

Abstract
Each year, tobacco use causes over 4 million deaths worldwide and billions of dollars are spent on treatment for tobacco-related illness. Bupropion, an atypical antidepressant, improves the rates of successful smoking cessation, however, the mechanisms by which bupropion reduces cigarette smoking and depression are unknown. Here we show that clinical concentrations of bupropion inhibit nicotine’s stimulatory effects on brain reward areas. Many drugs of abuse, including nicotine, stimulate dopamine (DA) release in the mesoaccumbens reward system. Nicotinic acetylcholine receptors in the ventral tegmental area (VTA) mediate nicotine’s stimulation of DA release, as well as its rewarding effects. Nicotinic receptors are expressed by excitatory and inhibitory neurons that control DA neuron excitability, and by the DA neurons themselves. Bupropion is a broad-spectrum non-competitive nicotinic receptor antagonist. Here we report that pre-treatment of brain slices with a clinically relevant concentration of bupropion dramatically reduces the effects of nicotine on DA neuron excitability. Nicotinic receptors on VTA DA neurons and their synaptic inputs are inhibited by 75 – 95% after bupropion treatment. We also find that bupropion alone reduces GABAergic transmission to DA neurons, thereby diminishing tonic inhibition of these neurons. This increases DA neuron excitability during bupropion treatment in the absence of nicotine, and may contribute to bupropion’s antidepressant actions.
Keywords: Acetylcholine, Dopamine, Glutamate, GABA, Synaptic Transmission, Nicotinic
Tobacco use is the largest preventable cause of death in developed countries, and despite significant public education efforts the number of smokers continues to increase worldwide [1]. Over 70% of adult smokers express a desire to quit, but only a small fraction succeed each year [2]. Nicotine replacement treatments for smoking cessation roughly doubles quit rate compared to placebo, but a large percentage of that group relapses to smoking within 6 to 12 months [3, 4]. The atypical antidepressant, bupropion (Zyban™) significantly improves the quitting success rate, even after one year [3], and until recently was the only approved smoking cessation aid other than nicotine replacement [5, 6]. Treatment for smoking cessation with bupropion lasts 7 to 12 weeks and starts while patients are still smoking. Typically, the target date for quitting is in the second week of treatment when patients lose their appetite for smoking [3, 5]. Nicotine self-administration studies in rats demonstrate that repeated bupropion administration decreases responding for nicotine, similar to its effects on smoking cessation in humans [7].
Bupropion was introduced clinically as the antidepressant Wellbutrin™ in the 1980s, but its mechanism of action is not completely understood [8-10]. Bupropion lacks interaction with most major receptor classes [8], but it inhibits [3H]-dopamine uptake (IC50 = 2 μM) and [3H]-norepinephrine uptake (IC50 = 5 μM) by rat brain synaptosomes [9]. In addition, bupropion is a broad-spectrum antagonist of nicotinic acetylcholine receptors (nAChRs) in heterologous expression studies [11], and in both in vitro and in vivo assays of native nAChR function [12-14]. Despite these important advances, bupropion’s mechanism of action and the importance of its interaction with nicotinic receptors in smoking cessation remains unclear.
Nicotine reinforces behavior by acting on nAChRs in the mesolimbic dopamine system [15, 16]. As with many drugs of abuse, nicotine increases dopamine release in the nucleus accumbens (NAcc) from midbrain dopamine neurons located in the ventral tegmental area (VTA) [17, 18]. A single exposure to nicotine increases dopamine release in the NAcc for over an hour in vivo [18, 19]. The rewarding effects of nicotine, as assayed by self-administration of the drug, along with nicotine-induced dopamine overflow in the NAcc are dependent upon activation of nAChRs within the VTA [15, 16]. It is important to note that although bupropion has been reported to block native nAChRs [13, 14] these studies have not examined the effects of the drug within the VTA.
We have shown that nicotine modulates VTA synaptic transmission in a persistent manner, which likely contributes to the prolonged excitation of dopamine neurons [20, 21]. Investigating the effects on excitatory transmission, nicotine promoted long-term potentiation of inputs onto dopamine neurons [20]. In addition, nicotine exposure caused a transient enhancement followed by a depression of inhibitory GABAergic inputs to the dopamine neuron, and the latter effect is due to desensitization of nAChRs that mediate tonic inhibitory drive to the dopamine neuron [22]. Here we investigate whether clinically relevant concentrations of bupropion affect cellular responses to nicotine within the VTA. We report that bupropion dramatically reduces nicotine’s effects on the mesolimbic dopamine system. In addition, bupropion treatment alone decreases the inhibitory drive to this system.
Materials and Methods
Horizontal brain slices were prepared from Sprague-Dawley rats (10–14 days of age). Following rapid decapitation, the brain was removed, the olfactory bulbs were cut away, and the midbrain was cut at the level of the 4th ventricle. Then the brain was placed in ice-cold artificial cerebrospinal fluid (aCSF) containing (in mM) NaCl 125, KCl 2.5, MgCl2 1, CaCl2 2.5, Glucose 20, NaH2PO4 1, NaHCO3 25, ascorbic acid 1; bubbled continuously with 95% O2/5% CO2, pH 7.3. From each brain, two or three slices (250–300 μm) were cut in cold aCSF and incubated for at least 1 hr (32–34°C). Slices were pretreated with bupropion at the test concentration during this incubation. For recording, the slice was transferred to a chamber superfused (~2 ml min−1) with aCSF lacking ascorbic acid at room temperature. Bupropion was included in all perfusion solutions at the test concentration.
