

Abstract
A 6,7-diaryl-2,3,8,8a-tetrahydroindolizin-5(1H)-one library was constructed and tested against the colon cancer cell line HCT-116 as an initial screen for cytotoxic properties. Of this library, the parent compound, in which the southern aromatic ring remains unsubstituted, and the northern aromatic ring carries a 4-methoxy group, exhibited the most potent cytotoxicity with an IC50 value of 0.39 μM and displayed promising activity in vivo in the NCI’s mouse hollow fiber assay.
As cancer cells adapt to various treatment modalities and subsequently become resistant to modern and conventional chemotherapy, the discovery and exploitation of new chemical classes of compounds that avoid the various cancer resistance mechanisms become an increasingly crucial endeavor. Moreover, the discovery of small molecules that exert cytotoxic properties via novel mechanisms of action would be of paramount importance. Such a discovery could also identify a novel cancer target and thus provide a lead for the development of a new class of anticancer agents. As such, our continuing search for the discovery and development of small molecule anticancer agents that operate through an unknown mechanism of action led us to our studies of the alkaloid tyloindicine family.
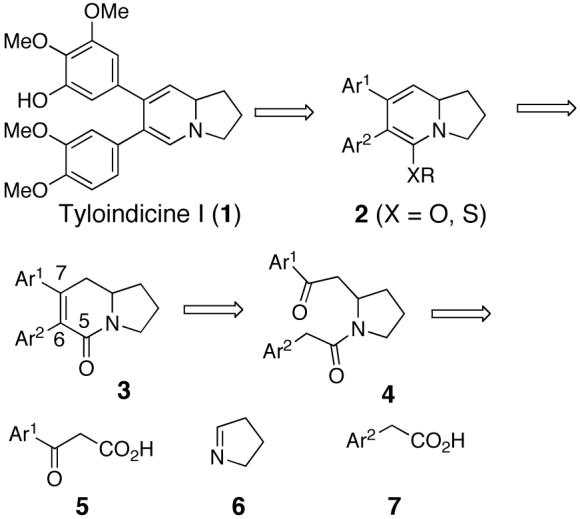
Tyloindicine I (1, Scheme 1) was isolated in 1991 in minute quantities from the aerial parts of Tylophora indica and falls into the family of seco-phenanthroindolizidine alkaloids.1,2 Our interest in tyloindicine I is a result of its potent nanomolar and cancer cell-selective cytotoxic properties and therefore, we set out to synthesize 1 and related analogs to pursue additional biological studies.2
Scheme 1.
Although no total syntheses exist to date for tyloindincine I, synthetic endeavors towards the related alkaloids ipalbidine3-13 and septicine11,14-24 have been utilizing synthetic methods that have been incorporated into this work.20,21 Research focusing on analogs related to tyloindicine I, ipalbidine, or septicine is sparse in the literature with the notable research in this area coming from the laboratories of Sharma et al. and focusing on 6-aryl-7-(4-(methythio)phenyl)-2,3,8,8a-tetrahydroindolizin-5(1H)-ones.25
In an attempted route to 1, for which the retrosynthesis is shown in Scheme 1, we devised a synthesis starting from β-keto acid 5, pyrrole 6, and arylacetic acid 7. These building blocks could be assembled to provide intermediate pyrrolidine 4, which upon aldol condensation would provide bicyclic lactam 3. Alkylation of the O- or S-enol amide of 3, followed by deoxygenation or desulfurization of intermediate 2 could lead to the targeted tyloindicine I. A model study was initiated as shown in Scheme 2, and the synthesis of the desired lactam intermediate 12 was accomplished as planned. Saponification of the commercially available β-keto ester 8 furnished the β-keto acid 9. A subsequent addition of 9 to (di-(3,4-dihydro-2H-pyrrole))diiodozinc, followed by decarboxylation, constructed the pyrrolidine 10.26 The amine in 10 was then acylated with phenylacetyl chloride to provide 11, which was subjected to KOH in refluxing ethanol to provide the target indolizidine lactam 12.27 The subsequent steps, the conversion of 12 to the corresponding tyloinidicine I analogue did not proceed as planned and that route towards tyloindicine I was abandoned.
