

Abstract
The azetidinone LY307174 (1) was identified as a screening lead for the vasopressin V1a receptor (IC50 45 nM at the human V1a receptor) based on molecular similarity to ketoconazole (2), a known antagonist of the luteinizing hormone releasing hormone receptor. Structure-activity relationships for the series were explored to optimize receptor affinity and pharmacokinetic properties, resulting in compounds with Ki values < 1 nM and brain levels after oral dosing ~100-fold higher than receptor affinities.
The neurohypophysial hormones vasopressin1 and oxytocin exert a wide range of physiological effects through binding to specific membrane receptors belonging to the G protein-coupled receptor (GPCR) superfamily. To date, three vasopressin receptor subtypes and one oxytocin receptor have been pharmacologically and functionally described1. V1a, V1b, and oxytocin receptors activate phospholipase C, resulting in the production of inositol 1,4,5-trisphosphate and diacylglycerol, mobilization of intracellular calcium, and activation of protein kinase C. V2 receptors stimulate adenylyl cyclase, resulting in the accumulation of cyclic AMP and activation of protein kinase A. All four receptor subtypes from several mammalian species have been recently cloned2–5, as well as closely related receptors from bony fishes and invertebrates6, 7. Although vasopressin is perhaps best-known for its role in the cardiovascular system, it also has actions in the central nervous system (CNS), and several CNS applications of vasopressin receptor antagonists have been suggested (reviewed in references 8 and 9). A number of research groups have prepared antagonists directed at the vasopressin V1 receptor10–15. While V1a antagonists have been made, none of these have been reported to penetrate the CNS efficiently.
Our vasopressin antagonist program was initiated to identify a CNS-active V1a antagonist: one with potent affinity for the human V1a receptor (IC50 < 10 nM), good oral availability, and ability to penetrate the blood brain barrier - in short, a candidate for human clinical development targeting CNS disorders. The program began at Lilly in 1990 with the selection of a 1,500-compound “Neuropeptide Cassette” - a library intended to identify nonpeptide ligands for neuropeptide receptors. The library applied the concept of receptor crosstalk – previously well documented for the biogenic amine subfamily of GPCRs – to the neuropeptide receptors. The underlying concept is illustrated in Figure 1. Suppose there are two related receptors, A and B. Over the course of evolution, the two receptors have remained identical in part of the binding site (shown as the semicircular portion), while in the rest of the binding site the two receptors have drifted apart due to mutations (shown as the triangular portion in A and the rectangular portion in B). Furthermore, suppose that alpha, a high-affinity ligand for A, is already known, and we wish to find beta, a high-affinity ligand for B. One approach would be to screen all available analogs of alpha in the hope that a mutation in alpha would complement the mutation in A, resulting in a compound with high affinity for B, i.e., beta16, 17.
Figure 1.

Receptor crosstalk as a screening tool
In the construction of the Neuropeptide Cassette, 26 known nonpeptide ligands for neuropeptide receptors (cholecystokinin, substance P, angiotensin II, opiate, leuteinizing hormone releasing hormone (LHRH), and motilin receptors) were used in the role of alpha, and molecular similarity searches in MACCS against the 26 alphas were used to identify “mutated” versions as candidates for beta. Similarity thresholds were adjusted to obtain ~300 matches for each of the 26 query structures, and the ~5,000 unique structures selected by this method were reduced to 1,500 using the leader clustering method.
Among the 26 query structures was ketoconazole, a marketed antifungal agent known to cause reproductive side-effects due to antagonism of the LHRH receptor18. The azetidinone LY307174 (1, Figure 2) was selected for screening based on 59% similarity to ketoconazole (2). Compound 1 was found to have an affinity of 45 nM for the cloned human V1a receptor. In agreement with the rationale in Figure 1, some features of 2 such as the dioxolane ring and terminal phenyl are conserved in 1, while others are totally replaced.
Figure 2.

Beyond the simple issue of affinity, 1 was considered an attractive lead for several reasons. LY307174 is a monocyclic beta-lactam (monobactam). Unlike fused-ring beta-lactams such as penicillins and cephalosporins, simple monobactams such as 1 are highly stable to chemical or enzymatic hydrolysis of the 4-membered azetidinone ring. The cis geometry of the rigid four-membered ring forces the three side-chains together into a fixed geometric configuration, presumably enabling complementarity with sub-pockets of the receptor. Although the molecular weight of 1 is relatively high (568.6), its compactness provided some hope that the series might show significant oral absorption.
After our lead compound was identified, we undertook a structure activity relationship (SAR) study utilizing the chiral Staudinger 2+2 cycloaddition reaction as depicted with a representative example in Scheme 1.
Scheme 1.

In the sequence in Scheme 1, trans-cinnamaldehyde is combined with glycine benzyl ester to form the imine 3, which is then reacted with the chiral auxiliary acid chloride 4, to form the 4-membered ring 5. In the example given in Scheme 1, the S-chiral auxiliary induces formation of the R-cis configuration in the azetidinone product. The methylene group of the glycine constituent could be acylated, producing 6, which displayed potent activity (V1a IC50 = 6.5 nM). The absolute stereochemistry of the chiral methine carbons of the azetidinone ring of 6, depicted in Scheme 1, was assigned based on earlier reports on the absolute configuration of these azetidinone carbons produced through the Staudinger 2+2 reaction with the Evans’ chiral auxiliary19,20. Confirming this assignment, the analog 7 was crystallized and the absolute configuration determined by X-ray analysis21 as shown in Figure 3.
Figure 3.

X-ray structure/absolute configuration of 7
The SAR strategy was based on the modification of four zones, A, B, C, and D, of the azetidinone molecule as depicted in Figure 4. A limited SAR explored effects of substitution in Zone A. For chemical accessibility, the dioxolane ring was replaced with isobutyl in many of the analogs (i.e., L-leucine benzyl ester was used in the 2 + 2 reaction). The affinity of the parent isobutyl analog, 18, was 39 nM (see Figure 6, below). Several alterations afforded a variety of azetidinones with a significant loss of V1a binding activity as represented in Figure 4. Similarly, a limited SAR at Zone B demonstrated that several modifications afforded derivatives with reduced V1a binding activity. Interestingly, replacing the phenyl with the 2-furyl substitution afforded a compound, 14, with comparable activity, V1a IC50 = 2 nM (Figure 5).
Figure 4.

SAR overview of Zone A, 20 compounds prepared
Figure 6.

SAR overview of Zone C, 35 compounds prepared
Figure 5.

SAR overview of Zone B, 3 compounds prepared
Zones C and D emerged as the most important points of structural modification. Removal of the Zone C substituent, as illustrated by the simple benzyl glycine derivative 5, resulted in 10-fold loss of V1a activity, IC50 = 400 nM. Interestingly, acylation of 5 with base and pivaloyl chloride afforded 6 with significantly enhanced potency (V1a IC50 = 5 nM). Several Zone C modifications and attendant human IC50 values are shown in Figure 6.
In 1, Zone D is occupied by the benzyl ester. The ester functionality was considered a potential metabolic weak point, so the first priority was to ascertain whether a more stable group such as amide could substitute for the ester linkage. As an intermediate, the trimethylsilyl ester derivative, 19, was prepared. Despite having equivalent bulk and hydrophobicity to the benzyl group, the trimethylsilyl substitution resulted in reduced activity (V1a IC50 = 405 nM), indicating that the shape of the benzyl group was important. The trimethylsilyl ester, in turn, was transformed into the acid 20, and subsequently coupled with benzylamine to afford the benzyl amide, 21 (V1a IC50 = 43 nM). Thus, the ester functionality of 1 could be converted to the corresponding amide, 21, without loss of binding affinity (V1a IC50 = 45 nM vs. V1a IC50 = 43 nM, respectively, see Figure 7).
Figure 7.
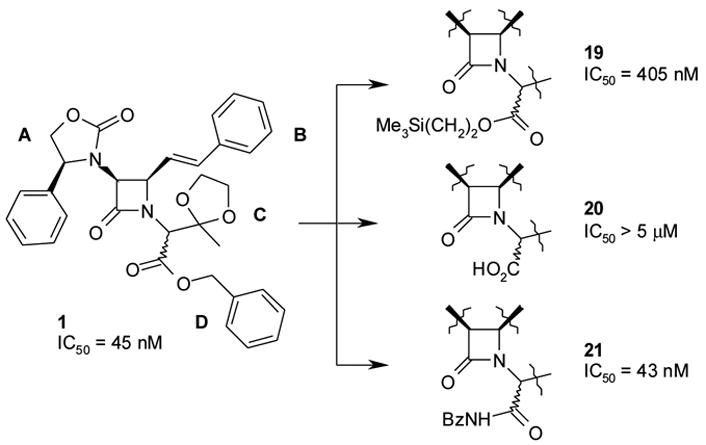
Acceptability of benzylamide in Zone D
At this point, the next step for this project was to find a lead structure that combined high receptor affinity (IC50 < 10 nM) with useful brain levels (>10X the receptor affinity). Although 6 had good V1a binding affinity (IC50 = 5 nM), it provided minimal brain penetration in dogs treated orally with 10 mg/kg (dog brain concentration of 6, 10 nM at 4 hrs). In an effort to find an amide substitution that would enhance oral absorption and CNS penetration, the L-leucyl platform was constructed (Figure 8). Although not optimal for affinity, this platform was used as the basis for SAR exploration of Zone D because of its stability under conditions used for synthesis. The Zone D explorations had two objectives: to find the optimal substitution pattern for the benzylamide group, and to find other groups that would be compatible with high affinity, especially groups with charged functions that would improve water solubility. Rapid solution phase amide syntheses mediated by solid phase carbodiimide afforded over 50 amides22, some of which are illustrated in Figure 8.
Figure 8.
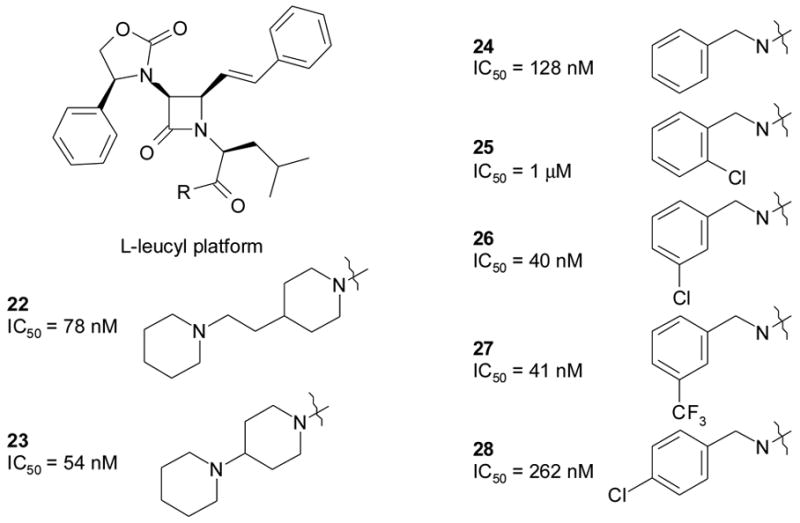
SAR overview of Zone D
A small substituent at the 3-position of the benzylamide (e.g., Cl or CF3) was found to provide optimal affinity. Among groups other than benzyl, piperidylpiperidine and piperidyl-ethyl-piperidine provided relatively good affinities. These groups would be positively charged at physiological pH, potentially leading to improved solubility. Interestingly, the piperidylpiperidine group was previously shown to provide optimal in vivo activity in an NK-1 clinical candidate23.
The piperidyl-ethyl-piperidine 22 was evaluated in dogs dosed orally at 10 mg/kg. Its brain level at 4 hrs post administration was 100 nM, 10-fold better than 6 (see Figure 9). Because the isobutyl substitution in Zone C was known to be suboptimal for V1a affinity, the corresponding compound with pivaloyl in Zone C was synthesized. Based on previous results, the pivaloyl group was expected to provide a roughly 8-fold boost in affinity, resulting in a predicted affinity ~10 nM for the combination. Instead, affinity was 85 nM (Figure 9).
Figure 9.

Non-additivity of the Zone C pivaloyl and Zone D piperidyl-ethyl-piperidine substitutions.
In searching for an explanation of the non-additivity seen in 29, it occurred to us that the binding mode of the molecule may have changed when benzyl was replaced with piperidyl-ethyl-piperidine. The two groups are after all completely different in shape and charge. With a proximally branched alkyl structure, the piperidyl-ethyl-piperidine group actually bears more resemblance to a typical Zone C substituent, such as methyldioxolane, pivaloyl, or isobutyl. We conjectured that the piperidyl-ethyl-piperidine bound into the Zone C pocket of the receptor, forcing the pivaloyl group into the free aqueous phase or into an unfavorable interaction with the Zone D pocket of the receptor. By this logic, reintroducing the benzylamide group in place of the pivaloyl should reoptimize binding in the Zone D pocket, creating a large boost in affinity. At this point, the vasopressin project was deprioritized, so confirmation of this hypothesis was put on hold pending completion of a successful out-licensing campaign.
After a four-year search for a partner, the program was reconstituted as a joint venture with Serenix Pharmaceuticals (later renamed Azevan), a start-up company created for this purpose. The initial strategy was to construct a scaffold with aminomalonic acid replacing L-leucine as depicted in Figure 10. If the piperidyl-ethyl-piperidine binds into Zone C according to the previous hypothesis, the binding pockets in Zones C and D should each tolerate a side-chain anchored by an amide coupling, allowing the piperidyl-ethyl-piperidinamide and benzylamide to be present simultaneously. With the correct stereochemistry, each group should be able to bind to its preferred pocket. The relevant aminomalonate platform was synthesized as illustrated in Scheme 2. Gratifyingly, 33 showed a dramatic enhancement of human V1a binding affinity (V1a Ki = 1.17 nM, Figure 11).
Figure 10.

Rationale for aminomalonate platform
Scheme 2.

