were suspended in Buffer B with the same proteinase inhibitors. The enriched membrane fractions were stable for at least 3 weeks under these conditions.
Membrane proteins and purified receptor were resolved by SDS-PAGE (12 % Bis-Tris gels, Invitrogen). For Western blots, the gel was transferred to Immobilon-P membrane (Millipore Corporation, Bedford, MA). The blot was probed with 1D4 antibody (a monoclonal antibody for rho-tag, purchased from Flintbox, British Columbia, Canada) and bands were visualized with West Pico chemiluminescent detection system (Pierce Biotechnology, Inc., Rockford, IL).
Membrane solubilization and Ste2p-rho purification
The membrane proteins were diluted to a concentration of 0.5 mg/mL with Buffer B with 0.5 % DM and were then solubilized by gentle shaking for 2 hrs at 4 °C. A small amount of insoluble material was removed by ultracentrifugation at 100,000 g for 30 min at 4 °C. The supernatant was then mixed with antibody 1D4-coupled Sepharose 4B and incubated on a rotary shaker for 16 hrs at 4 °C. The resin slurry was batch-loaded onto a column, and the column was washed with at least 100 column volumes of Buffer B. During the washing step, the concentration of DM was gradually decreased to 0.025 % before elution. The elution of bound receptor was accomplished by addition of the 9-amino acid rhodopsin peptide (100 molar excess in Buffer B with 0.025 % DM). The eluted receptor was washed and concentrated using an Ultra centrifugal filter with a 50 kDa MW cut-off value.
Chemical Digestion of the Purified Protein
For CNBr digestion, the final elution sample was dissolved in 70 % formic acid, and 200 mg/mL of CNBr were added. All digestion reactions took place under nitrogen and complete darkness at 37 °C overnight. After digestion, samples were dried by vacuum centrifugation, resuspended in 1ml H2O, and re-dried twice. Finally the sample was resuspended in 50 mL H2O and used for analysis.
Mass Spectrometry
a. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS)
MALDI-TOF experiments were performed on a PerSeptive Biosystems Voyager Elite MALDI-TOF, equipped with a 337 nm nitrogen laser, operating in the positive ion mode (delayed extraction/linear mode). Spectra were obtained in reflector mode, with +20 kV total acceleration voltage, +18 kV applied to the grid, +10 V applied to the guide wire, and 210 ns acceleration delay. Spectra were averaged from up to 256 individual laser pulses, obtained from several locations on each sample spot. Digested protein aliquots were applied to a pre-spotted thin-layer matrix (α-cyano-4-hydroxycinnamic acid, CHCA or Sinapinic acid, SA). External calibration was performed using gramicidin S and bovine insulin β-chain. Expected masses were calculated using Protein Prospector [19].
b. Nanospray mass spectrometry (NS-MS)
Nano LC-MS/MS analyses of the digested protein samples were performed on a integrated Famos/Switchos/Ultimate 1D switching HPLC system (LC Packings, a division of Dionex, San Francisco, CA) coupled with a nanospray ionization source on a LCQ-DECA quadrupole ion trap instrument (Thermo Finnigan) operating in the positive ion mode (all experiments performed at Oak Ridge National Laboratory, TN). Briefly, peptide samples were injected onto a C18 trap cartridge with the Famos autosampler, washed and backed flushed onto a VYDAC C18 column (75 μm id × 15 cm, 300Å with 5 μm particles), separated by a reverse phase separation and ionized (2.0kV) into the ion trap mass spectrometer. For all experiments, the MS was operated in the data dependent MS/MS mode, where the four most abundant peaks in every full MS scan were subjected to MS/MS analysis. All MS/MS spectra were analyzed by DBDigger algorithm [20] against all open reading frames from S. cerevisiae as well as the tagged plasmid sequence for Ste2p-rho. Spectra were searched against the composite database with following parameters: enzyme type, non-specific; Parent Mass Tolerance, 3.0; Fragment Ion Tolerance, 0.5. Variable modifications of −30 daltons and −48 daltons on C-terminal methionine residues were considered as well as methionine oxidations (+16 daltons). The output data files were then filtered and sorted with the DTASelect algorithm [21] using the following parameters: delCN of at least 0.08 and cross-correlation scores (Xcorrs) of at least 23 (+1), 28 (+2) and 43 (+3) and at least four unique peptides per protein. All peptide MS/MS spectra were manually verified after identification by search engine.
Results and Discussion
Expression and signaling activity of the non-glycosylated, rho epitope-tagged α-factor receptor
For Ste2p purification, an expression vector pBKY1 was constructed. This vector was derived from the 2μ-based p424GPD plasmid and contains the STE2 gene under the constitutive GPD promoter with multiple modifications: two cysteine residues (Cys59 and Cys252) were replaced by Ser, two glycosylation sites (Asn25 and Asn32) were mutated to Gln, and an epitope tag comprising the 9 amino acid sequence of rhodopsin (rho-tag) was introduced at the C-terminal end of the receptor sequence. The resulting modified Ste2p was designated as Ste2p-rho. The plasmid pBKY1 was transformed into the LM102 strain to test expression and biological activities. Ste2p with double Cys mutations (C59S/C252S) was used as a wild type control. Previously it had been shown that the double Cys mutation did not affect the receptor function in terms of ligand binding and signal transduction ability [22].