Scheme 2.
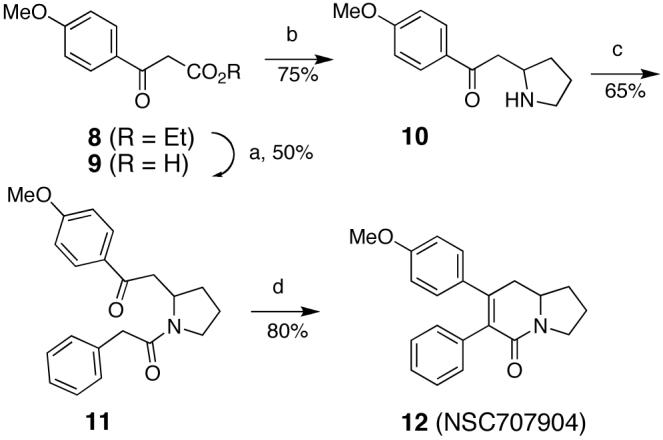
Reagents and conditions: a) 2.5% KOH(aq); b) (di-(3,4-dihydro-2H-pyrrole))diiodozinc, pH 7 phosphate buffer, MeOH; c) phenylacetyl chloride, TEA, DCM; d) 5% KOH(EtOH), reflux.
7-(4-Methoxyphenyl)-7-phenyl-2,3,8,8a-tetrahydroindolizin-5(1H)-one (12, NSC 707904) was submitted to the NCI for screening in their panel of 60 cancer cell lines and demonstrated selective cytotoxity at sub-micromolar concentrations towards colon cancer and leukemia cell lines. In one screen the concentration that inhibited growth of the HCT-116 colon tumor cell line was 0.024 μM. In another screen it was 0.53 μM.
The NCI’s COMPARE analysis of the 60 cell line assay results indicated that 12 acts by a different mechanism of action compared to all other anticancer agents in their database including tyloindicine I.28,29
As a result of these interesting findings, 12 (NSC 707904) was selected by the NCI for in vivo studies in the NCI’s mouse hollow fiber assay.30,31 In this assay, compounds are tested against a standard panel of 12 human cancer cell lines, including one colon cancer cell line (COLO 205). A point system is used to assess the activity of the test compounds. A value of 2 is assigned for each compound dose that results in 50% or greater reduction in viable cell mass either IP or SC. Compounds with a combined IP and SC score of 20, an SC score of 8, or a net cell kill of one or more cell lines are referred to the NCI’s Biological Evaluation Committee for Cancer Drugs. Compound 12 received an SC score of 8, demonstrating that 12 has promising activity according to this point system. Considering that only one colon cancer cell line was present in this assay panel, the SC in vivo activity is encouraging.
Because of these promising test results, we set out to prepare analogs of 12 so as to achieve an understanding of the structure-activity relationship for this class of compounds, to improve potency, and to prepare affinity analogues that could be used to identify the biological target or targets of 12. The analog library was generated utilizing the synthetic sequence shown in Scheme 2 from the appropriately substituted β-keto and aryl acetic acids and are shown in Table 1.
Table 1.
Cytotoxicity of 6,7-diaryl-2,3,8,8a-tetrahydroindolizin-5(1H)-one analogs 12-25.

| Analog | Northern Aryl Substitution R1 | Southern Aryl Substitution R2, R3, R4 | HCT-116 Cytotoxicity IC50, μM |
|---|---|---|---|
| 12 | OMe | H, H, H | 0.39 |
| 13 | OMe | Cl, H, H | 13 |
| 14 | OMe | OMe, H, H | 3.2a |
| 15 | OMe | Me, H, H | 10 |
| 16 | OMe | Cl, Cl, H | 4 |
| 17 | OMe | H, Cl, H | 8 |
| 18 | OMe | H, H, Cl | 8 |
| 19 | OMe | H, NH-LCBb, H | >25 |
| 20 | OMe | NH-LCBb, H, H | 25 |
| 21 | OMe | N3, H, H | 25 |
| 22 | O-LCBb | H, H, H | 17 |
| 23 | OMe | H, NH2, H | >25 |
| 24 | OH | H, H, H | 9 |
| 25 | OBn | H, H, H | 8.9 |
Assay conducted by National Cancer Institutes Developmental Therapeutics Program.