Synthesis of 33 and analogs
Figure 11.

D,L-aminomalonyl platform and attendant V1a affinity
Even though there was significant improvement in binding affinity in the aminomalonyl platform azetidinones (Figure 11), the compounds could only be obtained as diastereomeric mixtures, as a consequence of planarization of the malonate methine on dissociation of its acidic hydrogen. Consequently the L-aspartyl platform was prepared in the hope that this would retain the V1a binding affinity while obviating the issue of the labile methine hydrogen and the attendant diastereomic mixture. These synthetic transformations are illustrated in Scheme 3. The most potent compound synthesized, 37, had a V1a affinity comparable to that of 33 from the aminomalonyl platform (Figure 12).
Scheme 3.

Syntheses of L-aspartic and L-glutamic acid derivatives. R = −OSu, i: HNR1R2/THF; R = −OH, ii: HNR1R2/HOBT/EDC,HCl/CH2Cl2.
Figure 12.

L-aspartyl platform and attendant V1a affinity
Theorizing that the D-aspartyl platform would be more resistant to in vivo enzymatic hydrolysis, we prepared the platform with a D-aspartyl group in Zones C and D (see Scheme 4). Although in general the D-platforms tended to be less active than the L-platforms (see the D-glutamyl platform, below), this was less pronounced for the D-aspartyl platform. In the case of 41 (V1a Ki 1.49 nM, Figure 13), affinity of the D-aspartyl compound was actually slightly better than that of the corresponding L-aspartyl analog (42, V1a Ki = 1.89 nM). It is possible that the piperidylpiperidine side-chain may be able to adapt its conformation to match the receptor pocket in a unique way – a possibility that may also explain its superior pharmacokinetic properties in this series (vide infra) and the NK-1 series cited previously23.
Scheme 4.
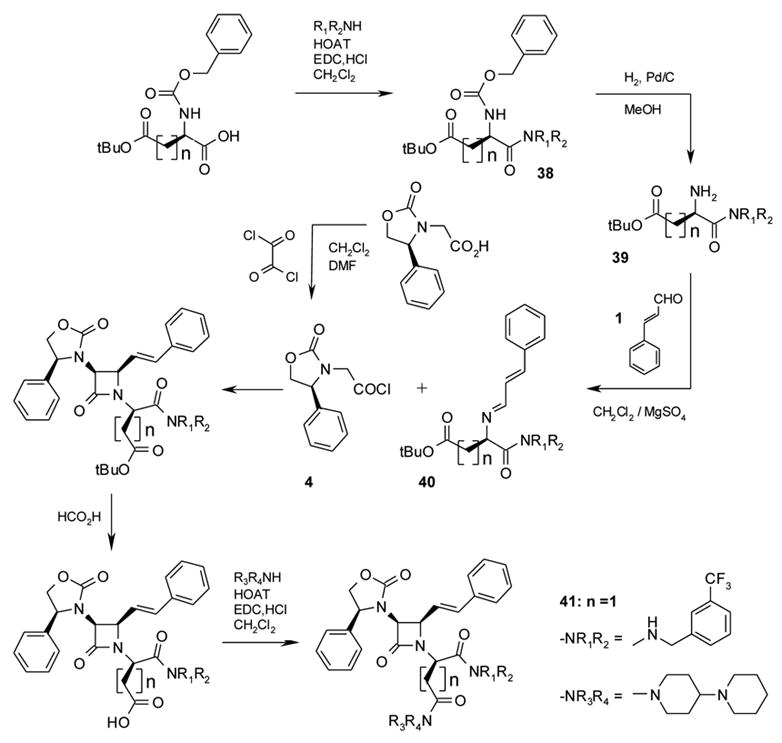
Syntheses of D-aspartic and D-glutamic acid derivatives
Figure 13.
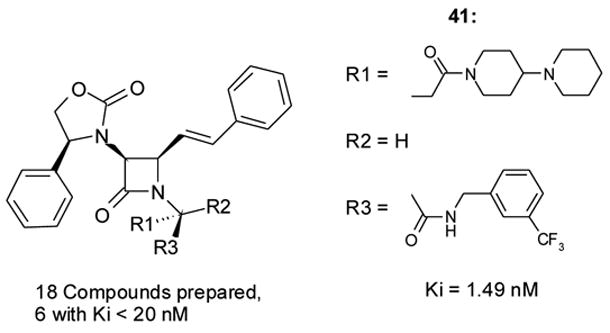
D-aspartyl platform and attendant V1a affinity
The same reasons that led to the investigation of the L- and D-aspartyl platforms also resulted in the preparation of a range of compounds derived from L- and D-glutamic acids. The syntheses of these L- and D-glutamyl platforms are also illustrated in Schemes 3, 4, and 5. The corresponding V1a binding affinities are shown (see Figures 14 and 15). For these two series, the most active compounds were found to be L-glutamate analogs, with the D-glutamate versions typically about 20-fold less active.
Scheme 5.

Synthesis of additional L-glutamic acid derivatives
Figure 14.

L-glutamyl platform and attendant V1a affinity
Figure 15.