To verify that the Ste2p-rho was expressed, total membrane protein was isolated, immunoblotted and probed by antibody to the N-terminus and 1D4, a monoclonal antibody to rho epitope (Figure 1). When analyzed by SDS-PAGE, wild type α-factor receptor, which is known to migrate slowly as a group of two or three bands showing heterogeneity of glycosylation that occurs at Asn25 and Asn32 residues [23, 24], was evident on the blot probed with antibody to the N-terminus of Ste2p, whereas Ste2p-rho was indicated by a single band of lower molecular weight indicating the non-glycosylated form of Ste2p (Figure 1A, left panel). As expected, an immunoblot analysis of membrane protein from the Ste2p-rho expressing strain probed with antibody to the rho epitope showed a single band indicating an unglycosylated α-factor receptor, and the Ste2p wild type receptor (not epitope tagged) was not detected by the 1D4 antibody (Figure 1A, right panel).
Figure 1. Expression and biological activity of Ste2p.
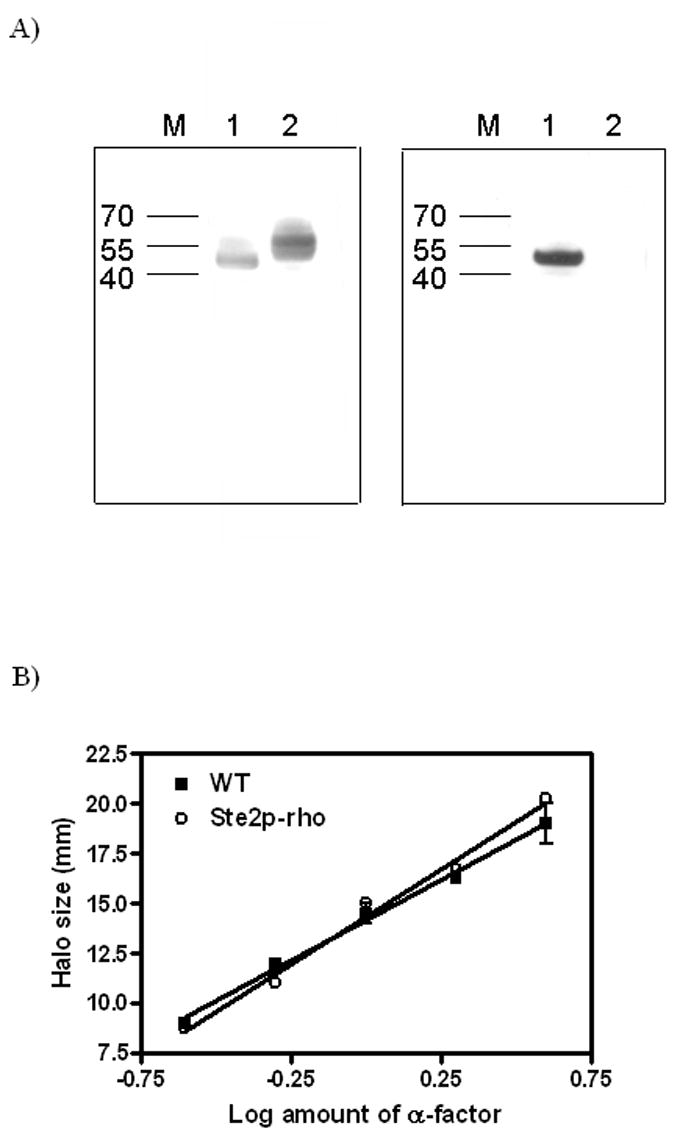
A) Western analysis of Ste2p. An equal amount of membranes from cells expressing either Ste2p-rho (non-glycosylated and rho-tagged, Lane 1) or wild-type, cys-less Ste2p (Lane 2) were analyzed in SDS-PAGE gel and probed with either antibody to the N-terminus (Left panel) or 1D4, a monoclonal antibody for the rho-tag (Right panel).
B) Growth arrest assay. Cells expressing Ste2p or Ste2p-rho were treated with various amounts of the α-factor agonist to induce growth arrest. The size of growth arrest zone (halo) was measured, and the data were plotted as halo size versus the amount of peptide and linearized by regression analysis.
After confirming the expression of the unglycosylated rho tagged-α factor receptor, the functional aspect of the Ste2p-rho was examined. Comparison of halo (zone of growth arrest) size demonstrated that the sequence modifications made to the receptor did not compromise its ability to mediate growth arrest in response to α-factor as demonstrated in this in vivo bioassay (Figure 1B). Previously the C59S/C252S receptor [22] and the N25Q/N32Q non-glycosylated receptor [24] were shown to retain full biological activity.