Neurons were visualized under infrared illumination using an upright microscope (Axioskop, Zeiss). When recording GABAergic transmission, electrodes (2.5–4 MΩ) contained (in mM): K-Gluconate 78, KCl 77, EGTA 1, HEPES 10, Glucose 10, ATP 5, GTP 100 μM (pH 7.4 with KOH). When recording glutamatergic transmission, electrodes were filled with (in mM): K-Gluconate 154, KCl 1, EGTA 1, HEPES 10, Glucose 10, ATP 5 (pH 7.4 with KOH). Standard whole-cell voltage clamp recordings were made using an Axopatch 200B amplifier, a Digidata 1320A interface, and pCLAMP 8 software (Molecular Devices Corp. Sunnyvale, CA). In whole-cell recordings, DA neurons in the VTA can be distinguished from other cell types in the nucleus by action potential firing rates, action potential duration, soma diameter and the presence of a prominent hyperpolarization activated current, Ih [23]. Recent findings suggest that non-DA neurons in the VTA may also express Ih [24, 25]. Cell morphology has some predictive value for identifying dopaminergic cells, but it is not entirely reliable [24], but we contend that the combination of these parameters yields a population of neurons that is predominantly dopaminergic. Neurons were held at −60 mV to assess the presence of Ih, but were held at −70 mV throughout the rest of the voltage clamp experiments. All experiments described here were performed on cells that expressed Ih > 30 pA at −120 mV. Spontaneous transmission was filtered at 1 kHz and digitized at 5 kHz. In experiments measuring membrane potential in current clamp recording mode, we used perforated patch recording methods with amphotericin B (660 μg/ml final concentration, Sigma) dissolved in DMSO was added to the pipette solution. These experiments were started when the series resistance dropped below 40 MΩ, but was typically 15–30 MΩ. In whole-cell recordings, series resistance values were 4-6 MΩ. To isolate GABAergic transmission, experiments were done in the presence of 10 μM 6,7-Dinitroquinoxaline-2,3-dione (DNQX) to block glutamate transmission [26]. Glutamatergic transmission was isolated by adding 20 μM bicuculline to the bath solution to block GABAergic synaptic inputs [27]. Nicotine tartrate, bicuculline methiodide, (both from Sigma, St Louis, MO), DNQX (Tocris, Ellisville, MO) and bupropion HCl (Sigma) were applied through bath perfusion during recording. Bicuculline and DNQX were present in the bath at least 15 minutes before the effect of nicotine was assessed. A new slice was used for each experiment, so that neurons were exposed only once to nicotine.
Spontaneous IPSC and EPSC were recorded continuously at a holding current of −70 mV and data analysis was carried out using MiniAnalysis software (Synaptosoft, Inc., Decatur, GA). Amplitude, rise-time and area thresholds were used to acquire events and the amplitude threshold was set to five times the RMS noise of each recording, as determined by the software. Each acquired event was visually inspected to protect against software errors. Graphical representations of the results were constructed with SigmaPlot 7.0 (SPSS, Inc.).
After plotting synaptic activity frequency histograms determination of ‘responsive’ cells involved comparing, via Student’s t-test, the baseline frequency for a 1 min period immediately prior to nicotine application with a 1 min period centered on the peak nicotine effect. In the absence of a clear nicotine-induced change in frequency, the frequency data were sampled from a 1 min window centered 1 min after the start of the nicotine application. This time point most commonly corresponds to the maximal effect of nicotine on synaptic transmission [20, 21]. The magnitude of each change in synaptic transmission was defined as the difference between the average frequency 1 min immediately prior to nicotine application and an average of 30 sec of frequency data sampled around the peak nicotine response. Again, in the absence of a clear effect of nicotine, the baseline frequency was compared with the frequency during a 30 sec window centered 1 min after the start of the nicotine application. In bupropion pretreated samples, the same methods were used for determining response prevalence and magnitude.
Action potential frequencies were also determined using MiniAnalysis software. For examining the effects of each drug treatment on action potential activity, baseline firing rate was sampled and averaged over 1 min immediately prior to drug application, for nicotine, the response magnitude was determined comparing the baseline frequency determination with the average frequency during a 30 sec window centered on the peak effect. In the absence of an effect on firing rate, the baseline was compared with a 30 sec window centered 30 sec after the beginning of the nicotine application, which was generally the peak of the nicotine effect. Bupropion’s effects on firing rate were much slower to develop, as such, we perfused bupropion for at least 20 min prior to sampling the frequency during a 30 sec window. Determination of significant differences in the modulation of action potential frequency in Figure 1c, each treatment condition (nicotine, bupropion, and the combination of the two drugs) was compared to its own baseline control period (no drug for nicotine and bupropion, bupropion alone for the combination of nicotine and bupropion) using paired t-tests.
Figure 1.
