

Summary of recent advances
Kinetic isotope effects are increasingly applied to investigate enzyme reactions and have been used to understand transition state structure, reaction mechanisms, quantum mechanical hydride ion tunneling and to design transition state analogue inhibitors. Binding isotope effects are an inherent part of most isotope effect measurements but are usually assumed to be negligible. More detailed studies have established surprisingly large binding isotope effects with lactate dehydrogenase, hexokinase, thymidine phosphorylase and purine nucleoside phosphorylase. Binding reactants into catalytic sites immobilizes conformationally flexible groups, polarizes bonds and distorts bond angle geometry, all of which generate binding isotope effects. Binding isotope effects are easily measured and provide high-resolution and detailed information on the atomic changes resulting from ligand-macromolecular interactions. Although binding isotope effects complicate kinetic isotope effect analysis, they also provide a powerful tool for finding atomic distortion in molecular interactions.
Introduction
Isotope effects for enzymatic reactions cause a change in rate or equilibrium as a result of an atomic substitution [1]. Enzymatic applications of isotope effects began in the early 1960's [2-4] and became a common tool of mechanistic enzymology following the Sixth Annual Harry Steenbock Symposium in 1976 [5]. Typically, 2H or 3H is substituted for 1H, 13C or 14C is substituted for 12C, 15N is substituted for 14N or 18O is substituted for 16O in otherwise normal substrates [6]. Reaction rates and binding equilibria are sensitive to isotopic substitution because increased atomic mass alters the bond vibrational environment of reactants [1]. Changes in bond vibrational status surrounding the isotopic substitution between free and bound states give rise to a binding preference for light or heavy isotope in the bound state and hence an experimentally accessible isotope effect. Likewise, differences between bond vibrational environments between free substrate and that at an enzymatic transition state report on bond vibrational environments at the transition state [7].
Experimental measurement of isotope effects typically uses a mixture of labeled and unlabeled reactants with individual isotopic substitutions at each atom of interest. The enzyme discriminates between the labeled and unlabeled reactants to generate an experimentally accessible isotope effect. For catalysis, competitive measurements of this type are called kcat/Km kinetic isotope effects (KIEs, or V/K KIE) and values obtained by competitive methods include every step between substrate binding and the first irreversible step in the reaction (Figure 1) [8]. Although V/K KIEs are a combination of binding and catalytic isotope effects, they are useful since they can be used to distinguish between the bond vibrational environment of the reactant free in solution compared to the enzyme-directed transition state [9]. By measuring binding isotope effects (BIEs) specifically [10], atomic distortion upon formation of the Michaelis complex can be understood. Binding isotope effects together with V/K KIEs can separate these distinct effects. As we shall see, BIEs can be insignificant, or can contribute in the same direction or in the opposite direction to the intrinsic KIEs that arise from transition state chemistry. A simple qualitative interpretation of BIEs is that binding of the molecule containing the heavier isotope will be diminished when bond(s) to the heavier isotopic atom are weaker in the bound complex (a normal isotope effect). Conversely, when bonds to the isotopic label are tighter in the bound complex, binding of the heavier isotopic ligand will be preferred (inverse isotope effect). Normal and inverse BIEs will be reported as positive and negative values expressed as percentages, that is, the percent preference for binding the labeled isotopomer. Recent reviews considering the atomic origins and kinetic effects of BIEs in catalysis are important contributions to this field [11•,12••,13••]
Figure 1.

The relationship between binding isotope effects (BIE), kinetic isotope effects measured by competition between isotopically labeled and unlabeled reactants (V/K KIE) and intrinsic kinetic isotope effects (intrinsic KIE) using the arsenolysis reaction of purine nucleoside phosphorylase as an example [39••]. In this reaction inosine (Ino) is converted to hypoxanthine (Hx) and ribosyl 1-arsenate (R1As) as immediate products. R1As is unstable and hydrolyzed rapidly. V/K KIE report on bond vibrational differences between the labeled reactants free in solution and those at the transition state when its formation is irreversible, thus mixing BIE and the isotopes from chemistry from the intrinsic KIE. BIE and V/K KIE measurement permits solution of the intrinsic KIE. Isotope effects from substrate distortion on binding can then be compared to those generated in the chemical step.