Ste2p-rho has similar affinity for α-factor to that of wild type receptor
A whole cell saturation binding assay with [3H]α-factor was performed on the LM102 strain expressing either wild type or Ste2p-rho (Figure 2 A). A Kd of 8.9 ± 0.6 nM was obtained for wild type receptor which is similar to that previously reported this receptor [16, 22]. The Kd value observed from Ste2p-rho was 12.7 ± 1.2 nM indicating that Ste2p-rho was fully active in terms of ligand binding. The radioligand binding assay is more quantitative than an immunoblot and has the added advantage of detecting only active cell surface molecules. Analysis of Bmax values from a saturation binding assay demonstrated that cell surface expression level of Ste2p-rho receptor was about 50 % of the wild type receptor level in LM102 strain (37,000 for Ste2p-rho versus 75,000 receptor molecules per cell for Ste2p) in agreement with the notion that, although glycosylation does not affect receptor function, this post-translational modification plays a role in regulating proper targeting of the receptor to the cell surface [23].
Figure 2. Whole cell [3H]α-factor binding assay.
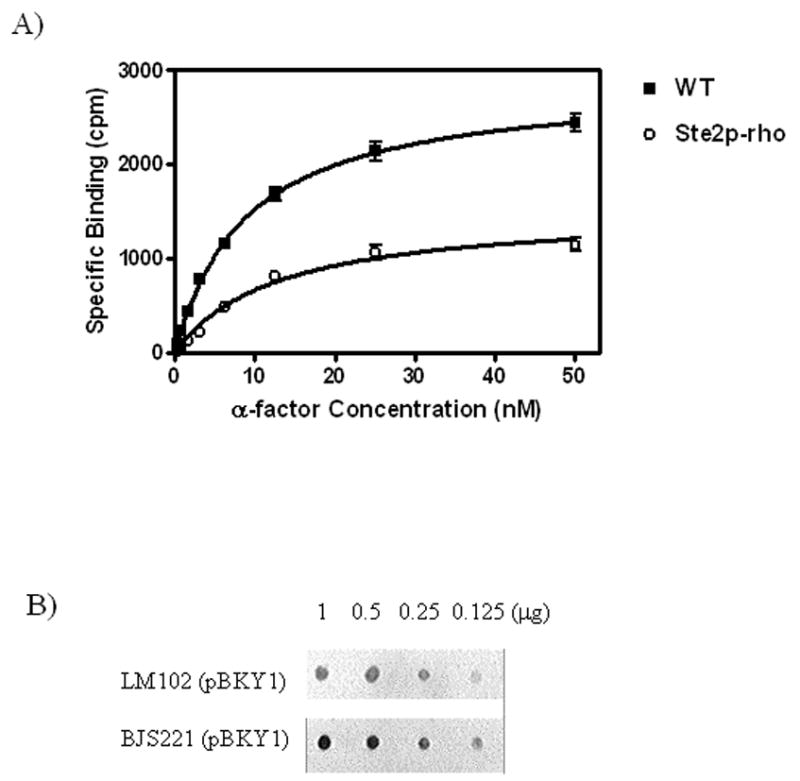
A) Binding of [3H]α-factor was performed as described under “Materials and Methods” to LM102 whole cells expressing either wild-type Ste2p or Ste2p-rho. The saturation isotherm shown is representative of three independent experiments. B) Dot blot quantification of Ste2p-rho expression level between LM102 and BJS strains. For the comparison of relative expression level of Ste2p-rho, membranes from LM102 and BJS strains were prepared, diluted, transferred to Nylon membranes, and probed with 1D4, a monoclonal antibody for the 9-amino acid rhodopsin tag. The blot shown is representative of three independent preparations with similar results obtained.
Expression of Ste2p-rho in a peptidase deficient yeast strain
For large scale purification of GPCRs, it is important to start with a system having a high level of receptor expression. To compensate for the lower expression level of Ste2p-rho in LM102 in comparison to expression of the wild type Ste2p, we transformed pBKY1 into the BJS21 strain, which is deficient for proteases Pep4p, Prc1p and Prb1p. Pep4p encodes a vacuolar protease essential for proper maturation of many vacuolar enzymes [25] and thus is critical for the down-regulation and degradation of Ste2p in the vacuolar compartment [26]. Although the BJS21 strain is less sensitive to α-factor arrest due to the presence of the BAR1 gene, an extracellular protease that digests α-factor [27], we reasoned that, in a Pep4p deficient cell, the level of receptor expression would be increased compared to LM012 strain. As expected, dot blot analysis of membrane proteins demonstrated that the level of Ste2p-rho expression in the BJS21 strain was higher than that in the LM102 strain (Figure 2 B).
Solubilization and purification of Ste2p-rho
BJS21 cell membranes expressing Ste2p-rho were solubilized in the presence of various concentrations of DM (0.1 % to 1 %) and applied to an affinity column pre-coupled with 1D4 antibody. The DM concentration was gradually decreased to from 0.5 to 0.001 % during purification. The preparation using 0.5 % DM for solubilization and 0.025% DM for elution was optimal for maximal yield and functionality of Ste2p (data not shown). Purified receptor showed a major band corresponding to an apparent molecular weight of 48 kDa on Coomassie blue-stained SDS-PAGE gels with some higher molecular weight bands present (Figure 3A, lane 5). Immunoblot analysis indicated that the higher molecular weight bands were Ste2p-rho oligomers or aggregates (Figure 3B, lane 5). Indeed, the approximate determination of molecular weight of those bands in comparison to marker proteins showed agreement with oligomeric forms of the receptor. These oligomers or aggregates may have been generated during the SDS-PAGE or be an indication that GPCRs form oligomers in vivo [28]. The final yield of purified receptor from a 1 L overnight culture (100 mg of crude membrane) was approximately 120 μg based on a protein assay (Table 1). This amount of Ste2p-rho was equivalent to 26 pmol of Ste2-rho/mg of membrane protein based on the molecular weight of Ste2p-rho (48 kDa).