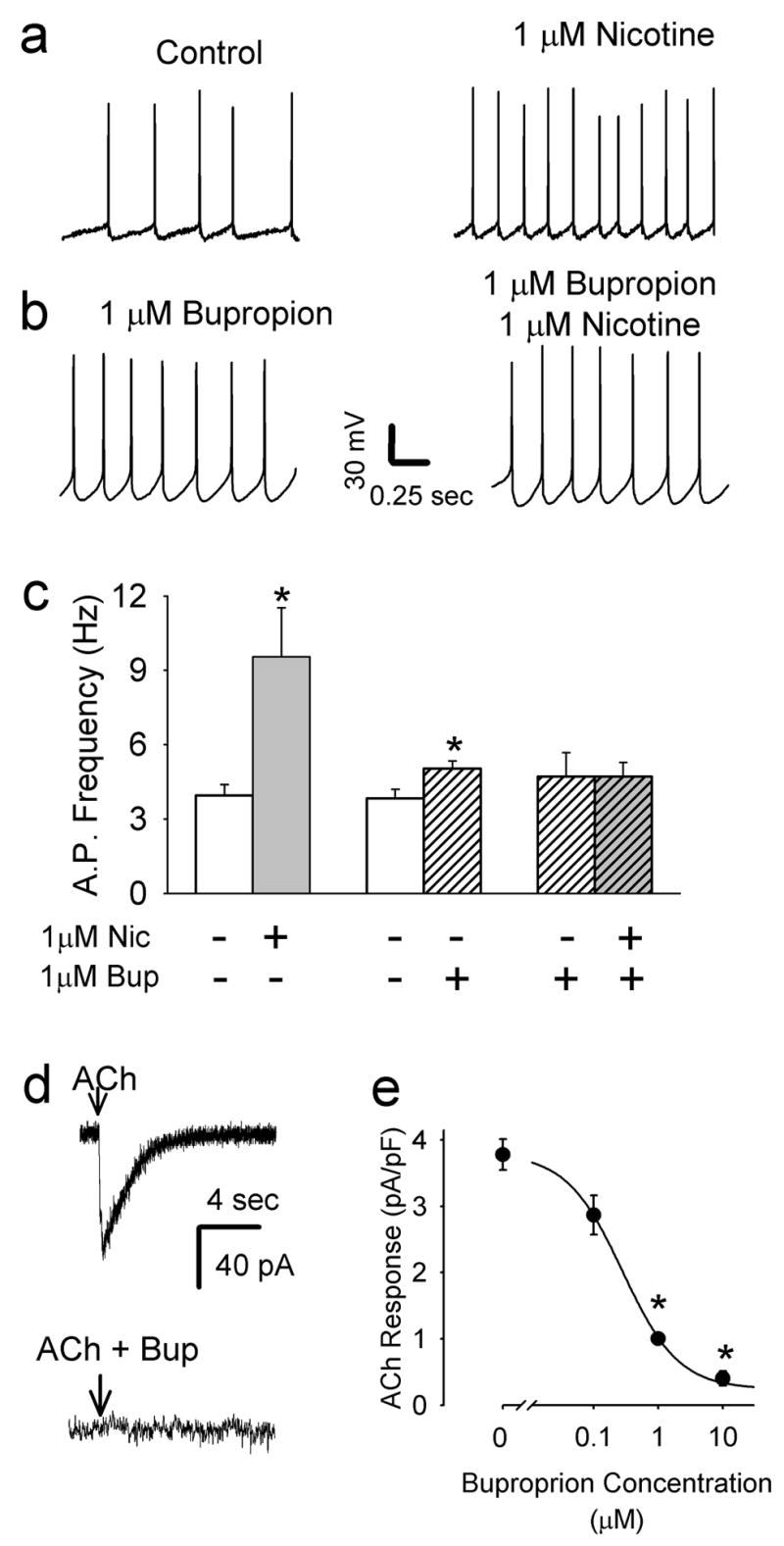
Bupropion inhibits nAChRs on VTA dopamine neurons. a) Bath application of 1 μM nicotine increases action potential frequency (n = 7). b) In slices pretreated with 1 μM bupropion nicotine does not alter action potential frequency in dopamine neurons (n = 4). c) Summary of average action potential firing rates in VTA dopamine neurons under different treatement paradigms. Acute application of nicotine (1μM) induced a significant increase in action potential activity relative to pre-nicotine baseline activity (n = 7, * p<0.05 by paired t-test). In acute tests of bupropion’s effects on action potential activity, baseline firing rate was sampled and averaged over 2 min immediately prior to 1 μM bupropion, which was perfused for at least 20 min prior to sampling the frequency for another 2 min. Under these conditions, bupropion also a significant increase in firing rate (n=6, * p<0.05 by paired t-test), but the magnitude of the change was smaller than nicotine. d) For assessing interaction of bupropion with nAChRs directly, responses to focal application of 1 mM ACh onto VTA dopamine neurons were used. In control cells without bupropion treatment, rapid inward currents were seen in the presence of inhibitors of synaptic transmission. The lower trace illustrates the lack of response from a neuron following pretreatment with 10 μM bupropion for > 2 hrs. e) To assess the sensitivity to bupropion, slices were pretreated with with various concentrations of the drug for > 2 hrs. Summary data of the inhibition of the ACh-induced inward current by a range of bupropion concentrations. (N= 23, 22, 17, 13 for 0, 0.1, 1, and 10 μM, respectively, * p < 0.01). Note that bupropion also blocks the increase in noise induced by nicotine, as with other nAChR antagonists [20], further supporting the idea that bupropion directly inhibits nAChRs on the dopamine neurons. Recordings of membrane potential were made in amphotericin perforated-patch whole-cell current clamp mode. Recordings of the current induced by nicotine were made in normal whole-cell voltage clamp.
Data are presented as mean ± standard error. Statistical comparison of response magnitudes in the presence and absence of bupropion was evaluated using Student’s t-test. Multiple comparisons in Figure 1d were done using Bonferroni protection, where the three comparisons required p < 0.16 by Student’s t-test.
Results
In brain slices containing the VTA, dopamine neurons that project to the nucleus accumbens are depolarized by nicotine concentrations experienced by smokers, which leads to increases in action potential frequency [28-30]. Using perforated-patch recording from VTA DA neurons, we found that bath application of 1 μM nicotine increased the action potential firing frequency to 242 ± 87 % of control (Fig 1a, 1c; n = 7; p<0.05 by paired t-test). Plasma levels of bupropion in humans treated with Zyban for as a smoking cessation aid can rise to a peak between 0.5 to 1 μM [31, 32]. It is reported that pre-application of bupropion reduces the apparent IC50 on cloned nAChRs [12] and patients on Zyban treatment have elevated bupropion plasma levels for weeks. When we tested acute application of clinically relevant bupropion concentrations we saw a weak effect on firing rate (Fig 1b, 1c; n=6; p< 0.05 by paired t-test). Acute bupropion application also inhibited currents induced by focal application of ACh, but both of these effects were slow to develop (~20 min for steady-state inhibition with 1 μM bupropion, data not shown), which complicated the data interpretation as the ACh responses can show varying degrees of run-down in whole cell recordings over a similar time scale. Therefore, we assessed nAChR antagonism in VTA slices that were pre-exposed to bupropion for a minimum of 2 hours (average pretreatment time: 186 ± 10 min, n = 40). As shown in Fig 1b, 1 μM bupropion pretreatment resulted in a small increase in action potential firing over baseline (138 ± 17% of pre-drug baseline, n = 6, p < 0.05 by paired t-test). In the presence of bupropion, nicotine did not alter the firing frequency of VTA dopamine neurons (n = 4; p = 0.92 by paired t-test; Fig 1b, 1c). Note that the weak excitatory effect of bupropion is not likely to occlude resolution of the strong effect of nicotine on firing rate. Bupropion pretreatment dose-dependently inhibited the depolarizing inward current induced by focal application of 100 μM acetylcholine (Fig 1d, 1e). Currents were normalized to cell capacitance and averaged from all recordings under each pretreatment condition. The bupropion concentration that caused 50% of the maximal inhibition (IC50) was 0.28 μM, with a Hill number of 0.99. Significant inhibition of the ACh induced currents by 1 μM and 10 μM bupropion (p < 0.01) and nearly complete inhibition of the currents with 10 μM bupropion. These data were averaged from both Ih-positive dopaminergic and Ih-negative non-dopaminergic neurons (n = 39 and 24, respectively). Plotting the data from Ih-positive neurons independently yielded an identical IC50.