BIE in Lactate Dehydrogenase
Anderson's laboratory pioneered the field of BIE by reporting a normal BIE (preferred binding of the light isotope) of 8.5% for the binding of [4-3H]NAD+ to lactate dehydrogenase [14]. Thus, interactions of the conjugated nicotinamide ring with the enzyme caused the C4-H bond (the site of hydride chemistry) to weaken in the Michaelis complex. Partial loss of the C4-H bond makes C4 more chemically reactive to the addition of a hydride ion, thus the Michaelis complex is distorted toward the transition state, even before the second substrate (the hydride donor) adds to the catalytic site (Figure 2). Binding of oxamate, an analogue of the lactate/pyruvate substrates is also influenced by a binding isotope effect such that [18O]oxamate gives an inverse BIE of (negative) −1.6%, thus the bond environment of the 18O is more constrained on the enzyme than in water. Computational analysis of bond vibrational frequencies of oxamate in water and on the enzyme revealed that a bifurcated hydrogen bond between the oxamate carboxylate and the guanidinium group of an active site histidine caused the inverse BIE (Figure 2) [15]. BIEs that occur with atoms near the reaction center can be interpreted in terms of electron distribution and bond vibrational modes being influenced by close amino acid contacts in the Michaelis complex [16••]. Thus, ionic, steric or H-bond contacts that weaken a covalent bond containing the isotopic label will cause a normal BIE. Contacts between the protein and ligand that constrain or strengthen such an atomic center will cause an inverse BIE, as in the case of [18O]oxamate (described above), and in one of the BIEs for glucose-hexokinase (described below).
Figure 2.
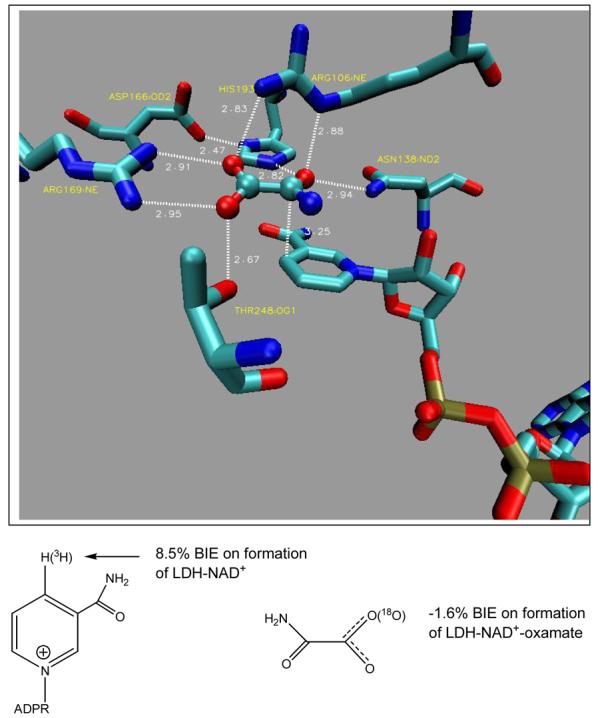
Crystal structure contacts between horse liver lactate dehydrogenase (LDH), NAD+ and oxamate (PDB ID 1I0Z) and illustrated in [16••]. The binding isotope effects are from [14, 15].
Nucleoside Hydrolase
A nucleoside hydrolase from Crithidia fasciculata (a protozoan) exhibited a 5.1% remote V/K KIE with [5'-3H]inosine reacting as the substrate (Figure 3). Although this was a V/K KIE experiment, the [5'-3H] bond is four bonds away from the reaction center, and the general rule for kinetic isotope effects is that effects are negligible more than two bonds away from the reaction center. This surprising isotope effect was interpreted as a BIE caused by distortion of sp3 geometry of the 5'-carbon [17]. An internal hydrogen bond was proposed to exist between the enzyme and the 5'-hydroxyl group such that the 5'-hydroxyl oxygen is placed near the 4'-ribosyl ring oxygen. In solution, the 5'-hydroxyl is free to rotate, but when fixed at the catalytic site, the fixed alignment of the hydroxyl weakens the C5'-H bonds to give the relatively large normal BIE. This interpretation was founded completely from KIE studies and subsequent x-ray crystallographic structures supported the proposal (Figure 3) [18]. The nucleoside hydrolase result suggested that remote BIEs might be a normal expectation for enzymes rather than isolated anomalies. This expectation was supported by computational and NMR studies on substituted alcohols [17,19].
Figure 3.

Kinetic isotope effects near the site of bond loss (the C-N bond connecting ribose and hypoxanthine) compared to the remote 5′-3H V/K KIE for nucleoside hydrolase from Crithidia fasciculata [17].