Figure 3. Solubilization and purification of Ste2p-rho.
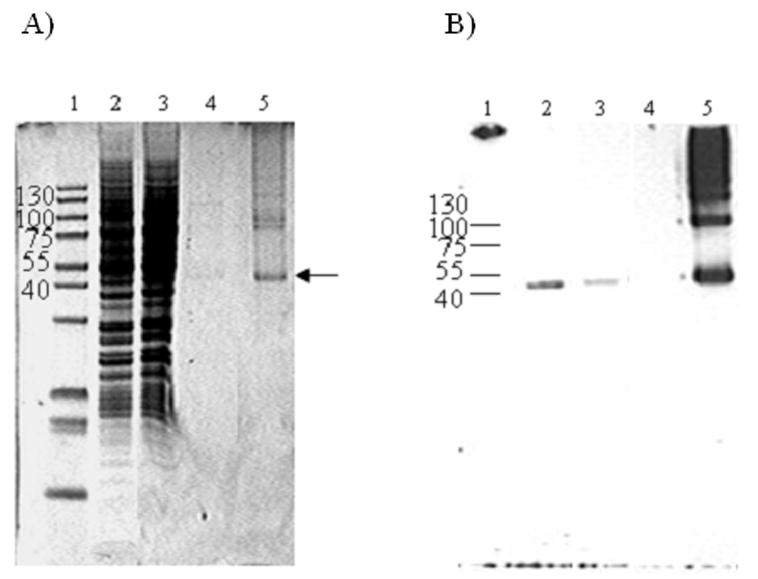
A, Coomassie Blue stain. B, Western blot analysis using 1D4 antibody. Lane 1, molecular weight marker; Lane2, solubilized membrane; Lane 3, fraction that did not bind to Sepharose-4B immobilized 1D4 immuno-affinity column; Lane 4, wash fraction; Lane 5, purified receptor eluted by competition with 9-amino acid rhodopsin peptide. An equal portion of the sample was loaded after concentration except for lane B5 in which one fourth was loaded to maximize the resolution of the monomer and oligomer bands.
Table 1.
Functionality and yield of purified Ste2p-rho from 1 L culture.
| Receptor and Strain | Growth arrest assay (%) | Kd (nM) | Bmax or Yield |
|---|---|---|---|
| WT (C59S, C252S) in LM102 | 100 ± 12 | 8.9 ± 0.6 | 75,300 ± 6800 per cell |
| Ste2p-rho in LM102 | 104 ± 7 | 12.7 ± 1.2 | 36,800 ± 3900 per cell |
| Purified Ste2p-rho from BJS21 | Not Applicable | 27.8 ± 3 | 120 μg/100 mg membrane (26 pmole/mg)
39 ± 7 % active receptora |
Active population of the purified receptor was calculated from the Bmax value in saturation binding assays. Specific activity of [3H]α-factor was 16 Ci/mmol and the average Bmax value of four separate binding assays was 6,000 cpm/40 ng receptor.
Ligand binding analysis
[3H]α-factor binding assay was performed to determine whether the purified receptor was correctly folded. The purified receptor, eluted in the presence of 0.025 % DM, bound α-factor with a Kd of 28 nM (Figure 4 and Table 1). This affinity was about 2-fold lower than that of Ste2p-rho as measured by a whole cell assay (Fig. 2A). A recent report on the purification of Ste2p heterologously expressed in HEK293 cells indicated that the purified receptor had a Kd of 34 nM for a fluorescent α-factor analog [9]. However, a direct comparison of affinity of the purified receptor between our study and that using a fluorescent ligand was not possible due to the difference in the ligands used and to the fact that the binding of the fluorescent analog was not measured in cell membranes from HEK293 cells or yeast. The difference in α-factor affinity for purified Ste2p-rho compared to its affinity for Ste2p-rho in cell membranes (Figure 2A) may result from either the absence of G-proteins, which are known to be required for the high affinity state of Ste2p [29] and other GPCRs such as the parathyroid hormone receptor [14, 30], in the purified Ste2p-rho sample, or differences in the environment between Ste2p solubilized in detergent as opposed to its phospholipid-embedded state in native membranes. Scatchard analysis of binding (data not shown) indicated that ~40 % of the purified receptor retained binding activity (Table 1), if it is assumed that each receptor has one ligand binding site. From two 3L cultures, 1.5 to 1.8 mg of Ste2p-rho has been repeatedly purified. This amount should provide sufficient material for attempting crystallization
Figure 4. [3H]α-factor binding to the purified Ste2p-rho in Dodecyl Maltoside micelles.