Excitatory glutamatergic transmission in the VTA regulates dopamine neuron excitability [33, 34] and it contributes to the nicotine-induced enhancement of excitation of these neurons both in vitro and in vivo [19, 20]. When recording from dopamine neurons in the presence of 20 μM bicuculline to block GABAergic transmission, the remaining fast synaptic transmission is glutamatergic and is blocked by 10 μM DNQX (not shown)[20]. Nicotine increased the frequency of spontaneous glutamatergic transmission by about 400% relative to pre-drug baseline in 63% of the dopamine neurons (Fig 2). This higher frequency of sEPSC was not associated with a shift in the amplitude of the events, supporting the idea that the nAChRs responsible are expressed on presynaptic terminals, as we have reported previously (data not shown) [20] After pretreatment of slices with 1 μM bupropion, nicotine still increased the spontaneous excitatory postsynaptic current (EPSC) frequency in 58% of the neurons, but the increase was reduced by 73% (Fig 2b,c). Thus, by inhibiting the nicotine-induced inward current in dopamine neurons and by reducing the nicotine-induced enhancement of excitatory transmission to these neurons, bupropion removes the excitatory effects of nicotine on the mesolimbic dopamine reward system.
Figure 2.

Enhancement of excitatory glutamatergic transmission by nicotine is inhibited by bupropion. a) Bath application of 1 μM nicotine enhances the frequency of spontaneous EPSCs. In addition, nicotine also increases the noise in the recording, due to the activation of postsynaptic nAChRs, as in the traces of Figure 1c. To reliably detect individual EPSCs, amplitude, surface area and rise time thresholds were set with the amplitude threshold at least 5 times the RMS noise measured in the presence of nicotine (see Methods)[20]. b) Histograms of EPSC frequency with and without 1 μM bupropion pretreatment. c) Summary graph illustrating the average % change in EPSC frequency following nicotine treatment with and without 1 μM bupropion. These averages include data from only those cells with statistically significant difference in frequency following nicotine treatment. Nicotine only n = 13 of 21; bupropion + nicotine, n = 7 of 12; * p < 0.05.
VTA dopamine neurons receive GABAergic inputs from local interneurons and projection fibers from the NAcc and the ventral pallidum [35]. In vivo biochemical data indicates that dopamine neurons are under tonic inhibitory control by GABA [36]. Recently, it was reported that non-dopamine neurons in the VTA express nAChRs that excite these neurons upon activation by nicotine [37]. Fast GABAergic transmission can be recorded selectively by bathing slices in 10 μM DNQX. The frequency of spontaneous inhibitory postsynaptic currents (sIPSCs) was increased by the application of nicotine to 318 % on average relative to pre-drug baseline (Fig 3). This increased frequency is associated with a shift to larger sIPSC amplitudes. Even though there is an effect on synaptic current magnitude, it is most likely mediated by nAChRs expressed presynaptically, as the effect is TTX sensitive [21]. This suggests that the larger amplitude events in the presence of nicotine are due to multi-vesicular, action potential dependent release events. Pretreatment of slices with 1 μM bupropion inhibited the effect of nicotine on IPSC frequency by 85% on average (Fig 3b,c). Thus, bupropion also inhibits the majority of nAChRs expressed by GABA neurons that project to the VTA dopamine neurons.
Figure 3.

Bupropion inhibits the enhancement of GABAergic transmission induced by nicotine. a) Nicotine (1 μM) increases the frequency of spontaneous IPSCs. b) Frequency histograms showing the increase by nicotine and the inhibition of nicotine’s enhancement by 1 μM bupropion. c) Summary graph illustrating the average % change in IPSC frequency following nicotine treatment with and without 1 μM bupropion. Averages are from all cells tested as nicotine induced statistically significant increases in IPSC frequency in all of the recordings. Nicotine only, n = 12; bupropion + nicotine, n = 12; * p < 0.001.
Interestingly, the baseline frequency of spontaneous IPSCs was significantly lower in slices that were pretreated with bupropion than in untreated control slices (Fig 4a,b). In contrast, the baseline spontaneous EPSC frequency was unaltered by the pretreatment with bupropion (Fig 4c). Possibly, the GABA neurons receive a tonic excitatory drive from cholinergic inputs from the laterodorsal tegmental and the pedunculopontine nuclei. It has been shown that the GABA neurons in the VTA receive cholinergic projections from these nuclei to the VTA [38]. Thus, inhibiting nicotinic receptors with bupropion could remove the excitatory cholinergic drive from GABA neurons, which would decrease inhibitory drive to VTA DA neurons. As a result, long-term exposure to bupropion may increase the basal level of excitation of dopamine neurons through prolonged disinhibition. Consequently, dopamine levels in the mesolimbic reward system may be increased in patients that are treated with Zyban, which may contribute to the antidepressant effects of the drug.
Figure 4.