D-glutamyl platform and attendant V1a affinity
It is of interest that the highest affinity in the glutamyl platform was seen with the 4-cyclohexylpiperazine side-chain, which is identical to the piperidylpiperidine side-chain that gave optimal activity in the aspartyl platform, except that the basic amino group has been moved one bond closer, offsetting the additional methylene in the glutamyl moiety. In both of these examples, as well as in 37, the positively charged group is 8 bonds away from the azetidine N. Thus it appears that optimal activity requires a positively charged group at a fixed distance from the azetidinone core. It is possible that the basic nitrogen binds to the same site as the guanidinium of arginine-vasopressin.
Five of the compounds with Ki <10 nM24 were tested for plasma and brain levels in rats after oral dosing (20 mg/kg). Azetidinone 22 was used as a comparator. In this screening experiment, three compounds were dosed per animal, and plasma and brain levels were measured by LC/MS. Compound 41 showed the highest levels in both plasma and brain; however, several-fold lower levels were seen in follow-up experiments where 41 was dosed alone, suggesting that the initial higher levels were due in part to co-dosing with 22. These results (plasma Cmax 50–80 ng/ml in two separate experiments) were considered encouraging but fell somewhat short of expectations for a clinical candidate.
Theorizing that facile enzymatic cleavage of the benzylamide bond in Zone D reduced levels of 41, we prepared 50 (SRX246) and 51 (SRX251) (see Figure 15 for structures and Scheme 4 for synthetic pathway). It was hoped that these molecules, the (R)-α-methylbenzyl and the N-methyl analogues of 41 respectively, would be more stable to enzymatic hydrolysis due to steric hindrance near the amide bond. Fortuitously, 50 and 51 proved to possess even higher in vitro V1a affinity than 41. Compound 52, the (S)-α-methylbenzyl diastereomer of 50, was 600-fold less potent (V1a Ki = 179 nM). Interestingly, 53, the trifluoromethyl analog of 50, had reduced V1a binding affinity (see Figure 15), suggesting that the α-methyl substitution shifted the binding mode of the adjacent phenyl ring. Azetidinone 54, the N-methyl derivative of 50, likewise had poor activity (V1a Ki = 9.8 nM), indicating that the N-methyl and alpha-methyl modifications also were incompatible.
Plasma levels for 50 and 51 were roughly double those of 41, to some degree confirming the original motivation for the construction of these two compounds. Cmax values were 100 ng/ml and 160 ng/ml, respectively, compared to 50–80 ng/ml for 41 (Figure 17). Plasma Tmax values for 50 and 51 were 4 hours. Plasma T1/2 values for 50 and 51 were 2.5 hrs and 4 hrs, respectively. The brain levels of 50 and 51 exceeded their Ki values by approximately 100 fold; brain Cmax values of 50 and 51 were 20 ng/ml and 48 ng/ml, respectively, equivalent to 28 nM and 62 nM. Brain T1/2 values of ~6 hr for both compounds suggest the possibility of once-a-day dosing.
Figure 17.
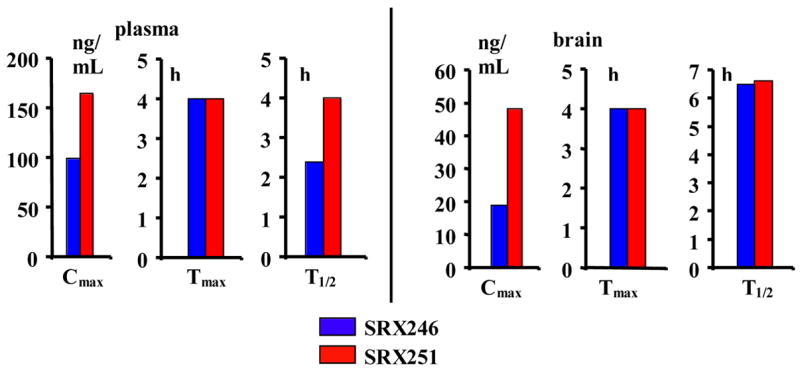
Oral pharmacokinetics of SRX246 and SRX251 in rats.
The binding affinity of all the reported compounds to other members of the vasopressin receptor family was also tested using cell lines that expressed human V1b and V2. The methods for human V1b and V2 binding assay are similar to that for V1a screening, namely the whole cell-based competitive binding assay, which is described in the section of Materials and Methods. The IC50 values of most Azevan compounds proved to be greater than 1 uM or 10 uM for both the V1b and V2 human receptor cell lines.
Conclusions
A novel series of vasopressin V1a antagonists has been designed from the unique monocyclic azetidinone platform. Subnanomolar affinities at the human V1a receptor have been achieved. On oral dosing, two members of the series, 50 (SRX246) and 51 (SRX251), reached brain levels ~100 times their in vitro receptor affinities. The candidate molecules are being further developed for human clinical evaluation.
Experimental
Chemistry:
The early experimental work that was initially performed at Eli Lilly (preparation and characterization of LY307174, etc.) is described in reference 22.
(4(S)-phenyloxazolidin-2-on-3-yl)acetyl chloride (4):
This material was not produced in one batch but rather prepared as required for each “chiral Staudinger 2+2 cycloaddition reaction”. Typically, a solution of 1 g (4.52 mmol) of (4(S)-phenyloxazolidin-2-on-3-yl)acetic acid (Evans, U.S. Patent No. 4,665,171) and 0.51 mL, (5.88 mmol) of oxalyl chloride in 150 mL of dichloromethane was treated with a catalytic amount of anhydrous dimethylformamide (85 μL/milliequivalent of acetic acid derivative) resulting in vigorous gas evolution. After 45 min, all gas evolution had ceased and the reaction mixture was concentrated under reduced pressure to provide the title compound as an off-white solid after drying for 2 h under vacuum. Compound 4 was used without purification for the “chiral Staudinger [2+2] cycloaddition reaction”.
General procedure for an amide formation from an activated ester derivative: N-Benzyloxycarbonyl-L-aspartic acid β-t-butyl ester α-(3-trifluoromethyl)benzylamide; (34a, 34 with HNR1R2 = 3-(trifluoromethyl)benzylamine and n = 1):
A solution of N-benzyloxycarbonyl-L-aspartic acid β-t-butyl ester α-N-hydroxysuccinimide ester (1.95 g, 4.64 mmol, Advanced ChemTech) in 20 mL of dry tetrahydrofuran was treated with 0.68 mL (4.74 mmol) of 3-(trifluoromethyl)benzylamine. Upon completion of the reaction as monitored by TLC (60:40 hexanes/ethyl acetate), the solvent was evaporated, and the resulting oil was partitioned between dichloromethane and a saturated aqueous solution of sodium bicarbonate. The organic layer was, dried over magnesium sulfate, filtered and evaporated to give 2.23 g (quantitative yield) of the title compound as a white solid; 1H NMR (CDCl3) δ 1.39 (s, 9H); 2.61 (dd, J=6.5 Hz, J=17.2 Hz, 1H); 2.98 (dd, J=3.7 Hz, J=17.0 Hz, 1H); 4.41 (dd, J=5.9 Hz, J=15.3 Hz, 1H); 4.50–4.57 (m, 2H); 5.15 (s, 2H); 5.96–5.99 (m, 1H); 6.95 (s, 1H); 7.29–7.34 (m, 5H); 7.39–7.43 (m, 2H); 7.48–7.52 (m, 2H).
The following compounds were obtained according to this procedure: N-Benzyloxycarbonyl-L-aspartic acid β-t-butyl ester α-[4-(2-phenylethyl)]piperazinamide; (34b, 34 with HNR1R2 = 4-(phenylethyl)piperazine and n = 1):
Use of N-benzyloxycarbonyl-L-aspartic acid β-t-butyl ester α-N-hydroxysuccinimide ester (5.0 g, 12 mmol, Advanced ChemTech) and 4-(phenylethyl)piperazine 2.27 mL (11.9 mmol) gave 5.89 g (quantitative yield) of 34b as a white solid; 1H NMR (CDCl3) δ 1.40 (s, 9H); 2.45–2.80 (m,10H); 3.50–3.80 (m, 4H); 4.87–4.91 (m, 1H); 5.08 (s, 2H); 5.62–5.66 (m, 1H); 7.17–7.33 (m, 10H).
N-Benzyloxycarbonyl-L-glutamic acid γ-t-butyl ester α-(3-trifluoromethyl)benzylamide; (34c, 34 with HNR1R2 = 3-(trifluoromethyl)benzylamine and n = 2):
Use of N -benzyloxycarbonyl-L-glutamic acid β-t-butyl ester α-N-hydroxysuccinimide ester (4.83 g, 11.1 mmol, Advanced ChemTech) and 3-(trifluoromethyl)benzylamine) 1.63 mL (11.4 mmol) gave 5.41 g (98%) of 34c as a white solid; 1H NMR (CDCl3) δ 1.40 (s, 9H); 1.88–1.99 (m, 1H); 2.03–2.13 (m, 1H); 2.23–2.33 (m, 1H); 2.38–2.47 (m,1H); 4.19–4.25 (s, 1H); 4.46–4.48 (m, 2H); 5.05–5.08 (m, 2H); 5.67–5.72 (m, 1H); 7.27–7.34 (m, 5H); 7.39–7.43 (m, 2H); 7.48–7.52 (m, 2H).
General procedure for amide formation from a carboxylic acid N-Benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-(3-trifluoromethyl)benzylamide; (38a, 38 with HNR1R2 = 3-(trifluoromethyl)benzylamine and n = 1):
A solution of 1 g (2.93 mmol) of N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate (Novabiochem) in 3–4 mL of dichloromethane was treated by sequential addition of 0.46 mL (3.21 mmol) of 3-(trifluoromethyl)benzylamine, 0.44 g (3.23 mmol) of 1-hydroxy-7-benzotriazole, and 0.62 g (3.23 mmol) of 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride. After at least 12 hours at ambient temperature or until complete as determined by thin layer chromatography (95:5 dichloromethane/methanol eluent), the reaction mixture was washed sequentially with a saturated aqueous sodium bicarbonate solution and with distilled water. The organic layer was dried over magnesium sulfate, filtered and evaporated to give 1.41 g (quantitative yield) of 38a as a white solid; 1H NMR (CDCl3) δ 1.39 (s, 9H); 2.61 (dd, J=6.5 Hz, J=17.2 Hz, 1H); 2.98 (dd, J=4.2 Hz, J=17.2 Hz, 1H); 4.41 (dd, J=5.9 Hz, J=15.3 Hz, 1H); 4.50–4.57 (m, 2H); 5.10 (s, 2H); 5.96–6.01 (m, 1H); 6.91–7.00 (m, 1H); 7.30–7.36 (m, 5H); 7.39–7.43 (m, 2H); 7.48–7.52 (m, 2H).
The following compounds were obtained according to this procedure N-Benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-(2-fluoro-3-trifluoromethyl)benzylamide; (38b, 38 with HNR1R2 = (2-fluoro-3-trifluoromethyl)benzylamine and n = 1):
Use of N -benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate (Novabiochem) (0.25 g, 0.73 mmol) and 0.12 mL of (2-fluoro-3-trifluoromethyl)benzylamine gave 0.365 g (quantitative yield) of 38b as a white solid; 1H NMR (CDCl3) δ 1.38 (s, 9H); 2.59 (dd, J=6.5 Hz, J=17.0 Hz, 1H); 2.95 (dd, J=4.3 Hz, J=17.0 Hz, 1H); 4.46–4.56 (m, 3H); 5.11 (s, 2H); 5.94–5.96 (m, 1H); 7.15 (t, J=8.0 Hz, 1H); 7.30–7.36 (m, 5H); 7.47–7.52 (m, 2H).
N-Benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-[(S)-α-methylbenzyl]amide; (38c, 38 with HNR1R2 = (S)-α-methylbenzylamine and n = 1):
Use of N -benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate (Novabiochem) (0.25 g, 0.73 mmol) and 0.094 mL of (S)-α-methylbenzylamine gave 0.281 g (90%) of 38c as a white solid; 1H NMR (CDCl3) δ 1.41 (s, 9H); 1.44 (d, J=7.0 Hz, 3H); 2.61 (dd, J=7.0 Hz, J=17.0 Hz, 1H); 2.93 (dd, J=4.0 Hz, J=17.5 Hz, 1H); 4.50–4.54 (m, 1H); 5.04–5.14 (m, 3H); 5.94–5.96 (m, 1H); 6.76–6.80 (m, 1H); 7.21–7.37 (m, 10H).
N-Benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-[(R)-α-methylbenzyl]amide; (38d, 38 with HNR1R2 = (R)-α-methylbenzylamine and n = 1):
Use of N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate (Novabiochem) (0.25 g, 0.73 mmol) and 0.094 mL of (R)-α-methylbenzylamine gave 0.281 g (90%) of 38d as a white solid; 1H NMR (CDCl3) δ 1.38 (s, 9H); 1.43 (d, J=6.9 Hz, 3H); 2.54 (dd, J=7.3 Hz, J=17.2 Hz, 1H); 2.87 (dd, J=4.1 Hz, J=17.2 Hz, 1H); 4.46–4.50 (m, 1H); 4.99–5.15 (m, 3H); 5.92–5.96 (m, 1H); 6.78–6.82 (m, 1H); 7.21–7.33 (m, 10H). 13CNMR (CDCl3) δ 21.99, 27.95, 37.58, 50.03, 50.92, 67.13, 81.87, 125.95, 126.72, 128.05, 128.21, 128.52, 128.58, 129.07, 136.08, 142.86, 155.93, 169.34, 171.31.
N-Benzyloxycarbonyl-D-aspartic acid γ-t-butyl ester α-[N-methyl-N-(3-trifluoromethylbenzyl)]amide; (38e, 38 with HNR1R2 = N-methyl-N-(3-trifluoromethylbenzyl)amine and n = 1):
Use of N-benzyloxycarbonyl-D-aspartic acid γ-t-butyl ester (0.303 g, 0.89 mmol, Novabiochem) and 0.168 g (0.89 mmol,) of N-methyl-N-(3-trifluoromethylbenzyl)amine gave 0.287 g (65%) of 38e as a white solid; 1H NMR (CDCl3) δ 1.40 (s, 9H); 2.55 (dd, J=5.8 Hz, J=15.8 Hz, 1H); 2.81 (dd, J=7.8 Hz, J=15.8 Hz, 1H); 3.10 (s, 3H); 4.25 (d, J=15.0 Hz, 1H); 4.80 (d, J=15.5 Hz, 1H); 5.01–5.13 (m, 3H); 5.52–5.55 (m, 1H); 7.25–7.52 (m, 10H).
N-Benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-[(S)-1-(3-trifluoromethylphenyl)ethyl]amide; (38f, 38 with HNR1R2 = (S)-1-(3-trifluoromethylphenyl)ethylamine and n = 1):
Use of N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate (Novabiochem) (84 mg, 0.25 mmol) and 47 mg of (S)-1-(3-trifluoromethylphenyl)ethylamine gave 122 mg (quantitative yield) of 38f as a white solid; 1H NMR (CDCl3) δ 1.38 (s, 9H); 1.43 (d, J=7.0 Hz, 3H); 2.59 (dd, J=6.7 Hz, J=17.3 Hz, 1H); 2.93 (dd, J=4.1 Hz, J=17.3 Hz, 1H); 4.49–4.53 (m, 1H); 5.05–5.28 (m, 3H); 5.91–5.95 (m, 1H); 6.84–6.87 (m, 1H); 7.29–7.52 (m, 9H).
N-Benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-[(R)-1-(3-trifluoromethylphenyl)ethyl]amide; (38g, 38 with HNR1R2 = (R)-1-(3-trifluoromethylphenyl)ethylamine and n = 1):
Use of N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate (Novabiochem) (150 mg, 0.44 mmol) and 83 mg of (R)-1-(3-trifluoromethylphenyl)ethylamine gave 217 mg (quantitative yield) of 38g as a white solid; 1H NMR (CDCl3) δ 1.38 (s, 9H); 1.43 (d, J=7.0 Hz, 3H); 2.54 (dd, J=7.4 Hz, J=17.3 Hz, 1H); 2.87 (dd, J=4.0 Hz, J=17.3 Hz, 1H); 4.47–4.51 (m, 1H); 5.01–5.16 (m, 3H); 5.92–5.96 (m, 1H); 6.90–6.94 (m, 1H); 7.26–7.49 (m, 9H).
N-Benzyloxycarbonyl-D-glutamic acid γ-t-butyl ester α-(3-trifluoromethyl)benzylamide; (38h, 38 with HNR1R2 = 3-(trifluoromethyl)benzylamine and n = 2):
Use of N-benzyloxycarbonyl-D-glutamic acid γ-t-butyl ester (1.14 g, 3.37 mmol) and 0.53 mL (3.70 mmol, Novabiochem) of 3-(trifluoromethyl)benzylamine gave 1.67 g (quantitative yield) of 38h as a white solid; 1H NMR (CDCl3) δ 1.40 (s, 9H), 1.89–1.97 (m, 1H); 2.04–2.12 (m, 1H); 2.24–2.31 (m, 1H); 2.39–2.46 (m,1H); 4.19–4.24 (s, 1H); 4.45–4.48 (m, 2H); 5.03–5.09 (m, 2H); 5.71–5.74 (m, 1H); 6.83–6.88 (m, 1H); 7.26–7.34 (m, 5H); 7.40–7.43 (m, 2H); 7.49–7.52 (m, 2H). 13C NMR (CDCl3) δ 27.86, 27.99, 31.73, 54.51, 67.11, 81.19, 122.90, 124.22 (q, 3JC-F, 4.0Hz, 1C), 124.31, (q, 3JC-F, 3.7Hz, 1C), 125.06, 128.03, 128.22, 128.52, 129.17, 130.88, 130.98 (q, 1JC–F, 32.3Hz, CF3), 136.07, 139.02, 171.43, 172.92.
N-Benzyloxycarbonyl-L-glutamic acid α-t-butyl ester γ-(4-cyclohexyl)piperazinamide: (43a, 43 with HNR1R2 = 1-cyclohexylpiperazine and n = 2):
Use of N-benzyloxycarbonyl-L-glutamic acid α-t-butyl ester (1.36 g, 4.03 mmol) and 0.746g (4.43 mmol) of 1-cyclohexylpiperazine gave 1.93 g (98%) of 43a as a white solid; 1H NMR (CDCl3) δ 1.02–1.12 (m, 5H); 1.43 (s, 9H), 1.60–1.64 (m, 1H); 1.80–1.93 (m, 5H); 2.18–2.52 (m, 8H); 3.38–3.60 (m,4H); 4.20–4.24 (m, 1H); 5.03–5.13 (m, 2H); 5.53–5.57 (m, 1H); 7.28–7.34 (m, 5H).
General procedure for hydrogenation of a benzyloxycarbonyl amine L-aspartic acid β-t-butyl ester α-(3-trifluoromethyl)benzylamide; (35a, 35 with HNR1R2 = 3-(trifluoromethyl)benzylamine and n = 1):
A suspension of 2.23 g (4.64 mmol) of N-benzyloxycarbonyl-L-aspartic acid β-t-butyl ester α-(3-trifluoromethyl)benzylamide 34a and palladium (5% wt. on activated carbon, 0.642 g) in 30 mL of methanol was held under an atmosphere of hydrogen until complete conversion as determined by thin layer chromatography (95:5 dichloromethane/methanol eluent). The reaction was filtered to remove the palladium over carbon and the filtrate was evaporated to give 1.52 g (96%) of 7a as an off–white solid; 1H NMR (CDCl3) δ 1.42 (s, 9H); 2.26 (brs, 2H); 2.63–2.71 (m, 1H); 2.82–2.87 (m, 1H); 3.75–3.77 (m, 1H); 4.47–4.50 (m, 2H); 7.41–7.52 (m, 4H); 7.90 (brs, 1H).
The following compounds were obtained according to this procedure L-aspartic acid β-t-butyl ester α-[4-(2-phenylethyl)]piperazinamide; (35b, 35 with HNR1R2 = 4-(phenylethyl)piperazine and n = 1):
Use of N-benzyloxycarbonyl-L-aspartic acid β-t-butyl ester α-[4-(2-phenylethyl)]piperazinamide (5.89 g, 11.9 mmol) 34b gave 4.24 g (98%) of 35b as an off-white solid; 1H NMR (CDCl3): δ 1.42 (s, 9H); 2.61–2.95 (m, 10H); 3.60–3.90 (m, 4H); 4.35–4.45 (m, 1H); 7.17–7.29 (m, 5H).
L-glutamic acid γ-t-butyl ester α-(3-trifluoromethyl)benzylamide; (35c, 35 with HNR1R2 = 3-(trifluoromethyl)benzylamine and n = 2):
Use of N-benzyloxycarbonyl-L-glutamic acid γ-t-butyl ester α-(3-trifluoromethyl)benzylamide (5.41 g, 10.9 mmol) 34c gave 3.94 g (quantitative yield) of 35c as an off-white solid; 1H NMR (CDCl3): δ 1.41 (s, 9H); 1.73–1.89 (m, 3H); 2.05–2.16 (m, 1H); 2.32–2.38 (m, 2H); 3.47 (dd, J=5.0 Hz, J=7.5 Hz, 1H); 4.47–4.49 (m, 2H); 7.36–7.54 (m, 4H); 7.69–7.77 (m, 1H).
D-aspartic acid β-t-butyl ester α-(3-trifluoromethyl)benzylamide; (39a, 39 with HNR1R2 = 3-(trifluoromethyl)benzylamine and n = 1):
Use of N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-(3-trifluoromethyl)benzylamide (1.41 g, 2.93 mmol) 38a gave 0.973 g (96%) of 39a an off-white solid; 1H NMR (CDCl3): δ 1.42 (s, 9H); 2.21 (brs, 2H); 2.67 (dd, J=7.1 Hz, J=16.8 Hz, 1H); 2.84 (dd, J=3.6 Hz, J=16.7 Hz, 1H); 3.73–3.77 (m, 1H); 4.47–4.50 (m, 2H); 7.41–7.52 (m, 4H); 7.83–7.87 (m, 1H).
D-aspartic acid β-t-butyl ester α-(2-fluoro-3-trifluoromethyl)benzylamide; (39b, 39 with HNR1R2 = (2-fluoro-3-trifluoromethyl)benzylamine and n = 1):
Use of N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-(2-fluoro-3-trifluoromethyl)benzylamide 38b (0.36 g, 0.72 mmol) gave 0.256 g (92%) of 39b as an off-white solid; 1H NMR (CDCl3) δ 1.39 (s, 9H); 2.50 (brs, 2H); 2.74 (dd, J=7.0 Hz, J=16.5 Hz, 1H); 2.86 (dd, J=4.8 Hz, J=16.8 Hz, 1H); 3.89 (brs, 2H); 4.47–4.57 (m, 2H); 7.16 (t, J=7.8 Hz, 1H); 7.48 (t, J=7.3 Hz, 1H); 7.56 (t, J=7.3 Hz, 1H); 7.97–8.02 (m, 1H).
D-aspartic acid β-t-butyl ester α-[(S)-α-methyl]benzylamide; (39c, 39 with HNR1R2 = (S)-α-methylbenzylamine and n = 1):
Use of N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-[(S)-α-methylbenzyl]amide (0.275 g, 0.65 mmol) 38c gave 0.17 g (90%) of 39c as an off-white solid; 1H NMR (CDCl3) δ 1.40 (s, 9H); 1.47 (d, J=6.9 Hz, 3H); 1.98 (brs, 2H); 2.49 (dd, J=7.9 Hz, J=17.7 Hz, 1H); 2.83 (dd, J=3.6 Hz, J=16.7 Hz, 1H); 3.69 (brs, 1H); 4.99–5.10 (m, 1H); 7.19–7.33 (m, 5H); 7.65–7.68 (m, 1H).
D-aspartic acid β-t-butyl ester α-[(R)-α-methylbenzyl]amide; (39d, 39 with HNR1R2 = (R)- α-methylbenzylamine and n = 1):
Use of N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-[(R)-α-methylbenzyl]amide (0.273 g, 0.64 mmol) 38d gave 0.187 g (quantitative yield) of 39d as an off-white solid; 1H NMR (CD3OD) δ 1.40 (s, 9H); 1.46 (d, J=7.0 Hz, 3H); 2.49 (dd, J=7.0 Hz, J=16.5 Hz, 1H); 2.61 (dd, J=5.5 Hz, J=16.0 Hz, 1H); 3.62 (dd, J=6.0 Hz, J=7.0 Hz, 1H); 4.98 (dd, J=7.0 Hz, J=14.0 Hz, 1H); 7.20–7.23 (m, 1H); 7.27–7.34 (m, 4H); 13 C NMR (CD3OD) δ 22.45, 28.31, 41.38, 50.17, 52.92, 82.17, 127.13, 128.09, 129.53, 144.96, 172.14, 175.11.
D-aspartic acid β-t-butyl ester α-[N-methyl-N-(3-trifluoromethylbenzyl)]amide; (39e, 39 with HNR1R2 = N-methyl-N-(3-trifluoromethylbenzyl)amine and n = 1):
Use of N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-[N-methyl-N-(3-trifluoromethylbenzyl)]amide 38e (0.282 g, 0.57 mmol) gave 0.195 g (95%) of 39e as an off-white solid. 10e exhibited a 1H NMR spectrum consistent with the assigned structure.
D-aspartic acid β-t-butyl ester α-[(S)-1-(3-trifluoromethylphenyl)ethyl]amide; (39f, 39 with HNR1R2 = (S)-1-(3-trifluoromethylphenyl)ethylamine and n = 1):
Use of N-Benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-[(S)-1-(3-trifluoromethylphenyl)ethyl]amide 38f (120 mg, 0.24 mmol) gave 80 mg (91%) of 39f as an an off-white solid; 1H NMR (MeOH-d4) δ 1.42 (s, 9H); 1.48 (d, J=7.1 Hz, 3H); 2.58 (dd, J=7.0 Hz, J=16.8 Hz, 1H); 2.69 (dd, J=5.8 Hz, J=16.8 Hz, 1H); 3.77–3.89 (m, 1H); 5.03–5.09 (m, 1H); 7.48–7.65 (m, 4H).
D-aspartic acid β-t-butyl ester α-[(R)-1-(3-trifluoromethylphenyl)ethyl]amide; (39g, 39 with HNR1R2 = (R)-1-(3-trifluoromethylphenyl)ethylamine and n = 1):
Use of N-Benzyloxycarbonyl-D-aspartic acid β-t-butyl ester α-[(R)-1-(3-trifluoromethylphenyl)ethyl]amide 38g (217 mg, 0.44 mmol) gave 157 mg (quantitative yield) of 39g as an off-white solid; 1H NMR (MeOH-d4) δ 1.39 (s, 9H); 1.49 (d, J=7.1 Hz, 3H); 2.55 (dd, J=6.6 Hz, J=16.4 Hz, 1H); 2.63 (dd, J=5.7 Hz, J=16.4 Hz, 1H); 3.60–3.64 (m, 1H); 5.00–5.08 (m, 1H); 7.47–7.67 (m, 4H).
D-glutamic acid γ-t-butyl ester α-(3-trifluoromethyl)benzylamide; (39h, 39 with HNR1R2 = 3-(trifluoromethyl)benzylamine and n = 2):
Use of N-benzyloxycarbonyl-D-glutamic acid γ-t-butyl ester α-(3-trifluoromethyl)benzylamide (1.667 g, 3.37 mmol) 38h gave 1.15 g (94%) of 39h as an an off-white solid; 1H NMR (CDCl3) δ 1.41 (s, 9H); 1.80–2.20 (m, 4H); 2.31–2.40 (m, 2H); 3.51–3.59 (m, 1H); 4.47–4.49 (m, 2H); 7.39–7.52 (m, 4H); 7.71–7.79 (m, 1H).
L-glutamic acid α-t-butyl ester γ-(4-cyclohexyl)piperazinamide; (44a, 44 with HNR1R2 = 1-cyclohexylpiperazine and n = 2):
Use of N-Benzyloxycarbonyl-L-glutamic acid α-t-butyl ester γ-(4-cyclohexyl)piperazinamide (1.93 g, 3.96 mmol) 43a gave 1.30 g (93%) of 44a as an off-white solid; 1H NMR (CDCl3) δ 1.02–1.25 (m, 5H); 1.41 (s, 9H); 1.45–1.50 (m, 1H); 1.56–1.60 (m, 1H); 1.69–1.80 (m, 6H); 3.30 (dd, J=4.8 Hz, J=8.5 Hz, 1H); 3.44 (t, J=9.9 Hz, 2H); 3.56 (t, J=9.9 Hz, 2H).
General procedure for formation of a 2-azetidinone from an imine and an acetyl chloride:
-
Step 1: General procedure for formation of an imine from an amino acid derivative:
A solution of 1 equivalent of an α-amino acid ester or amide (such as 35, 39 or 44) in dichloromethane was treated sequentially with 1 equivalent of an appropriate aldehyde (such as t-cinnamaldehyde), and a dessicating agent, such as magnesium sulfate or silica gel, in the amount of about 2 grams of dessicating agent per gram of starting α-amino acid ester or amide. The reaction was stirred at ambient temperature until all of the reactants were consumed as measured by thin layer chromatography (CH2Cl2 95%/MeOH 5%). When complete, the reaction mixture was then filtered, the filter cake was washed with dichloromethane, and the filtrate concentrated under reduced pressure to provide the desired imine that was used as is in the subsequent step.
-
Step 2: General procedure for the [2+2] cycloaddition of an imine and an acetyl chloride:
A dichloromethane solution of the imine (such as 36, 40, or 45) (10 mL dichloromethane/1 g imine) was cooled to 0°C. To this cooled solution was added 1.5 equivalents of an appropriate amine, typically triethylamine, followed by the dropwise addition of a dichloromethane solution of 1.1 equivalents of an appropriate acetyl chloride (such as 4) (10 mL of dichloromethane/1 g appropriate acetyl chloride). The reaction mixture was allowed to warm to ambient temperature over 1 h and was then quenched by the addition of a saturated aqueous solution of ammonium chloride. The resulting mixture was partitioned between water and dichloromethane. The layers were separated and the organic layer was washed successively with 1N hydrochloric acid, saturated aqueous sodium bicarbonate, and saturated aqueous sodium chloride. The organic layer was dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue was either used directly for further reactions, or purified by chromatography or by crystallization from an appropriate solvent system before use.
The following compounds were prepared according to this sequence of procedures: tert-Butyl [3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetate (55):
The imine prepared from 4.53 g (34.5 mmol) of glycine tert-butyl ester and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl chloride (4) to give 5.5 g (30%) of 55 as colorless crystals (recrystallized, n-chlorobutane); mp 194–195 °C. 1H NMR (CDCl3) δ 1.39 (s, 9H); 3.43 (d, J=18.0 Hz, 1H); 4.15 (dd, J=7.5 Hz, J=8.7Hz, 1H); 4.24 (d, J=18.0Hz, 1H); 4.55–4.63 (m, 3H); 4.80 (dd, J=7.7 Hz, J=8.5 Hz, 1H); 6.05–6.13 (m, 1H); 6.64 (d, J=16.0 Hz, 1H); 7.28–7.42 (m, 10H).
2(S)-(tert-Butoxycarbonylmethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(3-trifluoromethylbenzyl)amide (56):
Imine 36a prepared from 1.52 g (4.39 mmol) of L-aspartic acid β-t-butyl ester α-(3-trifluoromethyl)benzylamide (35a) and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl chloride (4) to give 2.94 g of an orange-brown oil that gave, after flash column chromatography purification (70:30 hexanes/ethyl acetate), 2.06 g (70%) of 56 as a white solid; 1H NMR (CDCl3) δ 1.39 (s, 9H); 2.46 (dd, J=11.1 Hz, J=16.3 Hz, 1H); 3.18 (dd, J=3.8 Hz, J=16.4 Hz, 1H); 4.12–4.17 (m, 1H); 4.26 (d, J=5.0 Hz, 1H); 4.45 (dd, J=6.0 Hz, J=14.9 Hz, 1H); 4.54 (dd, J=5.3 Hz, J=9.8 Hz, 1H); 4.58–4.66 (m, 3H); 4.69–4.75 (m, 1H); 4.81 (dd, J=3.8 Hz, J=11.1 Hz, 1H); 6.25 (dd, J=9.6 Hz, J=15.8 Hz, 1H); 6.70 (d, J=15.8 Hz, 1H); 7.14–7.17 (m, 2H); 7.28–7.46 (m, 11H); 7.62 (s, 1H); 8.27–8.32 (m, 1H).