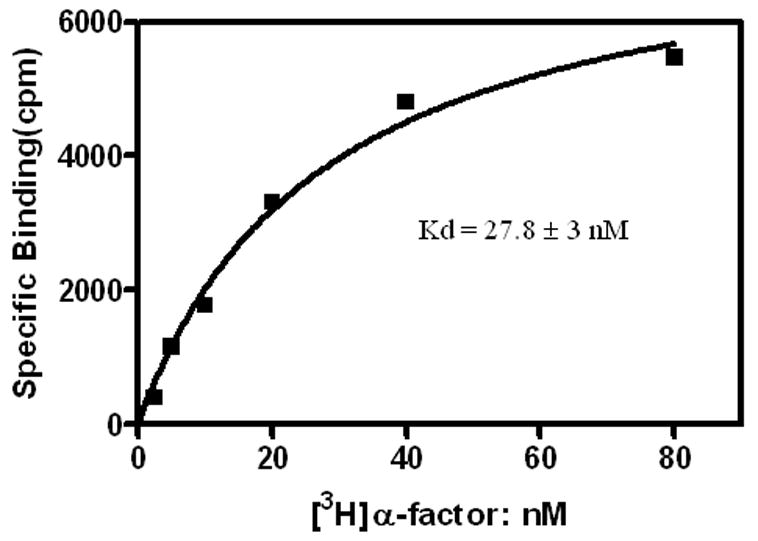
Binding of [3H]α-factor was performed as described under “Materials and Methods”. The data shown is a representative of four independent experiments with similar Kd values (25 to 33 nM).
Mass spectrometry analysis
To further confirm and characterize the purified Ste2p-rho, an aliquot the purified preparation was directly subjected to CNBr digestion and MALDI/TOF-MS and nanospray LC-MS/MS. As shown in Figure 5, a total of 8 peptide fragments that matched the calculated molecular weight of the peptides derived from Ste2p-rho were detected from MALDI/TOF spectra including a peptide containing the 9-amino acid rhodopsin sequence at the C-terminal end of the receptor (observed mass = 3255.5 Da). This covers 143 amino acids (32 %) out of 438 residues in Ste2p-rho (Table 2). In nanospray LC-MS/MS analysis, we retrieved and sequenced 9 unique peptides. Sequences from MS/MS results comprised over 40 % of the Ste2p-rho protein (Table 2); a representative tandem MS spectrum is shown in Figure 6. The CNBr digested peptides that were detected and identified by both MALDI-TOF and nanospray constitute 50 % coverage (223 amino acids) out of 438 total amino acids sequence of Ste2p-rho (Figure 7). Owing to difficulties in their purification, mass spectrometry studies of GPCR have rarely been reported [31]. Moreover all of the available MS spectra of GPCRs have been obtained by excision of targeted receptor bands from SDS-PAGE gels [31]. In contrast, the MS results shown in this study were acquired directly from the concentrated sample eluted from the affinity chromatography column. Considering the fact that the only available MS/MS spectrum of a GPCR is one of the C-terminal peptide of the opioid receptor [32], the MS methods and results described in this study are the most comprehensive mass spectrometric analyses of any GPCR obtained to date and indicate a preparation of high purity, in which using a strict filtering level no proteins besides Ste2p were unequivocally identified.
Figure 5. Identification of purified Ste2p-rho by MALDI-TOF-MS.
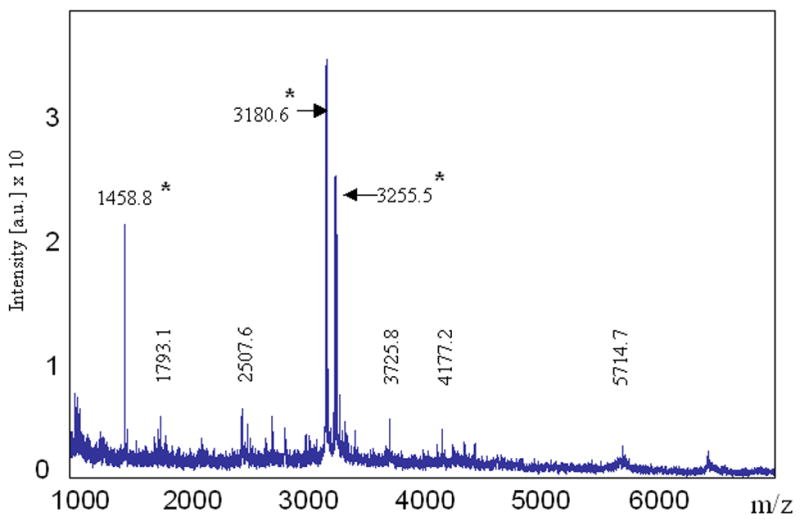
Peptide mixtures obtained after CNBr digestion of the elution sample were analyzed by MALDI-TOF-MS. The peptides identified by mass on the figure correspond to a portion of Ste2p-rho (Refer to Table 2). Peptides labeled with an asterisk are the fragments that were successfully identified by nanospray MS.
Table 2.