Hexokinase
The extent and magnitude of BIEs was tested systematically using binary and ternary complexes of human brain hexokinase [20,21]. Tritium was incorporated at every carbon-H bond to give a family of [1-3H]-, [2-3H]-, [3-3H]-, [4-3H]-, [5-3H]- and [6-3H2]glucoses [20]. Hexokinase-catalyzed phosphorylation of glucose occurs at the C6 primary hydroxyl group, making this the reaction center with all other BIEs being remote (more than two bonds away from the reacting atom, O6). Since distortion of sp3 centers and fixing rotational angles of primary and secondary hydroxyl groups are both known to contribute to bond vibrational changes that give rise to BIEs, isotope effects at each carbon-tritium bond indicate the local bond vibrational environment that is changed upon glucose binding. Tritium BIEs were observed from every carbon-tritium center of glucose in the binary complex of hexokinase•glucose [20]. The magnitude of the isotope effects varied from −7.3% for [2-3H]glucose to 6.5% for [6-3H2]glucose (Figure 4). Thus the C2-H bond becomes stiffer when glucose is bound to the enzyme but the C6-H bond become weaker upon glucose binding. Position-specific BIEs demonstrated partial deprotonation of O6 by the catalytic site Asp657 and steric compression of C2-H by contact with the carbonyl of Ser603 [20]. BIEs with the ternary complex of hexokinase•glucose•β-γ-CH2-ATP gave substantial changes at C6-H2, C5-H and C1-H (Figure 4). Using the crystal structure of human hexokinase, the changed BIEs for the ternary complex were interpreted in terms of conformational changes of Glu742 positioned near C1 and Asn683 located under the glucosyl ring near C5 (Figure 5) [21]. Computational modeling with 2-propanol was used to interpret isotope effects at each position. Significant BIEs occur with subtle bond length, angle and sp3 geometric distortions that are too small to be observed by crystallography, but when combined with crystallography, interpretation of the BIE effect changes is augmented [21]. The isotope effects on the α-, β-anomeric equilibrium of glucose were also considered and were shown to have an insignificant effect on the BIE shown in Figure 4. The transition state structure for the anomeric equilibrium of glucose has also been established based on intrinsic KIEs [22•].
Figure 4.
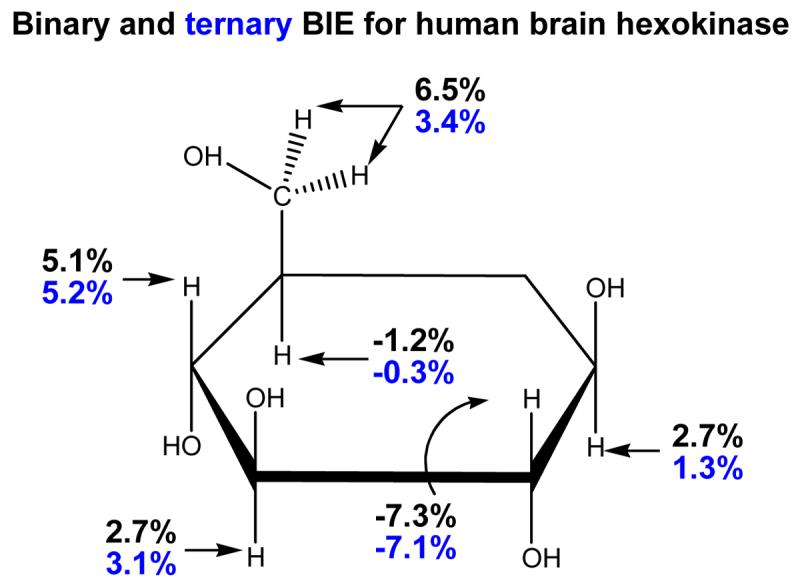
Binding isotope effects for individually labeled 3H-labeled glucose molecules binding to the catalytic domain of human brain hexokinase [20, 21]. The binary complex consists of glucose and hexokinase. The ternary complex includes the β,γ-methylene-ATP in complex with magnesium ion.
Figure 5.
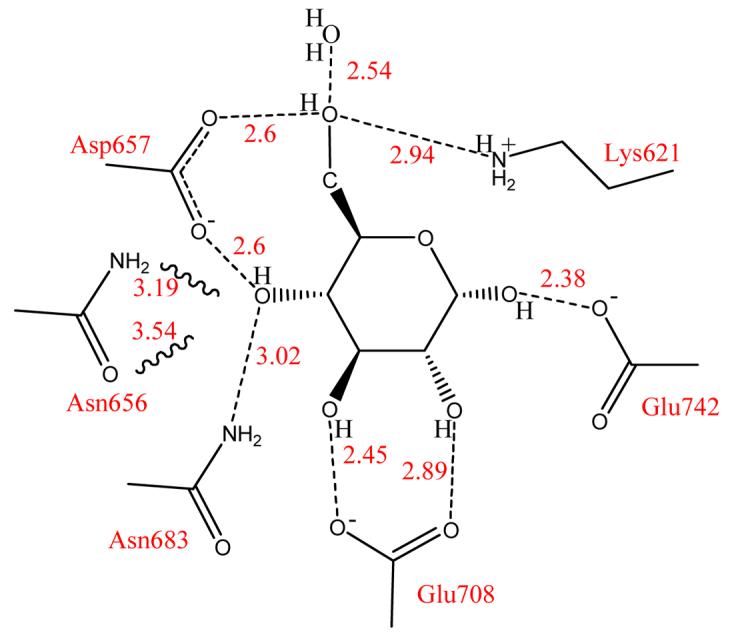
Amino acid contacts to glucose in the catalytic site of human brain hexokinase [21, 41]. Serine 603 is not shown here, but is in steric contact with the hydrogen at the 2-H of glucose, resulting in an inverse isotope effect for this position (Figure 4). The interactions of Asn656 to the 4-hydroxyl group are weak compared Asp657 and Asn683, indicated by the wavy lines.