2(S)-(tert-Butoxycarbonylmethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[4-(2-phenylethyl)]piperazinamide (57):
Imine 36b prepared from 4.20 g (11.6 mmol) of L-aspartic acid β-t-butyl ester α-[4-(2-phenylethyl)]piperazinamide (35b) and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl chloride (4) to give 4.37 g (55%) of 57 as a white solid after flash column chromatography purification (50:50 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.34 (s, 9H); 2.26–2.32 (m, 1H); 2.46–2.63 (m, 4H); 2.75–2.89 (m, 4H); 3.24–3.32 (m, 1H); 3.49–3.76 (m, 3H); 4.07–4.13 (m, 1H); 4.30 (d, J=4.6 Hz, 1H); 4.22–4.48 (m, 1H); 4.55–4.61 (m, 1H); 4.69–4.75 (m, 1H); 5.04–5.09 (m, 1H); 6.15 (dd, J=9.3 Hz, J=15.9 Hz, 1H); 6.63 (d, J=15.8 Hz, 1H); 7.18–7.42 (m, 15H).

(S)-(tert-Butoxycarbonylethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(3-trifluoromethylbenzyl)amide (58):
Imine 36c prepared from 3.94 g (10.93 mmol) of L-glutamic acid γ-t-butyl ester α-(3-trifluoromethyl)benzylamide (35c) and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl chloride (4) to give 5.53 g (75%) of 58 as a white solid after flash column chromatography purification (70:30 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.36 (s, 9H); 1.85–1.96 (m, 1H); 2.18–2.49 (m, 3H); 4.14–4.19 (m, 1H); 4.30 (d, J=4.9 Hz, 2H); 4.44 (dd, J=6.1 Hz, J=14.9 Hz, 1H); 4.56–4.67 (m, 4H); 4.71–4.75 (m, 1H); 6.26 (dd, J=9.6 Hz, J=15.8 Hz, 1H); 6.71 (d, J=15.8 Hz, 1H); 7.16–7.18 (m, 2H); 7.27–7.49 (m, 11H); 7.60 (s, 1H); 8.08–8.12 (m, 1H).

2(R)-(tert-Butoxycarbonylmethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(3-trifluoromethylbenzyl)amide (59):
Imine 40a prepared from 0.973 g (2.81 mmol) of D-aspartic acid β-t-butyl ester α-(3-trifluoromethyl)benzylamide (39a) and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl chloride (4) to give 1.53 g (82%) of 59 as a white solid after flash column chromatography purification (70:30 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.37 (s, 9H); 3.10 (dd, J=3.7 Hz, J=17.8 Hz, 1H); 3.20 (dd, J=10.7 Hz, J=17.8 Hz, 1H); 4.02 (dd, J=3.6 Hz, J=10.6 Hz, 1H); 4.11–4.17 (m, 1H); 4.24 (d, J=4.9 Hz, 1H); 4.46 (dd, J=5.8 Hz, J=15.1 Hz, 1H); 4.58–4.67 (m, 3H); 4.70–4.76 (m, 1H); 6.27 (dd, J=9.5 Hz, J=15.8 Hz, 1H); 6.79 (d, J=15.8 Hz, 1H); 7.25–7.50 (m, 13H); 7.63 (s, 1H); 8.50–8.54 (m, 1H).