LCB = (+)-biotinyl-6-aminohexanoic acid.
The cytotoxicity experiments were done as previously described32 except that the cells were incubated for 48 h after the addition of the compounds.
The analog library, along with compound 12, was tested against the HCT-116 colon cancer cell line as the initial screen to determine cytotoxicity (Table 1).
The results from the toxicity screen revealed that the activity we observed in our laboratory with 12 is consistent with the higher of the two values obtained in the NCI screen. A Topliss decision tree approach was applied towards the selection of the initial analogs 13-16 with the lead compound 12 being at the top of the schematic Topliss tree.33 With respect to structural modifications, the 4-chloro analog 13 was less active than 12 suggesting that the southern aryl ring has either stringent steric requirements or is controlled by either -σ or -π factors. In attempts to further define the southern aryl rings role, continuing down the Topliss tree to the 4-methoxy analog 14 revealed that it too was less active than the lead compound 12. This information indicates that the steric requirements of the southern ring may supercede any favorable or negative influences the -σ or -π factors may have been contributing. The other analogs in the Topliss series, 15-18, further suggested that regardless of the lipophilicity or the chlorine substitution patterns, overall changes in the southern ring are not well tolerated. Notwithstanding these results, one photoaffinity analog (21) and three biotinylated analogs (19, 20, 22) were generated in attempts to aid in the isolation of biological targets and allow for the determination of the mechanism of action of this class of compounds.34 These efforts were not successful with affinity analogs 19-22 showing poor cytotoxicity. Furthermore, aniline 23 had poor activity. These results further illustrate the steric sensitivity of the southern aromatic ring.
With respect to the northern ring, the phenol 24 as well as the benzyl protected analog 25 were prepared and tested. These compounds also showed reduced cytotoxicity. Phenol 24 was prepared in quantitative yield from 12 by treatment with BCl3, n-Bu4NI, CH2Cl2 (-78 °C to 0 °C) for 2 h. Analogue 25 was synthesized as shown in Scheme 2, using the O-benzyl analogue of 8. Thus, the northern aromatic ring appears to favor the 4-methoxy substitution over the 4-hydroxy substitution thereby suggesting that the electronic or lipophilic character of the northern ring affects the biological activity of these analogs.
Further efforts to investigate the steric and electronic parameters for the southern aryl ring were inspired by the nanomolar cytotoxicity of the related alkaloids tyloindicine I (Scheme 1)1,2 and tylophorine,24 that carry two methoxy groups on each aromatic ring. The cytotoxicity of these natural products suggests that electronic factors may serve to override the steric sensitivity of the southern aromatic ring inherent in analogs 13-21 and 23. As such, the more highly substituted analogs 26-28 were prepared following the synthesis shown in Scheme 2, using the appropriate β-keto esters and acyl chlorides. The IC50 values for analogs 26-28 are listed in Table 2. With the introduction of a second methoxy group at the southern ring, a comparison of 27 and 12 further confirms that substitution of the southern ring is detrimental to activity. In addition, the inferior cytotoxicity of the tri- and tetrasubstituted northern ring analogs 26 and 28, relative to the submicromolar activity of the parent compound 12, suggests that increasing the steric encumbrance around the northern aromatic ring has detrimental effects as well. It would seem that for this series, the electronics of the aryl rings might have a larger influence than previously anticipated.
Table 2.
Cytotoxicity of tylophorine analogs 26-28.

| Analog | R1, R2 | HCT-116 Cytotoxicity IC50, μM |
|---|---|---|
| 26 | H, OMe | >10 |
| 27 | H, H | 5.0a |
| 28 | OBn, OMe | 3.3 |
Assay conducted by the National Cancer Institute’s Developmental Therapeutics Program.