Bupropion reduces the basal frequency of spontaneous IPSCs. a) Examples of spontaneous IPSCs in control slice without drug treatment, and slice pretreated with 1 μM bupropion. b) Summary graph shows a significantly lower IPSC frequency in slices pretreated with 1 μM bupropion (* p < 0.03; no drug n = 12, bupropion, n = 12). c) EPSC frequency is unaffected by pretreatment with 1 μM bupropion (no drug n = 12 bupropion n = 12
Using extracellular recording configuration we monitored the acute effects of 1 μM bupropion treatment on action potential activity in VTA neurons and found an average increase in firing rate to 135 ± 11 % of the pre-drug firing rate, measured in the same neurons (n = 4; Figure 5A–C). This effect was consistent with the disinhibition of the DA neurons illustrated in Figure 4. While DA reuptake is only weakly affected by this low bupropion concentration [39], a low level of inhibition might alter local DA levels. Increases in extracellular DA within the VTA would be expected to activate D2 autoreceptors on the DA neurons, causing a decrease in firing frequency and an underestimation of the excitatory effects of bupropion. We tested the effects of nomifensine (10 μM), a selective DA transport inhibitor in the VTA slice and found no effect on DA neuron firing frequency, as shown in Figure 5C. Therefore, it is unlikely that the weak inhibition of DA transport by bupropion contributes significantly to the observed changes in VTA action potential activity.
Figure 5.

Bupropion increases action potential firing in VTA DA neurons. a) Example traces of spontaneous action potential firing in control and following treatment with bupropion (1 μM; left panels). The frequency histogram from another neuron shows a clear increase in firing rate following bath perfusion with bupropion (1 μM; right panel). Data were collected using extracellular recording. Scale bars 0.5 mV, 0.5 sec b) Interspike interval distributions before and 20 min after treatment with bupropion (1 μM). These data were collected from the same neuron shown in the histogram in (a). c) Summary data of the spike frequency effects of nomifensine (10 μM; n=3) and bupropion (1μM; n=4; * p < 0.05).
Discussion
Cigarette smoking is addictive, at least in part, through the effects of nicotine on midbrain dopamine reward centers [2, 40]. Nicotine acts directly on VTA dopamine neurons as well as the synaptic inputs to these neurons [20, 21, 29]. In this study we showed that bupropion exposure for more than one hour removes the excitatory actions of nicotine on the dopamine reward system. It dramatically diminishes the excitatory effects of nicotine on VTA dopamine neurons and on the glutamatergic and GABAergic synaptic inputs to these neurons.
Compared with other commonly used antidepressants, bupropion has relatively weak effects on dopamine re-uptake at clinical concentrations. Our findings suggest that bupropion’s inhibition of nicotine-induced excitation of the mesolimbic reward system may depend largely upon its inhibition of nAChRs within this circuitry. Although clinical tests of the non-competitive nicotinic antagonist mecamylamine has yielded negative results for smoking cessation [4]. Recently the partial agonist of nAChRs, varenicline, has been approved for smoking cessation [41, 42]. Administering this drug induces weak activation of nAChRs, which may enhance dopamine release and limit abstinence-induced craving. Varenicline would also antagonize the effects of nicotine at the same receptors, interfering with its reinforcing effects. Thus, antagonism of nAChRs may be a key element in the efficacy of bupropion in combatting nicotine addiction. In this context, the low affinity of bupropion may be an advantage relative to other more efficacious antagonists such as mecamylamine. We found that bupropion never inhibited nicotine’s effects completely, but a small response of 10 to 25% remained, suggesting that reward was reduced but not eliminated. In light of these observations, it would be interesting to test whether low concentrations of mecamylamine, mimicking the weaker antagonistic effects of bupropion, would be efficacious in the treatment of smoking cessation.
Bupropion alone reduced the inhibitory GABAergic input to dopamine neurons. We previously found that GABA neurons in the VTA experience a tonic excitatory drive by endogenous acetylcholine transmission [21], most likely originating from the cholinergic laterodorsal and the pedunculopontine tegmental nuclei. During smoking, desensitization of nAChRs on GABA neurons by nicotine removes this excitatory drive and reduces activity of GABA neurons, thereby diminishing inhibitory input to VTA dopamine neurons. Bupropion is an antagonist of nAChRs [11, 12] and inhibits the effects of nicotine on GABA neurons. This blockade of nAChRs will also remove the excitatory drive to GABA neurons by acetylcholine. As a result, GABAergic transmission to the VTA dopamine neurons is diminished and these neurons receive less inhibition. Thus, people treated with Zyban may experience elevated dopamine levels. This is consistent with the observation that nicotine and bupropion share similar discriminative effects in behavioral studies [43].
The reduction of GABAergic transmission in the VTA and the subsequent disinhibition of the dopamine system may contribute to the effectiveness of bupropion in smoking cessation. It is reported that monoamine oxidase (MAO) is chronically inhibited in the brains of smokers compared to non-smokers and former smokers [44], and MAO inhibitors help heavy smokers to successfully quit. Therefore, disinhibition of the dopamine system by bupropion may compensate for reduced dopamine levels when a person quits smoking. It is not known whether chronic MAO inhibition is part of smoking-related reward, but the disinhibition of the dopamine system by bupropion may reduce negative symptoms associated with abstinence from smoking. Abstinence symptoms are a major cause for relapse in unaided smokers. Indeed, it is reported that in addition to removing the appetite for smoking, bupropion also alleviates abstinence symptoms [45].
It should be emphasized that our investigation focused solely upon acute effects of bupropion in naïve animals that have not been previously exposed to nicotine or other addictive drugs. Thus, the effects of repeated, prolonged exposure to nicotine and bupropion may induce very different physiological effects in the VTA, as well as other brain areas. To better approximate the conditions of the human smoker, future studies will necessarily include investigating bupropion effects in animals that have been chronically exposed to nicotine. This is particularly important as upregulation of nAChR function is known to occur following chronic nicotine exposure [46, 47]. In this regard, it will also be interesting to assess possible changes in nAChR function following chronic bupropion treatment in animals.