2(R)-(tert-Butoxycarbonylmethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(2-fluoro-3-trifluoromethylbenzyl)amide (60):
Imine 40b prepared from 0.256 g (0.70 mmol) of D-aspartic acid β-t-butyl ester α-(2-fluoro-3-trifluoromethyl)benzylamide (39b) and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl (4) to give 0.287 g (60%) of 60 as a white solid after flash column chromatography purification (70:30 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.38 (s, 9H); 3.12 (dd, J=4.0 Hz, J=17.8 Hz, 1H); 3.20 (dd, J=10.4 Hz, J=17.8 Hz, 1H); 4.05 (dd, J=3.9 Hz, J=10.4 Hz, 1H); 4.14 (dd, J=J′=8.2 Hz, 1H); 4.25 (d, J=4.9 Hz, 1H); 4.59–4.67 (m, 4H); 4.74 (t, J=8.3 Hz, 1H); 6.36 (dd, J=9.6 Hz, J=15.8 Hz, 1H); 6.83 (d, J=15.8 Hz, 1H); 7.02–7.07 (m, 1H); 7.28–7.55 (m, 12H); 8.44–8.48 (m, 1H).

2(R)-(tert-Butoxycarbonylmethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(S)-α-methylbenzyl]amide (61):
Imine 40c prepared from 0.167 g (0.57 mmol) of D-aspartic acid β-t-butyl ester [(S)-α-methylbenzyl]amide (39c) and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl chloride (4) to give 0.219 g (63%) of 61 as a white solid after flash column chromatography purification (70:30 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.35 (s, 9H); 1.56 (d, J=7.0 Hz, 3H); 2.97 (dd, J=3.5 Hz, J=18.0 Hz, 1H); 3.15 (dd, J=11.0 Hz, J=17.5 Hz, 1H); 4.01 (dd, J=3.0 Hz, J=11.0 Hz, 1H); 4.14 (t, J=8.5 Hz, 1H); 4.24 (d, J=5.0 Hz, 1H); 4.57 (dd, J=5.0 Hz, J=9.5 Hz, 1H); 4.64 (t, J=8.8 Hz, 1H); 5.07 (t, J=8.5 Hz, 1H); 5.03–5.09 (m, 1H); 6.43 (dd, J=9.5 Hz, J=16.0 Hz, 1H); 6.83 (d, J=16.0 Hz, 1H); 7.16–7.20 (m, 1H); 7.27–7.49 (m, 14H); 8.07–8.10 (m, 1H).

2(R)-(tert-Butoxycarbonylmethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(R)-α-methylbenzyl]amide (62):
Imine 40d prepared from 0.187 g (0.46 mmol) of D-aspartic acid β-t-butyl ester [(R)-α-methylbenzyl]amide (39d) and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl chloride (4) to give 0.25 g (64%) of 62 as a white solid after flash column chromatography purification (70:30 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.36 (s, 9H); 1.59 (d, J=7.1 Hz, 3H); 3.10 (dd, J=3.5 Hz, J=17.8 Hz, 1H); 3.22 (dd, J=10.9 Hz, J=17.8 Hz, 1H); 3.93 (dd, J=3.5 Hz, J=10.8 Hz, 1H); 4.14 (t, J=8.1 Hz, 1H); 4.24 (d, J=5.0 Hz, 1H); 4.58 (dd, J=5.0 Hz, J=9.5 Hz, 1H); 4.65 (t, J=8.7 Hz, 1H); 4.74 (t, J=8.2 Hz, 1H); 5.06–5.14 (m, 1H); 6.32 (dd, J=9.5 Hz, J=15.8 Hz, 1H); 6.74 (d, J=15.8 Hz, 1H); 7.19–7.43 (m, 15H); 8.15–8.18 (m, 1H). 13C NMR (CDCl3) δ 21.84, 27.98, 35.44, 49.56, 54.81, 61.00, 61.98, 63.60, 71.00, 81.15, 121.25, 126.33, 126.80, 126.87, 127.18, 128.41, 128.75, 128.78, 129.69, 135.27, 135.91, 139.03, 143.99, 157.97, 162.99, 167.26, 170.94.

2(R)-(tert-Butoxycarbonylmethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-methyl-N-(3-trifluoromethylbenzyl)amide (63):
Imine 40e prepared from 0.195 g (0.41 mmol) of D-aspartic acid β-t-butyl ester α-[N-methyl-N-(3-trifluoromethylbenzyl)]amide (39e) and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl chloride (4) to give 0.253 g (69%) of 63 as a white solid after flash column chromatography purification (70:30 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.36 (s, 9H); 2.53 (dd, J=4.0 Hz, J=17.0 Hz, 1H); 3.06 (dd, J=10.8 Hz, J=16.8 Hz, 1H); 3.13 (s, 3H); 4.12 (dd, J=8.0 Hz, J=9.0 Hz, 1H); 4.26 (d, J=5.0 Hz, 1H); 4.38 (d, J=15.0 Hz, 1H); 4.46 (dd, J=5.0 Hz, J=9.5 Hz, 1H); 4.56 (t, J=6.8 Hz, 1H); 4.70–4.79 (m, 2H); 5.27 (dd, J=4.0 Hz, J=11.0 Hz, 1H); 6.22 (dd, J=9.3 Hz, J=15.8 Hz, 1H); 6.73 (d, J=15.8 Hz, 1H); 7.33–7.45 (m, 14H).

2(R)-(tert-Butoxycarbonylmethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(S)-1-(3-trifluoromethylphenyl)ethyl]amide (64):
Imine 40f prepared from 0.08 g (0.22 mmol) of D-aspartic acid β-t-butyl ester [(S)-α-methylbenzyl]amide (39f) and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl chloride (4) to give 53 mg (35%) of 64 as a white solid after flash column chromatography purification (70:30 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.36 (s, 9H); 1.59 (d, J=7.0 Hz, 3H); 3.00 (dd, J=3.4 Hz, J=17.8 Hz, 1H); 3.19 (dd, J=11.0 Hz, J=17.8 Hz, 1H); 4.01 (dd, J=3.4 Hz, J=11.0 Hz, 1H); 4.07–4.19 (m, 2H); 4.25 (d, J=5.0 Hz, 1H); 4.58–4.78 (m, 3H); 5.09–5.16 (m, 1H); 6.44 (dd, J=9.5 Hz, J=15.8 Hz, 1H); 6.83 (d, J=15.8 Hz, 1H); 7.19–7.47 (m, 13H); 7.63 (s, 1H), 8.26–8.29 (m, 1H).

2(R)-(tert-Butoxycarbonylmethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(R)-1 -(3-trifluoromethylphenyl)ethyl]amide (65):
Imine 40g prepared from 0.16 g (0.44 mmol) of D-aspartic acid β-t-butyl ester [(R)-α-methylbenzyl]amide (39g) and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl chloride (4) to give 0.166 g (55%) of 65 as a white solid after flash column chromatography purification (70:30 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.36 (s, 9H); 1.60 (d, J=7.1 Hz, 3H); 3.10 (dd, J=3.7 Hz, J=17.8 Hz, 1H); 3.19 (dd, J=10.6 Hz, J=17.8 Hz, 1H); 3.93 (dd, J=3.7 Hz, J=10.6 Hz, 1H); 4.16 (t, J=8.1 Hz, 1H); 4.25 (d, J=5.0 Hz, 1H); 4.58 (dd, J=5.0 Hz, J=9.5 Hz, 1H); 4.67 (t, J=8.6 Hz, 1H); 4.74 (t, J=8.2 Hz, 1H); 5.10–5.19 (m, 1H); 6.27 (dd, J=9.5 Hz, J=15.8 Hz, 1H); 6.74 (d, J=15.8 Hz, 1H); 7.17–7.59 (m, 13H); 7.75 (s, 1H), 8.25–8.27 (m, 1H).

2(R)-(tert-Butoxycarbonylethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(3-trifluoromethylbenzyl)amide (66):
Imine 40h prepared from 1.15 g (3.20 mmol) of D-glutamic acid γ-t-butyl ester α-(3-trifluoromethyl)benzylamide (39h) and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl chloride (4) to give 1.84 g (85%) of 66 as a white solid after flash column chromatography purification (70:30 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.37 (s, 9H); 2.23–2.39 (m, 4H); 3.71–3.75 (m, 1H); 4.13–4.18 (m, 1H); 4.31 (d, J=4.9 Hz, 1H); 4.44–4.51 (m, 2H); 4.56–4.68 (m, 2H); 4.71–4.76 (m, 1H); 6.26 (dd, J=9.5 Hz, J=15.8 Hz, 1H); 6.71 (d, J=15.8 Hz, 1H); 7.25–7.52 (m, 13H); 7.63 (s, 1H); 8.25–8.30 (m, 1H).

tert-Butyl 2(S)-(2-(4-cyclohexylpiperazin-1-ylcarbonyl)ethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetate (67)
Imine 45a prepared from 1.282 g (3.63 mmol) of L-glutamic acid α-t-butyl ester γ-(4-cyclohexyl)piperazinamide (44a) and t-cinnamaldehyde was combined with 2-(4(S)-phenyloxazolidin-2-on-3-yl) acetyl chloride (4) to give 1.946 g (80%) of 67 as a white solid after flash column chromatography purification (50:50 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.15–1.26 (m, 6H); 1.39 (s, 9H); 1.55–1.64 (m, 2H); 1.77–1.83 (m, 3H); 2.22–2.35 (m, 2H); 2.40–2.50 (m, 6H); 2.75–2.79 (m, 1H); 3.43–3.48 (m, 1H); 3.56–3.60 (m, 2H); 3.75–3.79 (m, 1H); 4.10 (t, J=8.3 Hz, 1H); 4.31–4.35 (m, 2H); 4.58 (t, J=8.8 Hz, 1H); 4.73 (t, J=8.4 Hz, 1H); 6.17 (dd, J=8.6 Hz, J=16.0 Hz, 1H); 6.65 (d, J=16.0 Hz, 1H); 7.27–7.42 (m, 10H).

General procedure for acylation of an (azetidin-2-on-1-yl)acetate:
A solution of (azetidin-2-on-1-yl)acetate in tetrahydrofuran (0.22 M in azetidinone) is cooled to −78 °C and treated with lithium bis(trimethylsilyl)amide (2.2 equivalents). The resulting anion is treated with an appropriate acyl halide (1.1 equivalents). Upon complete conversion of the azetidinone, the reaction is quenched with saturated aqueous ammonium chloride and partitioned between ethyl acetate and water. The organic phase is washed sequentially with 1N hydrochloric acid, saturated aqueous sodium bicarbonate, and saturated aqueous sodium chloride. The resulting organic layer is dried (magnesium sulfate) and evaporated. The residue is purified by silica gel chromatography with an appropriate eluent, such as 60:40 hexanes/ethyl acetate.
The following compound was prepared according to this procedure: 2,2,2-Trichloroethyl 2(RS)-(tert-butoxycarbonyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetate (30):
Azetidinone 55, 9.0 g (20 mmol), was acylated with 4.2 g (20 mmol) of trichloroethylchloroformate to give 7.0 g (56%) of 30 as a white solid; mp 176–178 °C. 1H NMR (CDCl3) δ 1.31–1.34 and 1.43–1.46 (m and m’, 9H); 4.12–4.18 (m, 1H); 4.53–4.90 (m; 6H); 5.18–5.24 (m, 1H); 6.10–6.18 (m, 1H); 6.61–6.66 (m, 1H); 7.28–7.41 (m, 10H).

2(RS)-(tert-Butoxycarbonyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(3-trifluoromethylbenzyl)amide (31):
A solution of 0.20 g (0.32 mmol) of 30 and 52 μL (0.36 mmol) of (3-trifluoromethylbenzyl)amine in THF was heated at reflux. Upon complete conversion (TLC), the solvent was evaporated and the residue was crystallized (chloroform/hexane) to give 0.17 g (82%) of 31 as a white solid; mp 182–184 °C. 1H NMR (CDCl3) δ 1.36–1.44 (m, 9H); 4.13–4.14–4.91 (m; 8H); 6.23–6.31 (m, 1H); 6.65–6.78 (m, 1H); 7.20–7.60 (m, 14H); 7.88–7.92 and 8.08–8.12 (m and m’, 1H). Anal. Calcd for C35H34F3N3O6: C, 64.71; H, 5.28; N, 6.47. Found: C, 64.78; H, 5.31; N, 6.36.

General procedure for hydrolysis of a tert-butyl ester:
A solution of tert-butyl ester derivative in formic acid, typically 1 g in 10 mL, is stirred at ambient temperature until no more ester is detected by thin layer chromatography (dichloromethane 95%/methanol 5%), a typical reaction time being around 3 hours. The formic acid is then evaporated under reduced pressure to yield the corresponding carboxylic acid.
The following compounds were prepared according to this procedure: 2(R,S)-(Carboxy)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(3-trifluoromethylbenzyl)amide (32):
Compound 31 (0.30 g, 0.46 mmol) was hydrolyzed to give 0.27 g (quantitative yield) of 32 as an off-white solid; 1H NMR (CDCl3) δ 4.17–5.28 (m, 9H); 6.21–6.29 (m, 1H), 6.68–6.82 (m, 1H); 7.05–7.75 (m, 13H); 9.12–9.18 (m, 1H).

2(S)-(Carboxymethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(3-trifluoromethylbenzyl)amide (68):
Compound 56 (1.72 g, 2.59 mmol) was hydrolyzed to give 1.57 g (quantitative yield) of 68 as an off-white solid; 1H NMR (CDCl3) δ 2.61 (dd, J=9.3 Hz, J=16.6 Hz, 1H); 3.09–3.14 (m, 1H); 4.10–4.13 (m, 1H); 4.30 (d, J=4.5 Hz, 1H); 4.39–4.85 (m, 6H); 6.20 (dd, J=9.6 Hz, J=15.7 Hz, 1H); 6.69 (d, J=15.8 Hz, 1H); 7.12–7.15 (m, 2H); 7.26–7.50 (m, 11H); 7.61 (s, 1H); 8.41–8.45 (m, 1H).