Thymidine Phosphorylase
Human thymidine phosphorylase (TP) catalyzes the reversible phosphorylation of thymidine to give thymine and α-D-2-deoxyribosyl 1-phosphate (Figure 6). The enzyme is an angiogenic factor and is involved in the metabolism of 5-F-uracil [23]. Unlike most N-ribosyl transferases, the arsenolysis reaction catalyzed by TP has a near-symmetrical nucleophilic displacement that characterizes its transition state [24]. Although this conclusion has been disputed by computational proposals [25-27], there is no other logical interpretation for the large (13.9%) [1'-14C]thymidine kinetic isotope effect [24,28]. A remote kinetic isotope effect for the [5'-3H2]thymidine was 6.1%, similar to that reported earlier for nucleoside hydrolase and attributed to a BIE [17,28]. BIEs are readily measured for two-substrate reactions but not for hydrolases like nucleoside hydrolase. Thymidine phosphorylase provided an opportunity to distinguish BIEs from V/K KIEs. A BIE for [5'-3H2]thymidine was measured to be 6.0%, within experimental error of the V/K KIE. Therefore, the remote [5'-3H2]thymidine V/K KIE is a BIE developed fully on formation of the Michaelis complex and independent of transition state formation. As the reaction proceeds from the Michaelis complex to the transition state, no additional changes occur to the electronic environment of the C5'-H bonds. The C5'-H bonds are weaker in both the Michaelis complex and at the transition state than in thymidine free in solution [13,28]. Crystal structures of human TP are not sufficiently developed to provide geometric information on the disposition of the 5'-hydroxyl at the catalytic site [29,30], but distortion of the sp3 center at C5' to increase freedom in the out of plane bending modes for the H5' atoms or freezing of the hydroxyl geometry to increase the length of the C5'-H bonds can cause these remote effects (Figure 7) [17,31].
Figure 6.

The reaction catalyzed by human thymidine phosphorylase [24]. The transition state is unusual for an N-ribosyltransferase reaction by featuring a nearly symmetrical nucleophilic displacement reaction. Other enzymatic N-ribosyltransferases feature dissociative transition states, as does the chemical reaction [42•].
Figure 7.
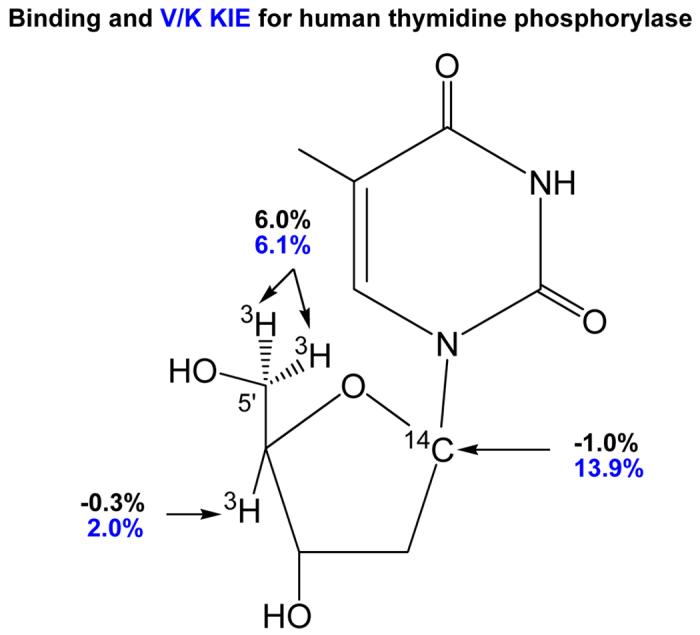
Comparison of BIE (in black) and V/K KIE (in blue) for human thymidine phosphorylase. The KIE reported here are for arsenolysis of thymidine. Use of arsenate as the nucleophile introduces an irreversible step following the transition state formation. Binding isotope effects were measured in the presence of sulfate as a non-reactive phosphate mimic [28].
Purine Nucleoside Phosphorylase
Genetic deficiency of human purine nucleoside phosphorylase (PNP) causes apoptosis in dividing T-cells making PNP a target for T-cell leukemias and autoimmune diseases [32,33]. The transition state structures for mammalian PNPs have been used to design transition state analogues with pM affinity [34,35•]. Two of these PNP inhibitors are in clinical trials for T-cell diseases [33,36]. Analysis of the V/K KIEs for arsenolysis of [5'-3H2]inosine indicated a 5.4% KIE [37]. As the C5'-H bonds are four bonds from the reaction center, it was assumed that these isotope effects arise primarily from binding effects as was found for the remote BIE for TP (see above). Crystallography of mammalian PNPs with substrate nucleosides or transition state analogues has shown the 5'-hydroxyl to be in a hydrogen bond with His257 to locate the hydroxyl oxygen within 2.8 Å of the O4' in the ribosyl ring in both the Michaelis complex and in complexes with transition state analogues [38] (Figure 8). Direct BIE experiments established that only 1.5% of the V/K KIE is generated in the Michaelis complex [39••]. An additional 4.6% isotope effect occurs as the Michaelis complex is converted to the transition state. Both BIE and V/K KIE are in the same direction, indicating partial weakening of C5'-H bonds in the Michaelis complex and additional bond vibrational freedom for these bonds at the transition state [39••].