Bupropion was the first antidepressant that clearly improved smoking cessation [5]. Other antidepressants have been tested more recently, with some encouraging results with the tricyclic antidepressant, nortriptyline [47, 48]. Mood and affect are strong motivators for smoking. Symptoms of affective disorders are more abundant among smokers than among non-smokers. In addition, people are more likely to smoke when they have a negative affect and these individuals find quitting to be extremely difficult [49]. Bupropion effectively reduces smoking in patients with a history of depression [50]. Disinhibition of the dopamine system by bupropion that we describe here, may also contribute to the antidepressant actions and its success in patients with a history of depression.
A recent behavioral study of in rats suggests that inhibition of dopamine reuptake does contribute to bupropion-induced attenuation of anhedonic effects during nicotine withdrawal [52]. In that study, the effects of nicotine on extracellular dopamine levels were not significantly attenuated by bupropion administration, but it is not clear the extent to which bupropion disinhibition of dopamine neurons might contribute to the observed effects, by the mechanisms explored in our experiments. It is possible that the efficacy of bupropion as a smoking cessation aid derives from the combined inhibition of nAChRs and dopamine reuptake.
In human adolescents the initial symptoms of nicotine dependence can already be present after smoking only a few cigarettes [53]. Recently, we reported that small doses of nicotine, similar to that received from one cigarette, induce a long-term enhancement of dopamine neuron excitability through synaptic mechanisms [20, 21]. These persistent changes in excitability within the brain reward system may reflect the early steps of nicotine dependence. As shown in this study, clinical concentrations of bupropion dramatically reduce the effects of nicotine on synaptic transmission in the VTA. Therefore, pretreatment with bupropion in an early stage of nicotine exposure may help protect individuals from addiction to tobacco.
Figure 6.
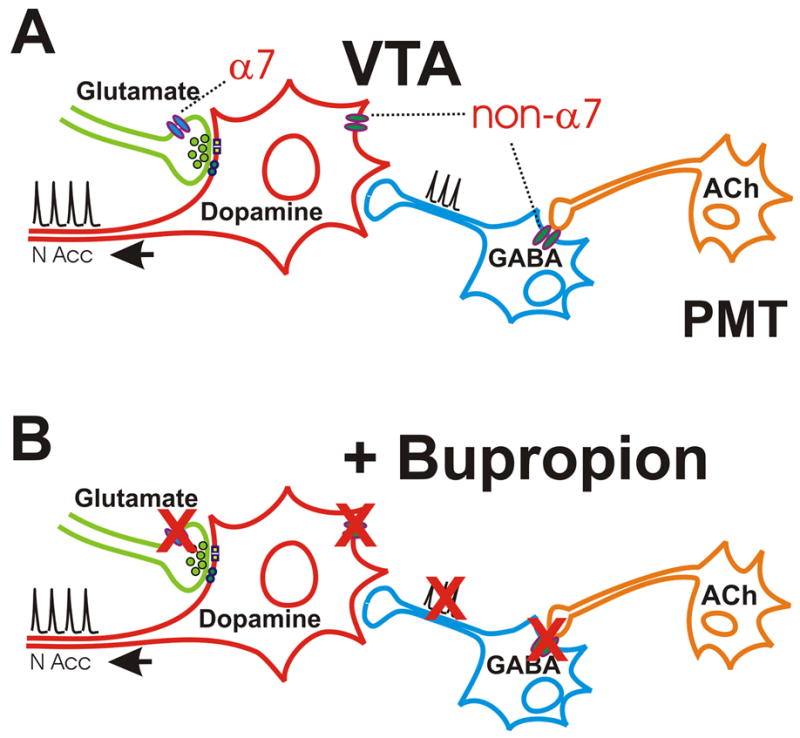
Summary diagram of nAChR effects in VTA and their inhibition by bupropion. A. An illustration of the proposed pattern of nAChR expression on VTA dopamine neurons as well as the glutamatergic and GABAergic inputs to these cells. Presynaptic α7 nAChRs enhance glutamate inputs to VTA dopamine neurons. Non-α7 nAChRs on the cell body of the dopamine neuron increase excitability of the dopamine neurons directly. Non-α7 nAChRs in the preterminal, somatic and dendritic regions of the GABA neurons help set the basal inhibitory tone to the dopamine neuron via cholinergic input from the pontomesencephalic tegmental nuclei (PMT). The receptor localization derives from this study and numerous previous investigations [11, 15, 16, 19-22, 28-30, 36-40].
B. In the presence of bupropion, the effects of nicotine on each of these receptor classes is inhibited. Blockade of the nAChRs on the GABA neurons by bupropion may be particularly important as it disinhibits the dopamine neuron by limiting cholinergic excitatory drive to the GABA neurons [21, 22]. This effect may enhance dopamine release and contribute to the antidepressant effects of the drug.