2(S)-(Carboxymethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[4-(2-phenylethyl)]piperazinamide (69):
Compound 57 (1.88 g, 2.78 mmol) was hydrolyzed to give 1.02 g (60%) of 69 as an off-white solid; 1H NMR (CDCl3) δ 2.63 (dd, J=6.0 Hz, J=16.5 Hz, 1H); 2.75–2.85 (m, 1H); 3.00 (dd, J=8.2 Hz, J=16.6 Hz, 1H); 3.13–3.26 (m, 4H); 3.37–3.56 (m, 4H); 3.86–4.00 (m, 1H); 4.05–4.11 (m, 1H); 4.24 (d, J=5.0 Hz, 1H); 4.46–4.66 (m, 1H); 4.65–4.70 (m, 1H); 5.10–5.15 (m, 1H); 6.14 (dd, J=9.3 Hz, J=15.9 Hz, 1H); 6.71 (d, J=15.9 Hz, 1H); 7.22–7.41 (m, 15H); 12.02 (s, 1H).

2(S)-(Carboxyethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(3-trifluoromethylbenzyl)amide (70):
Compound 58 (4.97 g, 7.34 mmol) was hydrolyzed to give 4.43 g (97%) of 70 as an off-white solid; 1H NMR (CDCl3) δ 1.92–2.03 (m, 1H); 2.37–2.51 (m, 3H); 4.13–4.19 (m, 1H); 3.32 (d, J=4.9 Hz, 1H); 4.35–4.39 (m, 1H); 4.44 (dd, J=5.9 Hz, J=14.9 Hz, 1H); 4.50–4.57 (m, 2H); 4.61–4.67 (m, 1H); 4.70–4.76 (m, 1H); 6.24 (dd, J=9.6 Hz, J=15.8 Hz, 1H); 6.70 (d, J=15.8 Hz, 1H); 7.18–7.47 (m, 14H).

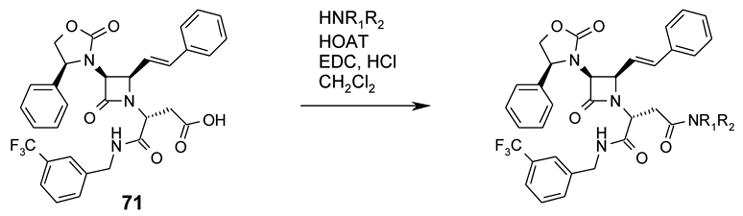
2(R)-(Carboxymethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(3-trifluoromethylbenzyl)amide (71):
Compound 59 (1.51 g, 2.27 mmol) was hydrolyzed to give 1.38 g (quantitative yield) of 71 as an off-white solid; 1H NMR (CD3OD) δ 2.93 (dd, J=5.5 Hz, J=17.0 Hz, 1H); 2.99 (dd, J=9.0 Hz, J=17.0 Hz, 1H); 4.17 (dd, J=7.0 Hz, J=9.0 Hz, 1H); 4.36 (dd, J=5.5 Hz, J=9.0 Hz, 1H); 4.40–4.45 (m, 2H) 4.55 (d, J=15.5 Hz, 1H); 4.65 (dd, J=5.0 Hz, J=9.0 Hz, 1H); 4.70 (dd, J=J’=9.0 Hz, 1H); 4.89 (dd, J=7.0 Hz, J=9.0 Hz, 1H); 6.26 (dd, J=9.0 Hz, J=16.0 Hz, 1H); 6.83 (d, J=16.0 Hz, 1H); 7.24–7.32 (m, 5H); 7.38–7.48 (m, 6H); 7.51–7.57 (m, 2H); 7.64 (s, 1H). 13C NMR (CD3OD) δ 43.82, 55.92, 62.01, 63.34, 64.52, 72.58, 123.03, 124.97 (q, 3JC-F, 3.8Hz, 1C), 125.54, (q, 3JC-F, 3.8Hz, 1C), 127.82, 128.55, 129.66, 129.81, 130.34, 130.44, 130.54, 131.77 (q, 1JC-F, 32.3Hz, CF3), 132.3–132.4 (m, 1C), 137.23, 138.42, 139.71, 141.19, 160.28, 166.12, 171.23.

2(R)-(Carboxymethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(2-fluoro-3-trifluoromethylbenzyl)carboxamide (72):
Compound 60 (0.26 g, 0.38 mmol) was hydrolyzed to give 0.238 g (quantitative yield) of 72 as an off-white solid; 1H NMR (CDCl3) δ 3.27 (d, J=7.2 Hz, 1H); 4.06 (t, J=7.2 Hz, 1H); 4.15 (t, J=8.1 Hz, 1H); 4.27 (d, J=4.8 Hz, 1H); 4.56–4.76 (m, 5H); 6.34 (dd, J=9.5 Hz, J=15.7 Hz, 1H); 6.80 (d, J=15.7 Hz, 1H); 7.06 (t, J=7.7 Hz, 1H); 7.31–7.54 (m, 12H); 8.58 (t, J=5.9 Hz, 1H).

2(R)-(Carboxymethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(S)-α-methylbenzyl]amide (73):
Compound 61 (0.215 g, 0.35 mmol) was hydrolyzed to give 0.195 g (quantitative yield) of 73 as an off-white solid; 1H NMR (CDCl3) δ 1.56 (d, J=7.0 Hz, 1H); 3.10 (dd, J=4.5 Hz, J=17.9 Hz, 1H); 3.18 (dd, J=9.8 Hz, J=17.9 Hz, 1H); 4.00 (dd, J=4.5 Hz, J=9.7 Hz, 1H); 4.14 (t, J=8.2 Hz, 1H); 4.26 (d, J=4.7 Hz, 1H); 5.02–5.09 (m, 1H); 6.41 (dd, J=9.4 Hz, J=15.8 Hz, 1H); 6.78 (d, J=15.8 Hz, 1H); 7.18 (t, J=7.3 Hz, 1H); 7.26–7.43 (m, 14H); 8.29 (d, J=8.2 Hz, 1H).

2(R)-(Carboxymethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(R)-α-methylbenzyl]amide (74):
Compound 62 (0.22 g, 0.35 mmol) was hydrolyzed to give 0.20 g (quantitative yield) of 74 as an off-white solid; 1 1.59 (d, J=7.0 Hz, 1H); 3.25 (d, J=7.0 Hz, 2H); 3.92 H NMR (CDCl3) δ (t, J=7.3 Hz, 1H); 4.15 (t, J=8.3 Hz, 1H); 4.26 (d, J=5.0 Hz, 1H); 4.52 (dd, J=4.8 Hz, J=9.3 Hz, 1H); 4.65 (t, J=8.8 Hz, 1H); 4.72 (t, J=8.3 Hz, 1H); 5.07–5.28 (m, 1H); 6.29 (dd, J=9.5 Hz, J=15.6 Hz, 1H); 6.71 (d, J=16.0 Hz, 1H); 7.20–7.43 (m, 15H); 8.31 (d, J=8.0 Hz, 1H). 13C NMR (CDCl3) δ 21.73, 34.23, 49.83, 54.37, 61.02, 62.04, 63.64, 71.10, 120.71, 126.36, 126.91, 126.96, 127.20, 128.49, 128.78, 128.89, 129.74, 135.16, 135.84, 139.43, 143.68, 158.06, 163.37, 167.38, 174.53.

2(R)-(Carboxymethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-methyl-N-(3-trifluoromethylbenzyl)amide (75):
Compound 63 (0.253 g, 0.37 mmol) was hydrolyzed to give 0.232 g (quantitative yield) of 75 as an off-white solid; 1H NMR (CDCl3) δ 3.07–3.15 (m, 4H); 4.13 (t, J=8.2 Hz, 1H); 4.30 (d, J=4.9 Hz, 1H); 4.46–4.78 (m, 5H); 5.23 (dd, J=4.6 Hz, J=9.7 Hz, 1H); 6.20 (dd, J=9.4 Hz, J=15.9 Hz, 1H); 6.73 (d, J=15.9 Hz, 1H); 7.25–7.43 (m, 14H).

2(R)-(Carboxylmethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(S)-1-(3-trifluoromethylpheny)ethyl]amide (76):
Compound 64 (38.5 mg, 0.06 mmol) was hydrolyzed to give 35 mg (quantitative yield) of 76 as an off-white solid. 1H NMR (CDCl3) δ 1.58 (d, J=7.0 Hz, 3H); 3.07–3.25 (m 2H); 4.02 (dd, J=4.8 Hz, J=9.5 Hz, 1H); 4.15 (t, J=8.2 Hz, 1H); 4.27 (d, J=4.8 Hz, 1H); 4.57 (dd, J=4.8 Hz, J=9.4 Hz, 1H); 4.61–4.77 (m, 2H); 5.07–5.29 (m, 1H); 6.41 (dd, J=9.4 Hz, J=15.8 Hz, 1H); 6.79 (d, J=15.8 Hz, 1H); 7.26–7.45 (m, 13H); 7.63 (s, 1H); 8.41 (d, J=8.1 Hz, 1H).

2(R)-(Carboxylmethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(R)-1-(3-trifluoromethylpheny)ethyl]amide (77):
Compound 65 (166 mg, 0.24 mmol) was hydrolyzed to give 152 mg (quantitative yield) of 77 as an off-white solid. 1 H NMR (CDCl3) δ 1.60 (d, J=7.0 Hz, 3H); 3.08–3.26 (m 2H); 3.93 (dd, J=4.7 Hz, J=9.5 Hz, 1H); 4.17 (t, J=8.1 Hz, 1H); 4.26 (d, J=4.7 Hz, 1H); 4.54 (dd, J=4.7 Hz, J=9.4 Hz, 1H); 4.62–4.76 (m, 2H); 5.06–5.28 (m, 1H); 6.24 (dd, J=9.4 Hz, J=15.8 Hz, 1H); 6.73 (d, J=15.8 Hz, 1H); 7.28–7.58 (m, 13H); 7.74 (s, 1H); 8.40 (d, J=7.4 Hz, 1H).

2(R)-(Carboxyethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(3-trifluoromethylbenzyl)amide (78):
Compound 66 (0.604 g, 0.89 mmol) was hydrolyzed to give 0.554 g (quantitative yield) of 78 as an off-white solid; 1H NMR (CDCl3) δ 2.27–2.34 (m, 4H); 4.10–4.30 (m, 2H); 4.30 (d, J=4.3 Hz, 1H); 4.40–4.64 (m, 4H); 4.47 (t, J=8.1 Hz, 1H); 6.22 (dd, J=9.4 Hz, J=15.7 Hz, 1H); 6.70 (d, J=15.7 Hz, 1H); 7.25–7.57 (m, 14H); 8.32–8.36 (m, 1H).

2(S)-(2-( -1-ylcarbonyl)ethyl)-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid (79):
Compound 67 (0.787 g, 1.28 mmol) was hydrolyzed to give 0.665 g (92%) of 79 as an off-white solid; 1H NMR (CDCl3) δ 1.05–1.13 (m, 1H); 1.20–1.40 (m, 5H); 1.60–1.64 (m, 1H); 1.79–1.83 (m, 2H); 2.00–2.05 (m, 2H); 2.22–2.44 (m, 3H); 2.67–2.71 (m, 1H); 2.93–3.01 (m, 4H); 3.14–3.18 (m, 1H); 3.38–3.42 (m, 1H); 3.48–3.52 (m, 1H); 3.64–3.69 (m, 1H); 4.06–4.14 (m, 2H); 4.34–4.43 (m, 2H); 4.56 (t, J=8.8 Hz, 1H); 4.73 (t, J=8.4 Hz, 1H); 6.15 (dd, J=9.1 Hz, J=16.0 Hz, 1H); 6.65 (d, J=16.0 Hz, 1H); 7.25–7.42 (m, 10H).

The compounds shown in Table 1/Figure 18, were prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 32, and 3-(trifluoromethyl)benzylamine was replaced with the appropriate amine HNR1R2; all compounds exhibited an 1H NMR spectrum consistent with the assigned structure.
Table 1.
Structures, HRMS (FAB), V1a Ki values.
| Cpd # | HNR1R2 | Calcd for: and equal to | Found: (m+H) | Ki (nM) |
|---|---|---|---|---|
| 33 | 4-[2-(1-Piperidinyl)ethyl]piperidine | C43H49F3N5O5 772.3686 | 772.3711 | 1.17 |
| 80 | 4-(1-Piperidinyl)piperidine | C41H45F3N5O5 744.3373 | 744.3399 | 3.39 |
| 81 | 4-Butylpiperazine | C39H43F3N5O5 718.3216 | 718.3194 | 6.5 |
| 82 | 1-Benzyl-4-aminopiperidine | C43H43F3N5O5 766.3216 | 766.3191 | 13.3 |
| 83 | 4-Isopropylpiperazine | C38H41F3N5O5 704.3060 | 704.3038 | 18.2 |
| 84 | 4-Cyclohexylpiperazine | C41H45F3N5O5 744.3377 | 744.3367 | 26.4 |
| 85 | 2-(1-Piperidinyl)ethylamine | C38H41F3N5O5 704.3060 | 704.3064 | 61 |
Figure 18.