Figure 8.

Alignment of inosine, phosphate and His257 near the transition state in the catalytic site of human PNP (lower figure). Vibrational modes of the protein are coupled into His 257 and to the phosphate to create periodic compression motions of the three oxygen atoms shown in bold [39,43••]. The effects of these compression modes is to destabilize electrons from the central oxygen, causing them to depart with the leaving group, creating a ribooxacarbenium ion at the transition state. The upper table resolves the relative contributions of BIE and the intrinsic KIE to the experimentally observed V/K KIE.
Mutations His257Phe, His257Gly and His257Asp were generated to probe the contributions of the C5'-hydroxyl His257 to BIE and KIE in human PNP. These three mutations altered the BIE values to 2%, 2% and −2%, respectively, while the transition state contribution further altered the C5'-H bond vibrational environment to contribute −3%, −14% and 7% from the intrinsic KIE, respectively [39••]. These surprising results establish that forces in the Michaelis complex, established from the BIEs, can be reversed or enhanced by the formation of subsequent forces developed in the transition state. Crystal structures of these mutants with transition state analogues at their catalytic sites and computational analysis of the BIE values indicated that polarization of the O5'-H bond both in the Michaelis complex and at the transition state is a dominant factor contributing to these effects [39••].
Adenosine Deaminase
Adenosine deaminase (ADA) uses a catalytic site Zn2+ to ionize a bound water molecule to a reactive hydroxyl ion that attacks C6 of adenosine to form a tetrahedral Meisenheimer intermediate with the subsequent protonation and loss of the amino group (Figure 9). The transition state structures of human, bovine and Plasmodium falciparum (a malaria parasite) demonstrate that the transition states are close to the covalent intermediate, but that the state of N1 protonation differs between these three enzymes [40••]. In the measurement of the V/K KIE values for atoms involved in the reaction coordinate, [1'-3H]adenosine, with the isotope located five bonds away from the reactive center, was used as a remote label. Surprisingly, this center showed remote isotope effects of 1.2% for P. falciparum ADA, 0.7% for human ADA and 0% for the bovine ADA. Possible origins for this unexpected remote isotope effect include rotation around the N-ribosidic bond, altering the electron distribution to C1'-H or sp3 geometric distortion arising from asymmetrical binding interactions of the adenine and ribosyl portions of the substrate. Binding interactions can differ between Michaelis and transition states and the origin of these recently observed isotope effects have not yet been assigned. However, they provide a potentially interesting tool to use in analysis of conformational interactions for nucleosides or oligonucleotides at the catalytic sites of the large number of enzymes using these structurally complicated substrates.
Figure 9.

The reaction catalyzed by adenosine deaminase involves a zinc-activated water and a short-lived covalent intermediate, the Meisenheimer complex (A). Coformycin is a tight-binding transition state analogue of the reaction. Kinetic isotope effects (V/K KIE as shown in Fig. 1) using the indicated atomic labels. KIE were measured with adenosine deaminases from Plasmodium falciparum (Pf ADA), human and bovine sources [40••]. The 1'-3H isotope effect is five bonds from the deamination chemistry and differs for the three enzymes. It is proposed to be a hyperconjugative binding isotope effect arising from rotation or distortion of the purine ring relative to the ribosyl ring when bound to the specific enzymes.