Acknowledgments
We thank Drs. J.R. Keath and J.R. Genzen for comments on earlier drafts of the manuscript. This work was funded by the National Science Foundation DGE0202337 to ZMF, the Netherlands Organization for Scientific Research NWO S93-334 to HDM, the National Institutes of Health DA07255 to BC, DA015918 and DA019695 to DSM, and by the Brain Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peto R, Chen ZM, Boreham J. Tobacco--the growing epidemic. Nat Med. 1999;5:15–7. doi: 10.1038/4691. [DOI] [PubMed] [Google Scholar]
- 2.Benowitz NL. Nicotine addiction. Primary Care. 1999;26:611–31. doi: 10.1016/s0095-4543(05)70120-2. [DOI] [PubMed] [Google Scholar]
- 3.Jorenby DE, Leischow SJ, Nides MA, Rennard SI, Johnston JA, Hughes AR, et al. A controlled trial of sustained-release bupropion, a nicotine patch, or both for smoking cessation. N Engl J Med. 1999;340:685–91. doi: 10.1056/NEJM199903043400903. [DOI] [PubMed] [Google Scholar]
- 4.Helge TD, Denelsky GY. Pharmacologic aids to smoking cessation. Cleve Clin J Med. 2000;67:81821–4. doi: 10.3949/ccjm.67.11.818. [DOI] [PubMed] [Google Scholar]
- 5.Hurt RD, Sachs DP, Glover ED, Offord KP, Johnston JA, Dale LC, et al. A comparison of sustained-release bupropion and placebo for smoking cessation. N Engl J Med. 1997;337:1195–202. doi: 10.1056/NEJM199710233371703. [DOI] [PubMed] [Google Scholar]
- 6.Hughes JR, Goldstein MG, Hurt RD, Shiffman S. Recent advances in the pharmacotherapy of smoking. Jama. 1999;281:72–6. doi: 10.1001/jama.281.1.72. [DOI] [PubMed] [Google Scholar]
- 7.Rauhut AS, Dwoskin LP, Bardo MT. Tolerance does not develop to the decrease in nicotine self-administration produced by repeated bupropion administration. Nicotine Tob Res. 2005;7:901–7. doi: 10.1080/14622200500381384. [DOI] [PubMed] [Google Scholar]
- 8.Ferris RM, Beaman OJ. Bupropion: a new antidepressant drug, the mechanism of action of which is not associated with down-regulation of postsynaptic beta-adrenergic, serotonergic (5-HT2), alpha 2-adrenergic, imipramine and dopaminergic receptors in brain. Neuropharmacology. 1983;22:1257–67. doi: 10.1016/0028-3908(83)90198-3. [DOI] [PubMed] [Google Scholar]
- 9.Ascher JA, Cole JO, Colin JN, Feighner JP, Ferris RM, Fibiger HC, et al. Bupropion: a review of its mechanism of antidepressant activity. J Clin Psychiatry. 1995;56:395–401. [PubMed] [Google Scholar]
- 10.Dwoskin LP, Rauhut AS, King-Pospisil KA, Bardo MT. Review of the pharmacology and clinical profile of bupropion, an antidepressant and tobacco use cessation agent. CNS drug reviews. 2006;12:178–207. doi: 10.1111/j.1527-3458.2006.00178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fryer JD, Lukas RJ. Noncompetitive functional inhibition at diverse, human nicotinic acetylcholine receptor subtypes by bupropion, phencyclidine, and ibogaine. J Pharmacol Exp Ther. 1999;288:88–92. [PubMed] [Google Scholar]
- 12.Slemmer JE, Martin BR, Damaj MI. Bupropion is a nicotinic antagonist. J Pharmacol Exp Ther. 2000;295:321–7. [PubMed] [Google Scholar]
- 13.Miller DK, Sumithran SP, Dwoskin LP. Bupropion inhibits nicotine-evoked [(3)H]overflow from rat striatal slices preloaded with [(3)H]dopamine and from rat hippocampal slices preloaded with [(3)H]norepinephrine. J Pharmacol Exp Ther. 2002;302:1113–22. doi: 10.1124/jpet.102.033852. [DOI] [PubMed] [Google Scholar]
- 14.Sidhpura N, Redfern P, Wonnacott S. Comparison of the effects of bupropion on nicotinic receptor-evoked [(3)H]dopamine release from rat striatal synaptosomes and slices. Eur J Pharmacol. 2007;567:102–9. doi: 10.1016/j.ejphar.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 15.Corrigall WA, Coen KM, Adamson KL. Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res. 1994;653:278–84. doi: 10.1016/0006-8993(94)90401-4. [DOI] [PubMed] [Google Scholar]
- 16.Nisell M, Nomikos GG, Svensson TH. Systemic nicotine-induced dopamine release in the rat nucleus accumbens is regulated by nicotinic receptors in the ventral tegmental area. Synapse. 1994;16:36–44. doi: 10.1002/syn.890160105. [DOI] [PubMed] [Google Scholar]
- 17.Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–8. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imperato A, Mulas A, Di Chiara G. Nicotine preferentially stimulates dopamine release in the limbic system of freely moving rats. Eur J Pharmacol. 1986;132:337–8. doi: 10.1016/0014-2999(86)90629-1. [DOI] [PubMed] [Google Scholar]
- 19.Schilstrom B, Nomikos GG, Nisell M, Hertel P, Svensson TH. N-methyl-D-aspartate receptor antagonism in the ventral tegmental area diminishes the systemic nicotine-induced dopamine release in the nucleus accumbens. Neuroscience. 1998a;82:781–9. doi: 10.1016/s0306-4522(97)00243-1. [DOI] [PubMed] [Google Scholar]
- 20.Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–57. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- 21.Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–19. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 22.Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53:606–17. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- 23.Johnson SW, North RA. Two types of neurons in the rat ventral tegmental area and their synaptic inputs. J Physiol (Lond) 1992;450:455–68. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006;577:907–24. doi: 10.1113/jphysiol.2006.117069. Epub 2006 Sep 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–97. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honore T, Davies SN, Drejer J, Fletcher EJ, Jacobsen P, Lodge D, et al. Quinoxalinediones: potent competitive non-NMDA glutamate receptor antagonists. Science. 1988;241:701–3. doi: 10.1126/science.2899909. [DOI] [PubMed] [Google Scholar]
- 27.Johnston GAR. Physiologic pharmacology of GABA and its antagonists in the vertebrate nervous system. In: Roberts E, Chase TN, Tower DB, editors. GABA in Nervous System Function. New York: Raven Press; 1976. pp. 305–17. [Google Scholar]