Overall, our results exhibit trends complementary to those outlined by Sharma et al. in which a 6,7-diaryl-2,3,8,8a-tetrahydroindolizin-5(1H)-one scaffold was employed with variously substituted northern and southern aromatic rings.25 Notably, Sharma found that more highly substituted northern rings led to a decrease in activity whereas the southern ring did retain moderate activity with mono- and di-substituted systems similar to analogs 13-18, 27 and 28. Although a southern ortho- and/or para-substituted methoxy group did not significantly affect activity, replacement with a hydroxyl group led to a significant decrease in activity suggesting H-bonding moieties were detrimental to activity similar to our aniline analog 23. Activities were tested against 8 cell lines, not including the HCT-116 cell line, thereby making direct comparisons with our results difficult.
In summary, a short synthesis was designed to construct 6,7-diphenyl-2,3,8,8a-tetrahydroindolizin-5(1H)-one (12) as well as the subsequent analogs 13-28. The lead compound 12 was shown to exhibit the most potent cytotoxicity towards the HCT-116 colon cancer cell line and showed promising activity in the mouse hollow fiber assay. There appear to be stringent steric requirements controlling the degree of aryl substitution in the southern and northern aromatic rings. Overall, the low molecular weight, potent and selective cytotoxicity towards colon cancer cell lines, and potential of ultimately determining a novel mechanism of action, make this collection of compounds an interesting avenue towards the discovery of a new class of anticancer agents worthy of further examination and scientific scrutiny.
Acknowledgments
These studies were supported by the National Cancer Institute (CA 90602 and N01-CM-67259). The authors wish to thank Dr. Ravi Varma (NCI, DTP) for carrying out the COMPARE analysis for analog 12 (NSC 707904) and for his support of this project. We also wish to thank the NCI’s DTP for carrying out cytotoxicity assays for compounds 12 (NSC 707904), 14 (NSC 707907), and 27 (NSC 707909). The complete cancer screening data for these compounds are available on the DTP website.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- (1).Ali M, Ansari SH, Qadry JS. J. Nat. Prod. 1991;54:1271. [Google Scholar]
- (2).Ali M, Ansari SH, Grever MR. Pharmazie. 2001;56:188. [PubMed] [Google Scholar]
- (3).Govindachari TR, Sidhaye AR, Viswanathan N. Tetrahedron. 1970;26:3829. [Google Scholar]
- (4).Wick AE, Bartlett PA, Dolphin D. Helv. Chim. Acta. 1971;54:513. [Google Scholar]
- (5).Howard AS, Gerrans GC, Michael JP. J. Org. Chem. 1980;45:1713. [Google Scholar]
- (6).Liu Z-J, Lu R-R, Hong H. Acta Chim. Sin. 1985;43:992. [Google Scholar]
- (7).Danishefsky SJ, Vogel C. J. Org. Chem. 1986;51:3915. [Google Scholar]
- (8).Jefford CW, Kubota T, Zaslona A. Helv. Chim. Acta. 1986;69:2048. [Google Scholar]
- (9).Sheehan SM, Padwa A. J. Org. Chem. 1997;62:438. doi: 10.1021/jo961690l. [DOI] [PubMed] [Google Scholar]
- (10).Ikeda M, Shikaura J, Maekawa N, Daibuzono K, Teranishi H, Teraoka Y, Oda N, Ishibashi H. Heterocycles. 1999;50:31. [Google Scholar]
- (11).Stevens RV, Luh Y. Tetrahedron Lett. 1977;18:979. [Google Scholar]
- (12).Padwa A, Sheehan SM, Straub CS. J. Org. Chem. 1999;64:8648. [Google Scholar]
- (13).Boto A, De León Y, Gallardo JA, Hernández R. Eur. J. Org. Chem. 2005:3461. [Google Scholar]
- (14).Russel JH, Hunziker H. Tetrahedron Lett. 1969;10:4035. [Google Scholar]
- (15).Govindachari TR, Viswanathan N. Tetrahedron. 1970;26:715. [Google Scholar]
- (16).Cragg JE, Herbert RB, Jackson FB, Moody CJ, Nicolson IT. J. Chem. Soc. Perkin Trans. 1. 1982:2477. [Google Scholar]
- (17).Iida H, Watanabe Y, Tanaka M, Kibayashi C. J. Org. Chem. 1984;49:2412. [Google Scholar]
- (18).Iwashita T, Suzuki M, Kusumi T, Kakisawa H. Chem. Lett. 1980;383 [Google Scholar]