Detection by MALDI-TOF and nanos pray LC-MS/MS of peptides from the final elution sample after CNBr digestion.
| Fragments (residue number in Ste2p-rho) | Calculated Massa | MALDI/TOF Observed Mass [M +H]+1 | Nanospray Observed Mass [M +H]+1 | Method of Detection |
|---|---|---|---|---|
| 181 – 189 | 953.1 | NDb | 953.6 | NSd |
| 155 – 165 | 1203.4 | ND | 1204.4 | NS |
| 398 – 409 | 1333.8 | ND | 1334.5 | NS |
| 55 – 69 | 1457.7 | 1458.8 | 1458.0 | Bothe |
| 295 – 311 | 1765.9 | ND | 1766.2 | NS |
| 55 – 71 | 1775.2 | 1793.1c | ND | MALDI |
| 389 – 410 | 2264.5 | ND | 2263.9 | NS |
| 166 – 189 | 2490.0 | 2507.6c | ND | MALDI |
| 190 – 218 | 3179.5 | 3180.6 | 3181.0 | Both |
| 410 – 438 | 3255.4 | 3255.5 | 3255.4 | Both |
| 219 – 250 | 3726.6 | 3725.8 | ND | MALDI |
| 181 – 218 | 4162.7 | 4177.2c | ND | MALDI |
| 251 – 294 | 4555.4 | ND | 4556.8 | NS |
| 166 – 218 | 5699.5 | 5714.7c | ND | MALDI |
The mass for the CNBr generated peptide fragments, some are partial cleavage products, were calculated using homoserine lactone as the C-terminal amino acid, except for 410-438, which is the C-terminus of Ste2p-rho.
Not detected.
Under the conditions of partial CNBr cleav age, internal methionine-sulfoxide is generated. Thus, the partially cleaved peptide fragments of Ste2p-rho are 16 Da larger than the size reported by the program for peptides.
Detected by nanospray-LC-MS/MS (NS).
Detected by both MALDI-TOF and nanospray.
Figure 6. MS/MS spectrum of a fragment of purified Ste2p-rho.
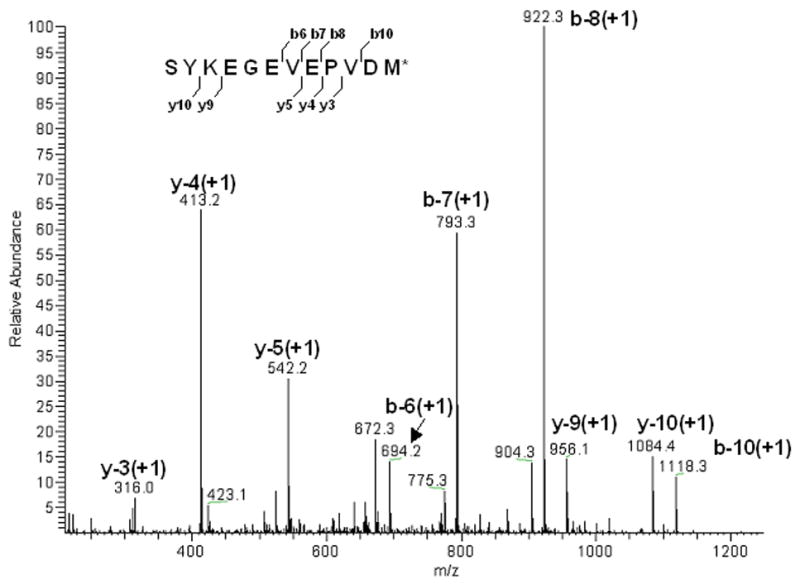
The fragment (inset) sequenced is located at the C-terminus of Ste2p-rho (S398 – M409). Observed mass was 1333.8 Da and calculated mass was 1334.5 Da.
Fig. 7. CNBr digested peptides identified by MS and MS/MS mass spectrometry.
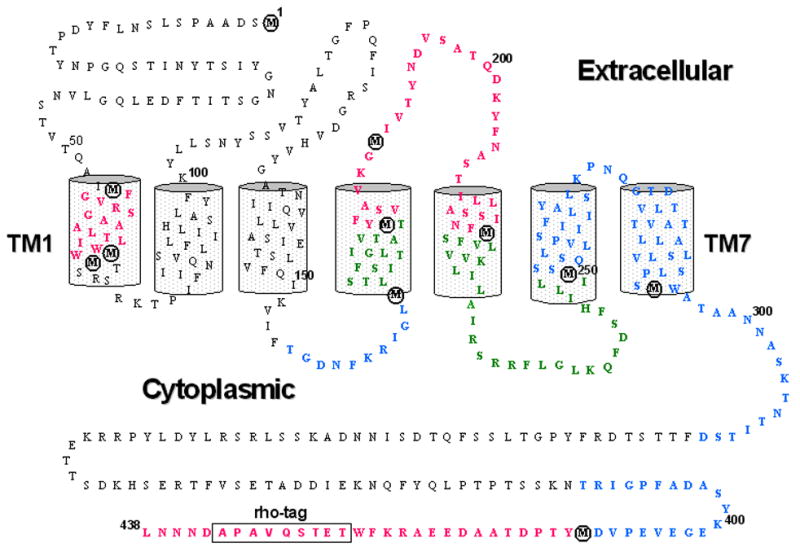
Amino acid sequence and transmembrane topology of Ste2p-rho is shown. A 9-amino acid rhodopsin sequence for affinity purification at the C-terminus is boxed and CNBr specific cleavage sites (Met residues) are circled. The peptides retrieved by MALDI-TOF, nanospray (MS/MS), or by both analyses (Table 2) are shown in green, blue, and red, respectively.