Conclusions
Although BIEs for enzyme-catalyzed reactions have been known since 1989, it is only in the last few years that attempts have been made to quantitate both BIEs and KIEs in specific reactions. When this is done, atomic alterations occurring in the Michaelis complex can be clearly distinguished from those in the transition state. From the few examples now available, the magnitudes of binding isotope effects are large enough to require consideration in full analysis of isotope effect investigation of enzymatic transition states. Importantly, BIEs from the Michaelis complex do not always cause isotope effects that are in the same direction as those generated at the transition state. Thus, competitive V/K KIEs can arise completely from BIEs or BIE values can be negligible, can contribute to or can decrease the observed value of the experimentally determined V/K KIE values. Despite the added complication BIE values add to the analysis of isotope effect studies, BIEs measured alone have potential for understanding geometric and electronic distortions of ligands interacting with macromolecules. Catalysis is not involved in BIEs, so binding inteactions can potentially be mapped for any ligand binding event, including interactions with receptors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Melander L, editor. Isotope Effects on Reaction Rates. Ronald Press; NY: 1960. [Google Scholar]
- 2.Bender ML, Hamilton GA. Kinetic isotope effects of deuterium oxide on several achymotrypsin-catalyzed reactions. J Am Chem Soc. 1962;84:2570–2577. [Google Scholar]
- 3.Thomson JF. Fumarase activity in D2O. Arch Biochem Biophys. 1962;90:1–6. doi: 10.1016/0003-9861(60)90602-0. [DOI] [PubMed] [Google Scholar]
- 4.Westheimer FH. The magnitude of the primary kinetic isotope effect for compounds of hydrogen and deuterium. Chem Rev. 1961;61:265–273. [Google Scholar]
- 5.Cleland WW, O'Leary MH, Northrop DB, editors. Isotope Effects on Enzyme-Catalyzed Reactions. University Park Press; Baltimore, MD: 1977. [Google Scholar]
- 6.Buddenbaum WE, Shiner VJ., Jr . Isotope Effects on Enzyme-Catalyzed Reactions. University Park Press; Baltimore, MD: 1977. pp. 1–36. [Google Scholar]
- 7.Berti PJ. Determining transition states from kinetic isotope effects. Meth Enzymol. 1999;308:355–397. doi: 10.1016/s0076-6879(99)08016-7. [DOI] [PubMed] [Google Scholar]
- 8.Northrop DB. The expression of isotope effects on enzyme-catalyzed reactions. Ann Rev Biochem. 1981;50:103–131. doi: 10.1146/annurev.bi.50.070181.000535. [DOI] [PubMed] [Google Scholar]
- 9.Cleland WW. Isotope effects: determination of enzyme transition state structure. Meth Enzymol. 1995;249:341–373. doi: 10.1016/0076-6879(95)49041-8. [DOI] [PubMed] [Google Scholar]
- 10.Schramm VL. Enzymatic Transition-State Analysis and Transition-State Analogs. Methods Enzymol. 1999;308:301–355. doi: 10.1016/s0076-6879(99)08015-5. [DOI] [PubMed] [Google Scholar]
- •11.Ruszczycky MW, Anderson VE. Interpretation of V/K isotope effects for enzymatic reactions exhibiting multiple isotopically sensitive steps. Jour of Theor Biol. 2006;243:328–342. doi: 10.1016/j.jtbi.2006.06.022. Realistic enzymatic complexity is examined for the contribution of individual steps in reaction to the experimentally measured V/K isotope effects. An essential primer for scientists in this field. [DOI] [PubMed] [Google Scholar]
- ••12.Lewis BE, Schramm VL. Enzymatic binding isotope effects and the interaction of glucose with hexokinase. In: Kohen A, Limbach H-H, editors. Isotope Effects in Chemistry and Biology. CRC Taylor & Francis; Boca Raton: 2006. pp. 1019–1953. This study examines atomic contributions to binding isotope effects using a combination of experimental and computational examples with the human brain hexokinase and its binding of specifically labeled glucoses. Effects of equilibrating substrate (α and β-anomers for glucose) is considered as a general case. [Google Scholar]
- ••13.Anderson VE. Quantifying energetic contributions to ground state destabilization. Arch of Biochem and Biophys. 2005;433:27–33. doi: 10.1016/j.abb.2004.09.026. Substrate binding to enzymes invariably causes conformational changes in both the protein and the reactant. The energetic changes can be estimated by a combination of spectroscopic and computational methods and pertinent examples are given. [DOI] [PubMed] [Google Scholar]
- 14.LaReau R, Wah W, Anderson V. Isotope effects on binding of NAD+ to lactate dehydrogenase. Biochemistry. 1989;28:3619–3624. doi: 10.1021/bi00434a070. [DOI] [PubMed] [Google Scholar]
- 15.Gawlita E, Paneth P, Anderson VE. Equilibrium isotope effect on ternary complex formation of [1-18O]oxamate with NADH and lactate dehydrogenase. Biochemistry. 1995;34:6050–6058. doi: 10.1021/bi00018a007. [DOI] [PubMed] [Google Scholar]