- (19).Mangla VK, Bhakuni DS. Ind. J. Chem. 1980;19B:931. [Google Scholar]
- (20).Mangla VK, Bhakuni DS. Ind. J. Chem. 1980;19B:748. [Google Scholar]
- (21).Iida H, Tanaka M, Kibayashi C. J. Chem. Soc., Chem. Commun. 1983:271. [Google Scholar]
- (22).Comins DL, Morgan LA. Tetrahedron Lett. 1991;32:5919. [Google Scholar]
- (23).Ciufolini MA, Roschangar F. J. Am. Chem. Soc. 1996;118:12082. [Google Scholar]
- (24).Comins DL, Chen X, Morgan LA. J. Org. Chem. 1997;62:7435. doi: 10.1021/jo9711495. [DOI] [PubMed] [Google Scholar]
- (25).Sharma VM, Adi Seshu KV, Krishna CV, Prasanna P, Sekhar VC, Venkateswarlu A, Rajagopal S, Ajaykumar R, Deevi DS, Rao Mamidi NVS, Rajagopalan R. Bioorg. Med. Chem. Lett. 2003;13:1679. doi: 10.1016/s0960-894x(03)00263-4. [DOI] [PubMed] [Google Scholar]
- (26).Baxter G, Melville J, Robins DJ. Synlett. 1991:359. [Google Scholar]
- (27).Synthesis of 9. To 8 (2.2 g, 10 mmol, neat) was added a freshly prepared KOH solution (2.5% aqu. 50 mL). The reaction mixture was stirred at room temperature for 40 h. Then the solution was extracted with ether (50 mL) and the ether layer was discarded. The aqueous layer was cooled to 0 °C and acidified to pH∼4 with cold sulphuric acid (1M). A white precipitate formed, which was taken into ether (50 mL). The aqueous layer was extracted with ether (2 × 20 mL). The combined ether extracts were dried over Na2SO4 and concentrated under reduced pressure, keeping the water bath at 20 °C. The crude acid (semi solid) was used immediately in the next step with out any purification (0.970 g, 50%). Synthesis of 10. To a solution of 9 (4.0 g, 20 mmol) in MeOH (10 mL) was added 1M phosphate buffer (5 mL) to a pH∼7.5 and a freshly prepared solution of (di-(3,4-dihydro-2H-pyrrole))diiodozinc (4.57 g, 10 mmol in water (60 mL). The reaction mixture was stirred for 3 days at room temperature and then cooled to 0 °C and acidified using Congo red as the indicator. The reaction mixture was extracted with ether and the ether layer was discarded. The water layer was cooled to 0 °C and basified carefully to pH∼8 using K2CO3 The water layer was extracted with CHCl3 (3 × 20 mL). The combined extracts were dried over Na2SO4 and concentrated to provide 10 as oil. The crude product (3 g, 75%) was used in the next step without additional purification. Synthesis of 11. To a solution of 10 (2.1 g, 10 mmol) in dry CH2Cl2 (10 mL), Et3N (1.5 mL, 15 mmol) was added and stirred for 5 min. To this solution phenylacetyl chloride (1.6 g, 15 mmol) was added slowly and stirred at room temperature for 12 h. The reaction mixture was extracted with CH2Cl2 (100 mL) washed with sat. NaHCO3 (20 mL), brine (10 mL) and water (10 mL), and dried over Na2SO4 The combined organic extracts were concentrated under reduced pressure. The crude product was purified twice by column chromatography on silica gel using 100% CH2Cl2 for the first column, followed by EtOAc: hexanes (7:3) for the second purification to provide 11 (2 g, 65%) as a semisolid. Synthesis of 12. To 11 (3.3 g, 10 mmol) was added ethanolic KOH (5%, 20 mL). After refluxing this reaction for 2 h, the solvent was removed under reduced pressure. The crude product was washed with 3% HCl and extracted with CH2Cl2 (2 × 50 mL), dried over Na2SO4, and concentrated under reduced pressure. Column chromatography on silica gel, using EtOAc: hexanes (1:1) provided 12 (2.7 g, 80%, mp = 179-182 °C) as colorless crystals. 1H NMR (400 MHz, CDCl3) δ 1.75 (m, 1H), 1.88 (m, 1H), 2.06 (m, 1H), 2.29 (m, 1H), 2.82 (m, 2H), 3.63-3.72 (m, 2H), 3.75 (s, 3H), 3.95 (m, 1H), 6.67 (d, J = 7.4 Hz, 2H), 6.98 (d, J = 8.6 Hz, 2H), 7.19 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 23.59, 34.22, 37.96, 45.21, 55.54, 56.02, 113.69, 127.04, 127.96, 130.29, 131.54, 132.77, 136.75, 145.10, 159.23, 164.53. HRMS (ES+) m/z calc’d for [M+H]+ C21H22NO2: 320.1651, found 320.1635.