Aminomalonic acid derivatives.
ASPARTIC ACID DERIVATIVES
The compounds shown in Table 2/Figure 19, were prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 68, and 3-(trifluoromethyl)benzylamine was replaced with the appropriate amine HNR1R2; all compounds exhibited an 1H NMR spectrum consistent with the assigned structure.
Table 2.
Structures, HRMS (FAB), V1a Ki values.
| Cpd # | HNR1R2 | Calcd for: and equal to | Found: (m+H) | Ki (nM) |
|---|---|---|---|---|
| 37 | 1-Benzyl-4-aminopiperidine | C44H45F3N5O5 780.3373 | 780.3356 | 1.13 |
| 42 | 4-(1-Piperidinyl)piperidine | C42H47F3N5O5 758.3529 | 758.3554 | 1.89 |
| 86 | (+)-3(S)-1-Benzyl-3-aminopyrrolidine | C43H43F3N5O5 766.3216 | 766.3201 | 3.60 |
| 87 | 4-Benzylhomopiperazine | C45H47F3N5O5 794.3524 | 794.3511 | 5.12 |
| 88 | 4-[2-(1-Piperidinyl)ethyl]piperidine | C44H51F3N5O5 786.3842 | 786.3860 | 6.00 |
| 89 | 1-Benzyl-4-aminopiperidine(*) (Zone B = 2-phenylethyl) | C44H47F3N5O5 782.3534 | 782.3535 | 23 |
| 90 | 4-(1-Pyrrolidinyl)piperazine | C41H45F3N5O5 744.3373 | 744.3355 | ~ 30 |
| 91 | 4-Cyclohexylpiperazine | C42H47F3N5O5 758.3529 | 758.3555 | ~ 30 |
| 92 | 4-Cyclopentylpiperazine | C41H45F3N5O5 744.3373 | 744.3397 | < 60 |
| 93 | 4-[2-(1-Pyrrolidinyl)ethyl]piperazine | C42H48F3N6O5 773.3638 | 773.3638 | < 60 |
A suspension of 0.05 g (0.064 mmol) of 37 and palladium (5% wt. on activated carbon, 0.024 g) in 5 mL of methanol was held under an atmosphere of hydrogen until complete disappearance of 37 as determined by thin layer chromatography (95:5 dichloromethane/methanol eluent). The reaction was filtered to remove the palladium over carbon and the filtrate was evaporated to give 0.04 g of crude 89. It was purified by column chromatography (95:5 dichloromethane/methanol eluent) to afford 0.011g (23%) of 89. HRMS (FAB) calcd for C44H47F3N5O5 782.3534, found 782.3535 (M+H)+.
Figure 19.

L-aspartic acid derivatives.
The compounds shown in Table 3/Figure 20, were prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 69, and 3-(trifluoromethyl)benzylamine was replaced with the appropriate amine HNR1R2; all compounds exhibited an 1H NMR spectrum consistent with the assigned structure.
Table 3.
Structures, HRMS (FAB), V1a Ki values.
| Cpd # | HNR1R2 | Calcd for: and equal to | Found: (m+H) | Ki (nM) |
|---|---|---|---|---|
| 94 | 3-(Trifluoromethyl)benzylamine | C44H45F3N5O5 780.3373 | 780.3344 | 6100 |
| 95 | 2-(Dimethylamino)ethylamine | C40H49F3N6O5 693.3764 | 693.3757 | > 600 |
| 96 | 3-(Dimethylamino)propylamine | C41H51N6O5 707.3921 | 707.3912 | > 600 |
Figure 20.

D-aspartic acid derivatives.
The compounds shown in Table 4/Figure 21, were prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 71, and 3-(trifluoromethyl)benzylamine was replaced with the appropriate amine HNR1R2; all compounds exhibited an 1H NMR spectrum consistent with the assigned structure.
Table 4.
Structures and HRMS (FAB), V1a Ki values.
| Cpd # | HNR1R2 | Calcd for: and equal to | Found: (m+H) | Ki (nM) |
|---|---|---|---|---|
| 41 | 4-(1-Piperidinyl)piperidine | C42H47F3N5O5 758.3529 | 758.3510 | 1.49 |
| 97 | 4-[(1-Piperidinyl)methyl]piperidine | C43H49F3N5O5 772.3686 | 772.3666 | 3.7 |
| 98 | 1-Benzyl-4-amino-piperidine | C44H45F3N5O5 780.3373 | 780.3353 | 5.58 |
| 99 | 4-[2-(1-Piperidinyl)ethyl]piperidine | C44H51F3N5O5 786.3842 | 786.3840 | 7.9 |
| 100 | 4-(1-Pyrrolidinyl)piperazine | C41H45F3N5O5 744.3373 | 744.3398 | 27.5 |
| 101 | 3-(S)-(1-Benzyl)-3-amino-pyrrolidine | C43H43F3N5O5 766.3216 | 766.3223 | > 30 |
Figure 21.
Additional D-aspartic acid derivatives
A typical example being 41: 2(R)-[[4-(Piperidin-1-yl)piperidin-1-yl]carbonylmethyl]-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(3-trifluoromethylbenzyl)amide:
Compound 41 was obtained as an off-white solid after flash silica gel column chromatography (CH2Cl2 99% and down to 90%/MeOH 1% and up to 10%, NH4OH < 1%). 1H NMR (CDCl3) δ 1.34–1.54 (m, 4H); 1.60–1.74 (m, 4H); 1.84–1.95 (m, 2H); 2.42–2.66 (m, 6H); 2.87–2.95 (m, 1H); 3.13–3.19 (m, 1H); 3.30–3.37 (m, 1H); 3.77–3.84 (m, 1H); 4.09–4.18 (m, 2H); 4.22 (dd, J=4.5 Hz, 1H); 4.43–4.56 (m, 2H); 4.58–4.66 (m, 2H); 4.70–4.74 (m, 2H); 6.25–6.32 (m, 1H); 6.80–6.85 (m, 1H); 7.25–7.53 (m, 13H); 7.64 (s, 1H); 8.62–8.69 (m, 1H). 13 C NMR (CDCl3) δ 24.02, 25.30, 27.10, 29.63, 33.72/33.75, 41.18/41.37, 43.15, 44.56/44.67, 49.90/49.98, 54.79/54.92, 61.16, 61.94/61.98, 62.50/62.62, 63.30/63.34, 71.06, 120.69, 123.05, 123.77 (q, 3JC-F, 3.5 Hz, 1C), 124.52 (q, 3JC-F, 3.8 Hz, 1C), 125.21, 126.78, 127.27, 128.77, 128.85, 128.92, 129.65, 129.74, 130.55, 130.76/130.79, 135.27, 135.68/135.72, 139.43, 139.48/139.53, 158.30, 163.18, 168.07/168.34, 169.41. HRMS (FAB) calcd for C42H47F3N5O5 758.3529, found 758.3510 (M+H)+.

2(R)-[[4-(Piperidin-1-yl)piperidin-1-yl]carbonylmethyl]-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-(2-fluoro-3-trifluoromethylbenzyl)amide (102):
Compound 102 was prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 72 and 3-(trifluoromethyl)benzylamine was replaced with 4-(1-piperidinyl)piperidine. 102 exhibited a 1H NMR spectrum consistent with the assigned structure. HRMS (FAB) calcd for C42H46F4N5O5 776.3435, found 776.3463 (M+H)+.

2(R)-[[4-(Piperidin-1-yl)piperidin-1-yl]carbonylmethyl]-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(S)-α-methylbenzyl]amide (52):
Compound 52 was prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 73 and 3-(trifluoromethyl)benzylamine was replaced with 4-(1-piperidinyl)piperidine. 52 exhibited a 1H NMR spectrum consistent with the assigned structure. HRMS (FAB) calcd for C42H50N5O5 704.3812, found 704.3806 (M+H)+.

2(R)-[[ 4-[2-(1-Piperidinyl)ethyl]piperidin -1-yl]carbonylmethyl]-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(S)-α-methylbenzyl]amide (103):
Compound 103 was prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 73 and 3-(trifluoromethyl)benzylamine was replaced with 4-[2-(1-piperidinyl)ethyl]piperidine. 103 exhibited a 1H NMR spectrum consistent with the assigned structure. HRMS (FAB) calcd for C44H54N5O5 732.4125, 732.4108 (M+H)+.

2(R)-[[4-(Piperidin-1-yl)piperidin-1-yl]carbonylmethyl]-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(R)-α-methylbenzyl]amide (50):
Compound 50 was prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 74 and 3-(trifluoromethyl)benzylamine was replaced with 4-(1-piperidinyl)piperidine. 50 was obtained as an off-white solid after flash silica gel column chromatography (CH2Cl2 99% and down to 90%/MeOH 1% and up to 10%, NH4OH < 1%). 1H NMR (CDCl3) δ 1.30–1.52 (m, 4H); 1.56–1.72 (m, 7H); 1.81–1.90 (m, 2H); 2.40–2.60 (m, 6H); 2.88–2.96 (m, 1H); 3.14–3.20 (m, 1H); 3.36–3.44 (m, 1H); 3.79–3.86 (m, 1H); 3.99–4.04 (m, 1H); 4.15 (t, J=8.3 Hz, 1H); 4.21 (dd, J=5.0 Hz, J=8.0 Hz, 1H); 4.44–4.55 (m, 1H); 4.60–4.75 (m, 3H); 5.07–5.14 (m, 1H); 6.29–6.36 (m, 1H); 6.74–6.79 (m, 1H); 7.19–7.46 (m, 15H); 8.23–8.28 (m, 1H). 13 C NMR (CDCl3) δ 21.87/21.90, 23.84, 24.90, 26.81, 33.56/33.59, 41.11/41.23, 44.52/44.57, 49.59, 49.67/49.75, 54.97, 55.16, 61.03, 61.77/61.81, 62.43/62.52, 63.37, 70.95, 121.16, 126.30, 126.79, 126.83, 127.25, 128.40, 128.70, 129.63, 129.67, 135.29/135.31, 135.80/135.83, 139.13, 143.93/143.96, 157.96, 163.29, 167.94/167.96, 168.23, 168.51. HRMS (FAB) calcd for C42H50N5O5 704.3812, found 704.3801 (M+H)+.

2(R)-[[ 4-[2-(1-Piperidinyl)ethyl]piperidin -1-yl]carbonylmethyl]-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(R)-α-methylbenzyl]amide (104):
Compound 104 was prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 74 and 3-(trifluoromethyl)benzylamine was replaced with 4-[2-(1-piperidinyl)ethyl]piperidine. 104 exhibited a 1H NMR spectrum consistent with the assigned structure. HRMS (FAB) calcd for C44H54N5O5 732.4125, 732.4127 (M+H)+.

2(R)-[[4-(Piperidin-1-yl)piperidin-1-yl]carbonylmethyl]-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-methyl-N-(3-trifluoromethylbenzyl)amide (51):
Compound 51 was prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 75 and 3-(trifluoromethyl)benzylamine was replaced with 4-(1-piperidinyl)piperidine. 51 was obtained as an off-white solid after flash silica gel column chromatography (CH2Cl2 99% and down to 90%/MeOH 1% and up to 10%, NH4OH < 1%). 51 exhibited a 1H NMR spectrum consistent with the assigned structure. HRMS (FAB) calcd for C43H49F3N5O5 772.3686, found 772.3674 (M+H)+.

2(R)-[[4-(Piperidin-1-yl)piperidin-1-yl]carbonylmethyl]-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(R)-α-methyl-3-trifluoromethylbenzyl]amide (53):
Compound 53 was prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 77 and 3-(trifluoromethyl)benzylamine was replaced with 4-(1-piperidinyl)piperidine. 53 was obtained as an off-white solid after flash silica gel column chromatography (CH2Cl2 99% and down to 90%/MeOH 1% and up to 10%, NH4OH < 1%). 53 exhibited a 1H NMR spectrum consistent with the assigned structure. HRMS (FAB) calcd for C43H49F3N5O5 772.3686, found 772.3703 (M+H)+.

2(R)-[[4-(Piperidin-1-yl)piperidin-1-yl]carbonylmethyl]-2-[3(S)-(4(S)-phenyloxazolidin-2-on-3-yl)-4(R)-(2-styryl)azetidin-2-on-1-yl]acetic acid N-[(S)-α-methyl-3-trifluoromethylbenzyl]amide (105):
Compound 105 was prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 76 and 3-(trifluoromethyl)benzylamine was replaced with 4-(1-piperidinyl)piperidine. 105 was obtained as an off-white solid after flash silica gel column chromatography (CH2Cl2 99% and down to 90%/MeOH 1% and up to 10%, NH4OH < 1%). 105 exhibited a 1H NMR spectrum consistent with the assigned structure. HRMS (FAB) calcd for C43H49F3N5O5 772.3686, found 772.3666 (M+H)+.

GLUTAMIC ACID DERIVATIVES
The compounds shown in Table 6/Figure 22, were prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 70, and 3-(trifluoromethyl)benzylamine was replaced with the appropriate amine HNR1R2; all compounds exhibited an 1H NMR spectrum consistent with the assigned structure.
Table 6.
[structures, HRMS (FAB), V1a IC50 and Ki values]
| Cpd # | HNR1R2 | Calcd for: and equal to | Found: (m+H) | Ki (nM) |
|---|---|---|---|---|
| 48 | 4-Cyclohexylpiperazine | C43H49F3N5O5 772.3686 | 772.3672 | 0.27 |
| 106 | 4-(Cyclohexylmethyl)piperazine | C44H51F3N5O5 786.3842 | 786.3862 | 0.46 |
| 46 | 4-(2-Phenylethyl)piperazine | C45H47F3N5O5 794.3529 | 794.3502 | 1.17 |
| 47 | 4-(1-Piperidinyl)piperidine | C43H49F3N5O5 772.3686 | 772.3685 | 1.45 |
| 107 | 4-Propylpiperazine | C40H45F3N5O5 732.3373 | 732.3371 | 1.19 |
| 108 | 4-[(1-Piperidinyl)methyl]piperidine | C44H51F3N5O5 786.3842 | 786.3821 | 1.95 |
| 109 | 2-(1-Piperidinyl)ethylamine | C40H45F3N5O5 732.3373 | 732.3374 | 9 |
| 110 | 4-[2-(1-Piperidinyl)ethyl]piperidine | C45H53F3N5O5 800.3999 | 800.4019 | 9.9 |
Figure 22.