- 28.Calabresi P, Lacey MG, North RA. Nicotinic excitation of rat ventral tegmental neurones in vitro studied by intracellular recording. Br J Pharmacol. 1989;98:135–40. doi: 10.1111/j.1476-5381.1989.tb16873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–4. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- 30.Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–7. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- 31.Findlay JW, Van Wyck Fleet J, Smith PG, Butz RF, Hinton ML, Blum MR, et al. Pharmacokinetics of bupropion, a novel antidepressant agent, following oral administration to healthy subjects. Eur J Clin Pharmacol. 1981;21:127–35. doi: 10.1007/BF00637513. [DOI] [PubMed] [Google Scholar]
- 32.Hsyu PH, Singh A, Giargiari TD, Dunn JA, Ascher JA, Johnston JA. Pharmacokinetics of bupropion and its metabolites in cigarette smokers versus nonsmokers. J Clin Pharmacol. 1997;37:737–43. doi: 10.1002/j.1552-4604.1997.tb04361.x. [DOI] [PubMed] [Google Scholar]
- 33.Johnson SW, Seutin V, North RA. Burst firing in dopamine neurons induced by N-methyl-D-aspartate: role of electrogenic sodium pump. Science. 1992;258:665–7. doi: 10.1126/science.1329209. [DOI] [PubMed] [Google Scholar]
- 34.Wang T, French ED. Electrophysiological evidence for the existence of NMDA and non-NMDA receptors on rat ventral tegmental dopamine neurons. Synapse. 1993;13:270–7. doi: 10.1002/syn.890130310. [DOI] [PubMed] [Google Scholar]
- 35.Kalivas PW, Churchill L, Klitenick MA. GABA and enkephalin projection from the nucleus accumbens and ventral pallidum to the ventral tegmental area. Neuroscience. 1993;57:1047–60. doi: 10.1016/0306-4522(93)90048-k. [DOI] [PubMed] [Google Scholar]
- 36.Westerink BH, Kwint HF, deVries JB. The pharmacology of mesolimbic dopamine neurons: a dual-probe microdialysis study in the ventral tegmental area and nucleus accumbens of the rat brain. J Neurosci. 1996;16:2605–11. doi: 10.1523/JNEUROSCI.16-08-02605.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin R, French ED. A comparison of the effects of nicotine on dopamine and non-dopamine neurons in the rat ventral tegmental area: an in vitro electrophysiological study. Brain Res Bull. 2000;51:507–14. doi: 10.1016/s0361-9230(00)00237-9. [DOI] [PubMed] [Google Scholar]
- 38.Fiorillo CD, Williams JT. Cholinergic inhibition of ventral midbrain dopamine neurons. J Neurosci. 2000;20:7855–60. doi: 10.1523/JNEUROSCI.20-20-07855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nomikos GG, Damsma G, Wenkstern D, Fibiger HC. In vivo characterization of locally applied dopamine uptake inhibitors by striatal microdialysis. Synapse. 1990;6:106–12. doi: 10.1002/syn.890060113. [DOI] [PubMed] [Google Scholar]
- 40.Koob GF. Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol Sci. 1992;13:177–84. doi: 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- 41.Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, et al. Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation. Journal of medicinal chemistry. 2005;48:3474–7. doi: 10.1021/jm050069n. [DOI] [PubMed] [Google Scholar]
- 42.Siu EC, Tyndale RF. Non-nicotinic therapies for smoking cessation. Annu Rev Pharmacol Toxicol. 2007;47:541–64. doi: 10.1146/annurev.pharmtox.47.120505.105354. [DOI] [PubMed] [Google Scholar]
- 43.Wiley JL, Lavecchia KL, Martin BR, Damaj MI. Nicotine-like discriminative stimulus effects of bupropion in rats. Exp Clin Psychopharmacol. 2002;10:129–35. doi: 10.1037//1064-1297.10.2.129. [DOI] [PubMed] [Google Scholar]
- 44.Fowler JS, Volkow ND, Wang GJ, Pappas N, Logan J, MacGregor R, et al. Inhibition of monoamine oxidase B in the brains of smokers. Nature. 1996;379:733–6. doi: 10.1038/379733a0. [DOI] [PubMed] [Google Scholar]
- 45.Balfour DJ. The pharmacology underlying pharmacotherapy for tobacco dependence: a focus on bupropion. Int J Clin Pract. 2001;55:53–7. [PubMed] [Google Scholar]
- 46.Alkondon M, Albuquerque EX. Nicotinic receptor subtypes in rat hippocampal slices are differentially sensitive to desensitization and early in vivo functional upregulation by nicotine and to block by bupropion. J Pharmacol Exp Ther. 2005 doi: 10.1124/jpet.104.081232. [DOI] [PubMed] [Google Scholar]
- 47.Buisson B, Bertrand D. Chronic exposure to nicotine upregulates the human (alpha)4((beta)2 nicotinic acetylcholine receptor function. J Neurosci. 2001;21:1819–29. doi: 10.1523/JNEUROSCI.21-06-01819.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hughes JR, Stead LF, Lancaster T. Nortriptyline for smoking cessation: A review. Nicotine Tob Res. 2005;7:491–9. doi: 10.1080/14622200500185298. [DOI] [PubMed] [Google Scholar]
- 49.Hughes JR, Stead LF, Lancaster T. Antidepressants for smoking cessation. Cochrane Database Syst Rev. 2003:CD000031. doi: 10.1002/14651858.CD000031. [DOI] [PubMed] [Google Scholar]
- 50.Lipkus IM, Barefoot JC, Williams RB, Siegler IC. Personality measures as predictors of smoking initiation and cessation in the UNC Alumni Heart Study. Health Psychology. 1994 Mar 1994;13 doi: 10.1037//0278-6133.13.2.149. [DOI] [PubMed] [Google Scholar]
- 51.Hayford KE, Patten CA, Rummans TA, Schroeder DR, Offord KP, Croghan IT, et al. Efficacy of bupropion for smoking cessation in smokers with a former history of major depression or alcoholism. Br J Psychiatry. 1999;174:173–8. doi: 10.1192/bjp.174.2.173. [DOI] [PubMed] [Google Scholar]
- 52.Paterson NE, Balfour DJ, Markou A. Chronic bupropion attenuated the anhedonic component of nicotine withdrawal in rats via inhibition of dopamine reuptake in the nucleus accumbens shell. Eur J Neurosci. 2007;25:3099–108. doi: 10.1111/j.1460-9568.2007.05546.x. [DOI] [PubMed] [Google Scholar]
- 53.DiFranza JR, Rigotti NA, McNeill AD, Ockene JK, Savageau JA, Cyr DS, et al. Initial symptoms of nicotine dependence in adolescents. Tob Control. 2000;9:313–9. doi: 10.1136/tc.9.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]