Summary
In the work reported here, non-glycosylated, Cys-less yeast α-factor receptor (Ste2p-rho) was purified by one-step antibody affinity chromatography. The purity, functionality and identity of the purified receptor were confirmed by immunodetection, ligand binding, and MALDI-TOF and LC-MS/MS analysis. We have demonstrated that milligram quantities of highly homogeneous Ste2p can be purified by a simple and cost effective procedure. Our results demonstrate that a comprehensive mass spectrometric analysis of GPCRs including MS/MS sequencing can be achieved. Overall the results presented here are a rare example of a GPCR purified to near homogeneity from its native system in which biological activity, purity and identity were analyzed. Further optimization to increase the proportion of the biologically active form of the receptor is being carried out, including treatment with agonist throughout the entire purification process and enrichment of active receptors using an α-factor-coupled affinity column. With a straightforward scale-up in yeast, we should be able to obtain sufficient material for crystallization attempts. The methods described herein can be directly applied to Ste2p mutants which are constitutively active [33], which selectively favor the inactive state of the receptor [18] or which may have evolved to thermal stability. Such receptors may have more favorable crystallization characteristics. Finally, our approach is applicable to other classes of GPCRs that have been expressed in biologically active states in S. cerevisiae.
Acknowledgments
This research was supported by grants GM 22086 (FN) and GM 22087 (JMB) from the National Institutes of Health and by the Joint Directed Laboratory Development program (JMB) from the University of Tennessee. NCV portion was sponsored by U.S. Department of Energy under contract DE-AC05-00OR22725 with Oak Ridge National Laboratory, managed and operated by UT-Battelle, LLC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Horn F, Bettler E, Oliveira L, Campagne F, Cohen FE, Vriend G. GPCRDB information system for G protein-coupled receptors. Nucleic Acids Res. 2003;31:294–297. doi: 10.1093/nar/gkg103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 3.Sarramegna V, Talmont F, Demange P, Milon A. Heterologous expression of G-protein-coupled receptors: comparison of expression systems from the standpoint of large-scale production and purification. Cell Mol Life Sci. 2003;60:1529–1546. doi: 10.1007/s00018-003-3168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 5.Sarramegna V, Muller I, Milon A, Talmont F. Recombinant G protein-coupled receptors from expression to renaturation: a challenge towards structure. Cell Mol Life Sci. 2006;63:1149–1164. doi: 10.1007/s00018-005-5557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naider F, Estephan R, Englander J, Suresh Babu VV, Arevalo E, Samples K, Becker JM. Sexual Conjugation in Yeast: A Paradigm to Study G Protein coupled Receptor Domain Structure. Biopolymers. 2004;76:119–128. doi: 10.1002/bip.10567. [DOI] [PubMed] [Google Scholar]
- 7.Ladds G, Davis K, Das A, Davey J. A constitutively active GPCR retains its G protein specificity and the ability to form dimers. Mol Microbiol. 2005;55:482–497. doi: 10.1111/j.1365-2958.2004.04394.x. [DOI] [PubMed] [Google Scholar]
- 8.David NE, Gee M, Andersen B, Naider F, Thorner J, Stevens RC. Expression and purification of the Saccharomyces cerevisiae alpha-factor receptor (Ste2p), a 7-transmembrane-segment G protein-coupled receptor. J Biol Chem. 1997;272:15553–15561. doi: 10.1074/jbc.272.24.15553. [DOI] [PubMed] [Google Scholar]
- 9.Shi C, Shin YO, Hanson J, Cass B, Loewen MC, Durocher Y. Purification and characterization of a recombinant G-protein-coupled receptor, Saccharomyces cerevisiae Ste2p, transiently expressed in HEK293 EBNA1 cells. Biochemistry. 2005;44:15705–15714. doi: 10.1021/bi051292p. [DOI] [PubMed] [Google Scholar]
- 10.Sen M, Marsh L. Noncontiguous domains of the alpha-factor receptor of yeasts confer ligand specificity. J Biol Chem. 1994;269:968–973. [PubMed] [Google Scholar]
- 11.Son CD, Sargsyan H, Naider F, Becker JM. Identification of ligand binding regions of the Saccharomyces cerevisiae alpha-factor pheromone receptor by photoaffinity cross-linking. Biochemistry. 2004;43:13193–13203. doi: 10.1021/bi0496889. [DOI] [PubMed] [Google Scholar]
- 12.Abel MG, Lee BK, Naider F, Becker JM. Mutations Affecting Ligand Specificity of the G Protein coupled Receptor for the Saccharomyces cerevisiae Tridecapeptide Pheromone. Biochim Biophys Acta. 1998;1448:12–26. doi: 10.1016/s0167-4889(98)00109-8. [DOI] [PubMed] [Google Scholar]
- 13.Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 14.Shimada M, Chen X, Cvrk T, Hilfiker H, Parfenova M, Segre GV. Purification and characterization of a receptor for human parathyroid hormone and parathyroid hormone-related peptide. J Biol Chem. 2002;277:31774–31780. doi: 10.1074/jbc.M204166200. [DOI] [PubMed] [Google Scholar]