L-glutamic acid derivatives.
The compounds shown in Table 7/Figure 23, were prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 79, and 3-(trifluoromethyl)benzylamine was replaced with the appropriate amine HNR1R2; all compounds exhibited an 1H NMR spectrum consistent with the assigned structure.
Table 7.
Structures, HRMS (FAB), V1a Ki values.
| Cpd # | HNR1R2 | Calcd for: and equal to | Found: (m+H) | Ki (nM) |
|---|---|---|---|---|
| 111 | 2-Fluoro-3-(trifluoromethyl)benzylamine | C43H48F4N5O5 790.3592 | 790.3572 | 0.20 |
| 112 | 2,3-Dichlorobenzylamine | C42H48Cl2N5O5 772.3033 | 772.3011 | 0.38 |
| 113 | 2-Fluoro-5-(trifluoromethyl)benzylamine | C43H48F4N5O5 790.3592 | 790.3568 | 0.41 |
| 114 | 3-Fluoro-5-(trifluoromethyl)benzylamine | C43H48F4N5O5 790.3592 | 790.3577 | 0.51 |
| 115 | 3-Chlorobenzylamine | C42H49ClN5O5 738.3422 | 738.3441 | 0.66 |
| 116 | 3,5-Difluorobenzylamine | C42H48F2N5O5 740.3624 | 740.3624 | 0.82 |
| 117 | 2-(Trifluoromethyl)benzylamine | C43H49F3N5O5 772.3686 | 772.3692 | 0.93 |
| 118 | 3,5-Bis-(trifluoromethyl)benzylamine | C44H48F6N5O5 840.3560 | 840.3583 | 4.45 |
| 119 | 4-Fluoro-3-(trifluoromethyl)benzylamine | C43H48F4N5O5 790.3592 | 790.3588 | < 30 |
Figure 23.

Additional L-glutamic acid derivatives.
The compounds shown in Table 8/Figure 24, were prepared using the “General procedure for amide formation from a carboxylic acid”, except that N-benzyloxycarbonyl-D-aspartic acid β-t-butyl ester monohydrate was replaced with 78, and 3-(trifluoromethyl)benzylamine was replaced with the appropriate amine HNR1R2; all compounds exhibited an 1H NMR spectrum consistent with the assigned structure.
Table 8.
Structures, HRMS (FAB), V1a Ki values.
| Cpd # | HNR1R2 | Calcd for: and equal to | Found: (m+H) | Ki (nM) |
|---|---|---|---|---|
| 49 | 4-Cyclohexylpiperazine | C43H49F3N5O5 772.3686 | 772.3707 | 5.84 |
| 120 | 4-Butylpiperazine | C41H47F3N5O5 746.3529 | 746.3503 | 10.5 |
| 121 | 4-Isopropylpiperazine | C40H45F3N5O5 732.3373 | 732.3347 | 13 |
| 122 | 4-(2-Phenylethyl)piperazine | C45H47F3N5O5 794.3529 | 794.3552 | 33 |
| 123 | 4-(Cyclohexylmethyl)- piperazine | C44H51F3N5O5 786.3842 | 786.3823 | > 12 |
| 124 | 4-(1-Piperidinyl)piperidine | C43H49F3N5O5 772.3686 | 772.3688 | ~ 30 |
Figure 24.

D-glutamic acid derivatives
Biological evaluation:
V1a Receptor Binding Assay (Azevan Pharmaceuticals Inc.):
A cell line expressing the human V1a receptor in CHO cells (henceforth referred to as the hV1a cell line) was obtained from Dr. Michael Brownstein, NIMH, Bethesda, MD, USA. The hV1a cell culture and cell-based hV1a receptor binding assay were performed according to the methods described by Thibonnier et al.26 with modifications. The hV1a cell line was grown in alpha-MEM with 10% fetal bovine serum and 250ug/ml G418 (Gibco, Grand Island, NY, USA). For competitive binding assay, hV1a cells were plated into 12-well culture plate at 1:16 dilution from a confluency flask, and maintained in culture for at least two days. Culture medium was then removed, cells were washed with 2ml binding buffer (25mM HEPES, 0.25% BSA, 1x DMEM, PH = 7.0). To each well, 990ul binding buffer containing 1nM 3H-arginine-vasopressin was added, and followed by 10ul series diluted test compounds dissolved in DMSO. All incubations were in triplicate, and dose-inhibition curves consisted of total binding (DMSO) and 5 concentrations (usually 0.1, 1.0, 10, 100, and 1000nm) of test agent encompassing the IC50. 100nM unlabeled arginine-vasopressin (Sigma) was used to assess non-specific binding. Cells were incubated for 30 minutes at 37oC, assay mixture was removed and each well was washed three times with PBS (PH = 7.4). 1ml 2% SDS was added per well and plates were let sit for 30 minutes. The whole content in a well was transferred to a scintillation vial. Each well was rinsed with 0.5ml PBS, which was then added to the corresponding vial. Scintillation fluid (Ecoscint, National Diagnostics, Atlanta, Georgia) was then added at 3ml per vial. Samples were counted in a liquid scintillation counter (Beckman LS6500). IC50 values were calculated by Prism Curve fitting software. The conversion factor for computing Ki from IC50 was 0.61.
V1a Receptor Binding Assay (Eli Lilly & Co):
The V1a binding assay employed at Eli Lilly used the same cell line expressing the human V1a receptor in CHO cells, but the assay differed inasmuch as binding was carried out on membranes using a filtration method22. IC50 values measured by the two methods usually differed by 2-fold or less.
Measurement of plasma and brain levels in dogs after oral dosing:
Samples were collected from a single dog dosed orally with compound at 10 mg/kg in 10% DMSO/90% PEG300. Blood and brain samples (cortex, brain stem, cerebellum, hypothalamus and hippocampus regions) were harvested and frozen in liquid nitrogen and stored at −70 °C prior to analysis by LC/MS mass spectrometry. Samples were thawed on ice, homogenized in pH 9.4 buffer on ice extracted with ethyl acetate after addition of a reference compound to measure extraction efficiency. Extracts were removed and taken to dryness, reconstituted in acetonitrile and injected on a Finnigan ESI LC/MS/MS system. Spiked control brain samples were prepared in similar fashion for use as quantitation standards.
Measurement of plasma and brain levels in rats after oral dosing:
Initial experiments showed that in most cases our compounds could be dissolved at 20 mg/ml in 23% hydroxypropyl beta-cyclodextrin with 0.68% lactic acid. Sprague-Dawley rats (Charles River Laboratory) were dosed by gavage with a combination of three compounds (20 mg/kg each): 22 + 41 + 48 or 42 + 46 + 47. Blood (approximately 3 mL) was collected from 3 animals per group per time point at 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours postdose. Blood was collected via exsanguination (cardiac puncture) under carbon dioxide anesthesia into tubes containing sodium heparin anticoagulant and plasma was separated by centrifugation within 1 h. Brains were collected at the same time points. Plasma and brain levels were determined by LC/MS/MS.
Figure 16.

Structures and human V1a binding affinities of 50, 51 and 53
Table 5.
HRMS (FAB), V1a Ki values.
| Cpd # | Calcd for: and equal to | Found: (m+H) | Ki (nM) |
|---|---|---|---|
| 50 | C42H50N5O5 704.3812 | 704.3801 | 0.30 |
| 51 | C43H49F3N5O5 772.3686 | 772.3674 | 0.66 |
| 102 | C42H46F4N5O5 776.3435 | 776.3463 | 1.13 |
| 53 | C43H49F3N5O5 772.3686 | 772.3703 | 1.27 |
| 104 | C44H54N5O5 732.4125 | 732.4127 | 8.7 |
| 105 | C43H49F3N5O5 772.3686 | 772.3666 | > 6 |
| 52 | C42H50N5O5 704.3812 | 704.3806 | > 30 |
| 103 | C44H54N5O5 732.4125 | 732.4108- | 179 |
Acknowledgments
We thank Norman Mason and Anne Van Abbema for carrying out vasopressin binding experiments at Lilly, and Elizabeth Lutz and Harlan Shannon for help with the cyclodextrin/lactic acid formulation. This work was supported by NIH grants HD37290 to ngs and MH063663 to gak.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jard S, Elands J, Schmidt A, Barberis C. In: Progress in Endocrinology. Imura H, Shizume K, editors. Elsevier: Amsterdam; 1988. pp. 1183–1189. [Google Scholar]
- 2.Birnbaumer M, Seibold A, Gilbert S, Ishido M, Barberis C, Antaramian A, Brabet P, Rosenthal W. Nature. 1992;357:333. doi: 10.1038/357333a0. [DOI] [PubMed] [Google Scholar]
- 3.Kimura T, Tanizawa O, Mori K, Brownstein MJ, Okayama H. Nature. 1992;356:526. doi: 10.1038/356526a0. [DOI] [PubMed] [Google Scholar]
- 4.Morel A, O'Carroll AM, Brownstein MJ, Lolait SJ. Nature. 1992;356:523. doi: 10.1038/356523a0. [DOI] [PubMed] [Google Scholar]
- 5.Sugimoto T, Saito M, Mochizuki S, Watanabe Y, Hashimoto S, Kawashima H. J Biol Chem. 1994;269:27088. [PubMed] [Google Scholar]
- 6.Mahlmann S, Meyerhof W, Hausmann H, Heierhorst J, Schönrock C, Zwiers H, Lederis K, Richter D. Proc Natl Acad Sci U S A. 1994;91:1342. doi: 10.1073/pnas.91.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Kesteren RE, Tensen CP, Smit AB, Van Minnen J, Van Soest PF, Kits KS, Meyerhof W, Richter D, Van Heerikhuizen H, Vreugdenhil E, Geraerts WPM. Neuron. 1995;15:897. doi: 10.1016/0896-6273(95)90180-9. [DOI] [PubMed] [Google Scholar]
- 8.Ring RH. Curr Pharm Des. 2005;11:205. doi: 10.2174/1381612053382241. [DOI] [PubMed] [Google Scholar]
- 9.Landgraf R. CNS Neurol Disord Drug Targets. 2006;5:167. doi: 10.2174/187152706776359664. [DOI] [PubMed] [Google Scholar]
- 10.Tahara A, Tsukada J, Tomura Y, Kusayama T, Wada K, Ishii N, Taniguchi N, Suzuki T, Yatsu T, Uchida W, Shibasaki M. Pharmacol Res. 2005;51:275. doi: 10.1016/j.phrs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Xiang MA, Chen RH, Demarest KT, Gunnet J, Look R, Hageman W, Murray WV, Combs DW, Patel M. Bioorg & Med Chem Let. 2004;14:2987. doi: 10.1016/j.bmcl.2004.02.103. [DOI] [PubMed] [Google Scholar]
- 12.Derdowska I, Prahl A, Kowalczyk W, Janecki M, Melhem S, Trzeciak HI, Lammek B. Eur J Med Chem. 2005;40:63. doi: 10.1016/j.ejmech.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 13.Serradeil-Le Gal C, Derick S, Brossard G, Manning M, Simiand J, Gaillard R, Griebel R, Guillon G. Stress. 2003;6:199. doi: 10.1080/1025389032000114524. [DOI] [PubMed] [Google Scholar]
- 14.Shimada Y, Taniguchi N, Matsuhisa A, Akane H, Kawano N, Suzuki T, Tobe T, Kakefuda A, Yatsu T, Tahara A, Tomura Y, Kusayama T, Wada K, Tsukada J, Orita M, Tsunoda T, Tanaka A. Bioorg & Med Chem Let. 2006;14:1827. doi: 10.1016/j.bmc.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 15.Galanski ME, Erker T, Studenik CR, Kamyar M, Rawnduzi P, Pabstova M, Lemmens-Gruber R. Eur J Pharm Sci. 2005;24:421. doi: 10.1016/j.ejps.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 16.This argument is very similar to the concept underlying “privileged fragments”. Evans BE, Rittle KE, Bock MFG, Dipardo RM, Friedinger RM, Whiter WL, Lundell GF, Veber DF, Anderson PS, Chang RSL, Lotti VJ, Cerino DJ, Chen TB, Kling PJ, Kunkel KA, Springer JP, Hirshfield J. J Med Chem. 1988;31:2235. doi: 10.1021/jm00120a002.
- 17.A second potential approach is implicit in the weak (suboptimal) binding of beta to A in Figure 1. By screening weak binders to a related receptor (A), one could find strong binders to the target receptor (B). Pairwise comparisons of results for NK-2, NPY1, BK2, and V1a screening of the Neuropeptide Cassette confirm this hypothesis: prescreening for weak activity at any one of the four targets resulted in 4- to 7-fold enrichments in hit rates for the other three targets (RFB, unpublished results). LY307174 itself had weak but detectable affinity (IC50 ~40 μM) at the hamster NK-2 receptor.
- 18.De B, Plattner JJ, Bush EN, Jae HS, Diaz G, Johnson ES, Perun TJ. J Med Chem. 1989;32:2036. doi: 10.1021/jm00129a003. [DOI] [PubMed] [Google Scholar]
- 19.Evans DA, Sjogren EB. Tet Let. 1985;26:3783. [Google Scholar]
- 20.Evans DA, Sjogren EB. Tet Let. 1985;26:3787. [Google Scholar]
- 21.X-ray resolution and ORTEP of 7 provided as a pdf file.
- 22.Bruns RF, Cooper RDG, Dressman BA, Hunden DC, Kaldor SW, Koppel GA, Rizzo JR, Skelton JJ, Steinberg MI. U.S. Patent 6,204,260. 2001. [Google Scholar]
- 23.Hipskind PA, Howbert JJ, Bruns RF, Foreman MM, Gehlert DR, Iyengar S, Phebus LA, Regoli D, Cho SSY, Crowell TA, Johnson KW, Krushinski JH, Li DL, Lobb KL, Mason NR, Muehl BS, Nixon JA, Simmons RM, Threlkeld PG, Waters DC, Gitter BD. J Med Chem. 1996;39:736. doi: 10.1021/jm950616c. [DOI] [PubMed] [Google Scholar]
- 24.The other four compounds tested for plasma and brain levels were 42, 46, 47, and 48.
- 25.Evans DA, Sjobren EB. U.S. Patent 4,665,171. 1987. [Google Scholar]
- 26.Thibonnier M, Auzan C, Madhun Z, Wilkins P, Berti-Mattera L, Clauser E. J Biol Chem. 1994;269:3304. [PubMed] [Google Scholar]