- ••16.Exequiel JR, Pineda T, Callender R, Schwartz SD. Ligand binding and protein dynamics in lactate dehydrogenase. Biophys J. 2007 doi: 10.1529/biophysj.107.106146. in press, 107.106146. Molecular dynamics calculations are used to explore the motions lactate dehydrogenase undergoes to permit substrate entry into the relatively closed catalytic site. Subtle brotein and solvent reorganization permits binding. The strucures shown in Fig. 2 are taken from this paper. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horenstein BA, Schramm VL. Correlation of the molecular electrostatic potential surface of an enzymatic transition state with novel transition-state inhibitors. Biochemistry. 1993;32:9917–9925. doi: 10.1021/bi00089a007. [DOI] [PubMed] [Google Scholar]
- 18.Degano M, Almo SC, Sacchettini JC, Schramm VL. Trypanosomal nucleoside hydrolase. A novel mechanism from the structure with a transition-state inhibitor. Biochemistry. 1998;37:6277–6285. doi: 10.1021/bi973012e. [DOI] [PubMed] [Google Scholar]
- 19.Gawlita E, Lantz M, Paneth P, Bell AF, Tonge PJ, Anderson VE. H-Bonding in alcohols is reflected in the C-H bond strength: Variation of C-D vibrational frequency and fractionation factor. J Am Chem Soc. 2000;122:11660–11669. [Google Scholar]
- 20.Lewis BE, Schramm VL. Binding equilibrium isotope effects for glucose at the catalytic domain of human brain hexokinase. J Am Chem Soc. 2003;125:4785–4798. doi: 10.1021/ja0298242. [DOI] [PubMed] [Google Scholar]
- 21.Lewis BE, Schramm VL. Glucose binding isotope effects in the ternary complex of brain hexokinase demonstrate partial relief of ground-state destabilization. J Am Chem Soc. 2003;125:4672–4673. doi: 10.1021/ja029852k. [DOI] [PubMed] [Google Scholar]
- •22.Lewis BE, Choytun N, Schramm VL, Bennet AJ. Transition States for glucopyranose interconversion. J Am Chem Soc. 2006;128:5049–5058. doi: 10.1021/ja0573054. One of the oldest questions in carbohydrate chemistry, is solved by the application of equilibrium and kinetic isotope effects. Transition states are formed by with multiple water molecules and a single water molecule spanning the hydroxyl groups on C1 and C5 cannot explain the results. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Temmink OH, de Bruin M, Turksma AW, Cricca S, Laan AC, Peters GJ. Activity and substrate specificity of pyrimidine phosphorylases and their role in fluoropyrimidine sensitivity in colon cancer cell lines. Int J Biochem Cell Biol. 2007;39:565–575. doi: 10.1016/j.biocel.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 24.Birck MR, Schramm VL. Nucleophilic participation in the transition state for human thymidine phosphorylase. J. Am. Chem. Soc. 2004;126:2447–2453. doi: 10.1021/ja039260h. [DOI] [PubMed] [Google Scholar]
- 25.Edwards PN. A kinetic, modeling and mechanistic re-analysis of thymidine phosphorylase and some related enzymes. J Enzyme Inhib Med Chem. 2006;21:483–499. doi: 10.1080/14756360600721075. [DOI] [PubMed] [Google Scholar]
- 26.McNally VA, Gbaj A, Douglas KT, Stratford IJ, Jaffar M, Freeman S, Bryce RA. Identification of a novel class of inhibitor of human and Escherichia coli thymidine phosphorylase by in silico screening. Bioorg Med Chem Lett. 2003;13:3705–3709. doi: 10.1016/j.bmcl.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Priego EM, Mendieta J, Gago F, Balzarini J, De Clercq E, Camarasa MJ, Perez-Perez MJ. Towards new thymidine phosphorylase/PD-ECGF inhibitors based on the transition state of the enzyme reaction. Nucleosides Nucleotides Nucleic Acids. 2003;22:951–953. doi: 10.1081/NCN-120022693. [DOI] [PubMed] [Google Scholar]
- 28.Birck MR, Schramm VL. Binding causes the remote [5′-3H]thymidine kinetic isotope effect in human thymidine phosphorylase. J Am Chem Soc. 2004;126:6882–6883. doi: 10.1021/ja0492642. [DOI] [PubMed] [Google Scholar]
- 29.El Omari K, Bronckaers A, Liekens S, Perez-Perez MJ, Balzarini J, Stammers DK. Structural basis for non-competitive product inhibition in human thymidine phosphorylase: implications for drug design. Biochem J. 2006;399:199–204. doi: 10.1042/BJ20060513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Norman RA, Barry ST, Bate M, Breed J, Colls JG, Ernill RJ, Luke RW, Minshull CA, McAlister MS, McCall EJ, McMiken HH, Paterson DS, Timms D, Tucker JA, Pauptit RA. Crystal structure of human thymidine phosphorylase in complex with a small molecule inhibitor. Structure. 2004;12:75–84. doi: 10.1016/j.str.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 31.Maiti NC, Zhu Y, Carmichael I, Serianni AS, Anderson VE. JCH Correlates with alcohol hydrogen bond strength. J Org Chem. 2006;71:2878–2880. doi: 10.1021/jo052389k. [DOI] [PubMed] [Google Scholar]
- 32.Ravandi F, Gandhi V. Novel purine nucleoside analogues for T-cell-lineage acute lymphoblastic leukaemia and lymphoma. Expert Opin Investig Drugs. 2006;15:1601–1613. doi: 10.1517/13543784.15.12.1601. [DOI] [PubMed] [Google Scholar]