- 15.Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 16.Lee BK, Khare S, Naider F, Becker JM. Identification of residues of the Saccharomyces cerevisiae G protein-coupled receptor contributing to alpha-factor pheromone binding. J Biol Chem. 2001;276:37950–37961. doi: 10.1074/jbc.M103579200. [DOI] [PubMed] [Google Scholar]
- 17.Raths SK, Naider F, Becker JM. Peptide analogues compete with the binding of alpha-factor to its receptor in Saccharomyces cerevisiae. J Biol Chem. 1988;263:17333–17341. [PubMed] [Google Scholar]
- 18.Lee BK, Lee YH, Hauser M, Son CD, Khare S, Naider F, Becker JM. Tyr266 in the sixth transmembrane domain of the yeast alpha-factor receptor plays key roles in receptor activation and ligand specificity. Biochemistry. 2002;41:13681–13689. doi: 10.1021/bi026100u. [DOI] [PubMed] [Google Scholar]
- 19.Chalkley RJ, Baker PR, Huang L, Hansen KC, Allen NP, Rexach M, Burlingame AL. Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting, quadrupole collision cell, time-of-flight mass spectrometer: II. New developments in Protein Prospector allow for reliable and comprehensive automatic analysis of large datasets. Mol Cell Proteomics. 2005;4:1194–1204. doi: 10.1074/mcp.D500002-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Tabb DL, Narasimhan C, Strader MB, Hettich RL. DBDigger: reorganized proteomic database identification that improves flexibility and speed. Anal Chem. 2005;77:2464–2474. doi: 10.1021/ac0487000. [DOI] [PubMed] [Google Scholar]
- 21.Tabb DL, McDonald WH, Yates JR., 3rd DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akal-Strader A, Khare S, Xu D, Naider F, Becker JM. Residues in the first extracellular loop of a G protein-coupled receptor play a role in signal transduction. J Biol Chem. 2002;277:30581–30590. doi: 10.1074/jbc.M204089200. [DOI] [PubMed] [Google Scholar]
- 23.Hirschman JE, De Zutter GS, Simonds WF, Jenness DD. The G beta gamma complex of the yeast pheromone response pathway. Subcellular fractionation and protein-protein interactions. J Biol Chem. 1997;272:240–248. doi: 10.1074/jbc.272.1.240. [DOI] [PubMed] [Google Scholar]
- 24.Mentesana PE, Konopka JB. Mutational analysis of the role of N-glycosylation in alpha-factor receptor function. Biochemistry. 2001;40:9685–9694. doi: 10.1021/bi0108507. [DOI] [PubMed] [Google Scholar]
- 25.Hicke L, Zanolari B, Riezman H. Cytoplasmic tail phosphorylation of the alpha-factor receptor is required for its ubiquitination and internalization. J Cell Biol. 1998;141:349–358. doi: 10.1083/jcb.141.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi L, Javitch JA. The Second Extracellular Loop of the Dopamine D2 Receptor Lines the Binding Site Crevice. Proc Natl Acad Sci U S A. 2004;101:440–445. doi: 10.1073/pnas.2237265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ballensiefen W, Schmitt HD. Periplasmic Bar1 protease of Saccharomyces cerevisiae is active before reaching its extracellular destination. Eur J Biochem. 1997;247:142–147. doi: 10.1111/j.1432-1033.1997.00142.x. [DOI] [PubMed] [Google Scholar]
- 28.Salim K, Fenton T, Bacha J, Urien-Rodriguez H, Bonnert T, Skynner HA, Watts E, Kerby J, Heald A, Beer M, McAllister G, Guest PC. Oligomerization of G-protein-coupled receptors shown by selective co-immunoprecipitation. J Biol Chem. 2002;277:15482–15485. doi: 10.1074/jbc.M201539200. [DOI] [PubMed] [Google Scholar]
- 29.Dosil M, Schandel KA, Gupta E, Jenness DD, Konopka JB. The C terminus of the Saccharomyces cerevisiae alpha-factor receptor contributes to the formation of preactivation complexes with its cognate G protein. Mol Cell Biol. 2000;20:5321–5329. doi: 10.1128/mcb.20.14.5321-5329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gan L, Alexander JM, Wittelsberger A, Thomas B, Rosenblatt M. Large-scale purification and characterization of human parathyroid hormone-1 receptor stably expressed in HEK293S GnTI- cells. Protein Expr Purif. 2006;47:296–302. doi: 10.1016/j.pep.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Alves ID, Sachon E, Bolbach G, Millstine L, Lavielle S, Sagan S. Analysis of an intact G-protein coupled receptor by MALDI-TOF mass spectrometry: molecular heterogeneity of the tachykinin NK-1 receptor. Anal Chem. 2007;79:2189–2198. doi: 10.1021/ac062415u. [DOI] [PubMed] [Google Scholar]
- 32.Christoffers KH, Li H, Howells RD. Purification and mass spectrometric analysis of the delta opioid receptor. Brain Res Mol Brain Res. 2005;136:54–64. doi: 10.1016/j.molbrainres.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 33.Sommers CM, Martin NP, Akal-Strader A, Becker JM, Naider F, Dumont ME. A limited spectrum of mutations causes constitutive activation of the yeast alpha-factor receptor. Biochemistry. 2000;39:6898–6909. doi: 10.1021/bi992616a. [DOI] [PubMed] [Google Scholar]