- (28).Paull KD, Hamel E, Malspeis L. In: In Cancer Chemotherapeutic Agents. Foye WO, editor. American Chemical Society; Washington, DC: 1995. [Google Scholar]
- (29).Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero SA, Rubinstein L, Plowman J, Boyd MR. J. Natl. Cancer Inst. 1989;81:1088. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- (30).Hollingshead MG, Alley MC, Canalier RF, Abbott BJ, Mayo JG, Malspeis L, Grever MR. Life Sci. 1995;57:131. doi: 10.1016/0024-3205(95)00254-4. [DOI] [PubMed] [Google Scholar]
- (31).Plowman J, Dykes DJ, Hollingshead M, Simpson-Herren L, Alley MC. In: Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval. Teicher B, editor. Humana; Towana, NJ: 1997. p. 101. [Google Scholar]
- (32).Spletstoser JT, Flaherty PT, Himes RH, Georg GI. J. Med. Chem. 2004;47:6459. doi: 10.1021/jm030581d. [DOI] [PubMed] [Google Scholar]
- (33).Topliss JG. J. Med. Chem. 1977;20:463. doi: 10.1021/jm00214a001. [DOI] [PubMed] [Google Scholar]
- (34).Synthesis of 19. With gentle heating and stirring, LC-biotin (0.070 g, 0.20 mmol) was solubilized in DMF (2 mL) after which was added EDCI (0.050g, 0.26 mmol). With the addition of DMAP (0.070 g, 0.57 mmol) the reaction mixture turned orange in color and the solution was stirred for 30 min. at ambient temperature. In a separate flask was dissolved aniline 23 (0.021 g, 0.063 mmol) in DMF (2 mL) and the LC-biotin mixture was subsequently added. The reaction was allowed to progress over 24 h after which it was quenched with the addition of water. EtOAc was added and used to extract the product into the organic phase. Equal amounts of water and EtOAc were added and the organic product was extract into the EtOAc layer. The aqueous and organic phases were separated and the aqueous phase was extracted an additional four times with EtOAc. The combined organic fractions were washed four times with water to remove excess DMF then once with brine. The organic phase was dried over sodium sulfate then condensed under reduced pressure. The biotinylated product 19 was purified via flash column chromatography using silica gel (9% MeOH/CH2Cl2) in 29% yield (0.029 g): 1H NMR (400 MHz, CDCl3) δ 1.20-2.35 (m, 20H), 2.60-2.95 (m, 4H), 3.10-3.35 (m, 3H), 3.60-3.73 (m, 2H), 3.73 (s, 3H), 4.00 (m, 1H), 4.28 (m, 1H), 4.42 (m, 1H), 6.36 (d, J = 19.58 Hz, 1H), 6.51 (d, J = 7.55 Hz, 1H), 6.65 (d, J = 7.56 Hz, 2H), 6.76 (d, J = 9.56 Hz, 1H), 6.96 (m, 4H), 7.39 (d, J = 9.73 Hz, 1H), 7.61 (m, 1H), 9.03 (d, J = 11.84, 1H); MS (FAB+) [M+H]+ C37H48N5O5S: m/z 674.