- 33.Galmarini CM. Drug evaluation: forodesine - PNP inhibitor for the treatment of leukemia, lymphoma and solid tumor. IDrugs. 2006;9:712–722. [PubMed] [Google Scholar]
- 34.Miles RW, Tyler PC, Furneaux RH, Bagdassarian CK, Schramm VL. Purine nucleoside phosphorylase. Transition state structure, transition state inhibitors and one-third-the-sites reactivity. In: Frey PA, Northrop DB, editors. Enzymatic Mechanisms. IOS Press; Washington DC: 1999. pp. 32–47. [Google Scholar]
- •35.Lewandowicz A, Taylor Ringia EA, Ting LM, Kim K, Tyler PC, Evans GB, Zubkova OV, Mee S, Painter GF, Lenz DH, Furneaux RH, Schramm VL. Energetic mapping of transition state analogue interactions with human and Plasmodium falciparum purine nucleoside phosphorylases. J Biol Chem. 2005;280:30320–30328. doi: 10.1074/jbc.M505033200. Binding interactions of individual atomic sites on transition state analogues are cooperative binding to their target enzymes. The sum of ΔΔG values for individual atomic substitutions is greater than for the intact inhibitor. These support remote BIE interactions from energetic interactions far from the site of chemistry. [DOI] [PubMed] [Google Scholar]
- 36.Balakrishnan K, Nimmanapalli R, Ravandi F, Keating MJ, Gandhi V. Forodesine, an inhibitor of purine nucleoside phosphorylase, induces apoptosis in chronic lymphocytic leukemia cells. Blood. 2006;108:2392–8. doi: 10.1182/blood-2006-03-007468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewandowicz A, Schramm VL. Transition state analysis for human and Plasmodium falciparum purine nucleoside phosphorylases. Biochemistry. 2004;43:1458–1468. doi: 10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- 38.Fedorov A, Shi W, Kicska G, Fedorov E, Tyler PC, Furneaux RH, Hanson JC, Gainsford GJ, Larese JZ, Schramm VL, Almo SC. Transition state structure of purine nucleoside phosphorylase and principles of atomic motion in enzymatic catalysis. Biochemistry. 2001;40:853–860. doi: 10.1021/bi002499f. [DOI] [PubMed] [Google Scholar]
- ••39.Murkin AS, Birck MR, Rinaldo-Matthis A, Shi W, Taylor EA, Almo Steven C, Schramm VL. Neighboring Group Participation in the Transition State of Human Purine Nucleoside Phosphorylase. Biochemistry. 2007;46:5038–5049. doi: 10.1021/bi700147b. BIEs from the 5'-3H remote position of PNP are measured and compared to intrinsic V/K KIE to fully resolve binding and chemical-step isotope effects. Mutation of His257 (in H-bond contact with the 5'-hydroxyl group) causes changes in all isotope effects, to demonstrate the broad range of normal and inverse BIE coupled to remote regions of the catalytic site. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••40.Luo M, Singh V, Taylor EA, Schramm VL. Transition state variation in human, bovine and Plasmodium falciparum adenosine deaminases. J Am Chem Soc. 2007;129:8008–8017. doi: 10.1021/ja072122y. An isotope effect is generated in the C1-3H1 (anomeric carbon) bond of adenosine in V/K isotope effect measurements. At 5 bonds remote from the site of chemistry, and differing for the three enzymes reported here, this remote effect has the hallmarks of a BIE. Not previously appreciated, this effect may have its origin in the glycosyl torsion angle between the purine and the ribosyl group, making it of potential use in nucleic acids. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aleshin AE, Zeng C, Bourenkov GP, Bartunik HD, Fromm HJ, Honzatko RB. The mechanism of regulation of hexokinase: new insights from the crystal structure of recombinant human brain hexokinase complexed with glucose and glucose-6-phosphate. Structure. 1998;6:39–50. doi: 10.1016/s0969-2126(98)00006-9. [DOI] [PubMed] [Google Scholar]
- •42.McCann JA, Berti PJ. Transition State Analysis of Acid-Catalyzed dAMP Hydrolysis. J Am Chem Soc. 2007;129:7055–7064. doi: 10.1021/ja067371l. Remote 5'-3H2 V/K kinetic isotope effects are shown to be small, 1.2% in the acid-catalyzed solvolysis of dAMP. Since geometric distortion is not anticipated in solvolysis, the result indicates that the relatively large 5'-3H2 V/K kinetic isotope effects seen in enzymes are a result of binding distortions combined with transition state interactions. [DOI] [PubMed] [Google Scholar]
- ••43.Nunez S, Wing C, Antoniou D, Schramm VL, Schwartz SD. Insight into catalytically relevant correlated motions in human purine nucleoside phosphorylase. J Phys Chem A. 2006;110:463–472. doi: 10.1021/jp051277u. Computational analysis of the dynamic vibrational patterns of human PNP involve coupling the protein motions to His257, which in turn, causes a compression dynamic mode between the 4'-ring oxygen and the 5'-hydroxyl oxygen of inosine at the catalytic site. [DOI] [PubMed] [Google Scholar]