

Abstract
Experimental data for the unfolding of cytochrome c and azurin by guanidinium chloride (GuHCl) are used to construct free-energy diagrams for the folding of the oxidized and reduced proteins. With cytochrome c, the driving force for folding the reduced protein is larger than that for the oxidized form. Both the oxidized and the reduced folded forms of yeast cytochrome c are less stable than the corresponding states of the horse protein. Due to the covalent attachment of the heme and its fixed tetragonal coordination geometry, cytochrome c folding can be described by a two-state model. A thermodynamic cycle leads to an expression for the difference in self-exchange reorganization energies for the folded and unfolded proteins. The reorganization energy for electron exchange in the folded protein is approximately 0.5 eV smaller than that for a heme in aqueous solution. The finding that reduced azurin unfolds at lower GuHCl concentrations than the oxidized protein suggests that the coordination structure of copper is different in oxidized and reduced unfolded states: it is likely that the geometry of CuI in the unfolded protein is linear or trigonal, whereas CuII prefers to be tetragonal. The evidence indicates that protein folding lowers the azurin reorganization energy by roughly 1.7 eV relative to an aqueous Cu(1, 10-phenanthroline)22+/+ reference system.
Keywords: protein folding, rack formation, cytochrome c, azurin, electron transfer
The folding of a protein to its native three-dimensional structure is a spontaneous process, driven by the tendency of the peptide chain to assume the conformation of minimum free energy. As first clearly enunciated by Lumry and Eyring in 1954 (1, 2), the universal minimum for a given protein (i.e., for a specific amino acid sequence) may be reached at the expense of some local energy maximum. They further suggested that evolution has availed itself of this so-called rack phenomenon to create strain and distortion in prosthetic groups or coenzymes, thereby tuning the electronic properties by the mechanical force. This idea also led to a visualization of evolutionary fine tuning of active-site properties in protein superfamilies by small variations in amino acid sequences.
The idea of conformationally induced strain in protein active sites was further developed both by Lumry himself (3) and by other authors. Vallee and Williams (4) stressed, in particular, how strain in the active site of the ground state of a catalytic metalloenzyme (e.g., a blue copper protein) can poise the metal ion for its reaction with substrate. The unique properties of blue copper were first described in 1960 (5), and they were attributed to a rack mechanism by one of us in 1964 (6). The first attempt to estimate the rack energy for blue copper, based on ligand-field considerations, was published in 1983 (7), and recently, Brill (8) has developed a model to calculate the mechanical energy associated with stress and strain and applied it to one specific blue protein, azurin. Interestingly, electronic structure calculations (9, 10) and spectroscopic experiments (9) have suggested that there is little if any strain on CuII in a blue copper site, but that the bonding of methionine sulfur to CuI is weakened by forced elongation in a folded cupredoxin. These findings raise the possibility that the main role of the rack is to shield the copper from water and other potential ligands (10). Two detailed reviews (11, 12), featuring rather disparate accounts of developments in the field, have been published in the last few years.
In this communication we will show how the properties of redox metalloproteins are related to the energetics of protein folding. We will discuss cytochrome c and azurin as examples, because the crystal structures of the wild-type (13, 14) as well as of several mutant proteins (15, 16) are available; there is a wealth of spectroscopic, thermodynamic, and kinetic data to draw upon (11, 15); and the folding of these proteins is being studied experimentally in our laboratories (17–19).
Rack Formation by Folding
In Fig. 1 we give a thermodynamic cycle for the folding of a redox protein (17). If the reduction potentials of the folded and unfolded protein are different, then the folding free energies of the oxidized and reduced proteins will differ by a corresponding amount. In a high-potential metalloprotein, the redox center in the folded state generally has a higher potential than in the unfolded protein, so that the driving force for folding is higher for the reduced protein. This can be due to the native fold destabilizing the oxidized metal or stabilizing the reduced center, or a combination of both. This is the essence of the rack concept (1, 2). Blue copper proteins provide good examples, in which both effects are operating (11).
Figure 1.

(Upper) Thermodynamic cycle and free-energy diagram for the oxidized (OX) and reduced (RED) states of unfolded (U) and folded (F) forms of a redox-active protein. (Lower) GuHCl denaturation curves and unfolding isotherms for oxidized (FeIII) and reduced (FeII) horse heart cytochrome c (22.5°C).
If the difference in folding free energies for the oxidized and reduced protein (Δ(ΔGf) ≡ ΔGf,OX − ΔGf,RED) is sufficiently large, it may be possible to find conditions in which the oxidized protein is completely unfolded, whereas the reduced one is fully folded. This is the basis for electron-transfer-initiated folding (17, 18), which has brought folding studies into a much shorter time regime (≥ nanoseconds) compared with that conventionally used (millisecond in stopped-flow dilution experiments). Recent theoretical work (20, 21) suggests that this is a necessity for studies of the initial collapse to a compact denatured state.
Cytochrome c
Cytochrome c (Cyt c) is a small (12.5 kDa) protein in which the heme is covalently bound to the peptide through thioether linkages with Cys14 and Cys17 residues (22). The imidazole Nɛ of His18 is bound at one axial heme site and is believed to remain coordinated to the iron atom except in strongly acidic solutions (pH ≤ 2.5). The sixth Fe ligand is the thioether side chain of Met80; this ligand is not as robust as His18, and is known to dissociate under mild denaturing conditions. The reduction potential of the heme in the folded protein [EF = 260 mV vs. normal hydrogen electrode (NHE)] (22) is much higher than that of an exposed heme in aqueous solution (EU ∼ −100 mV). This dramatic increase in potential upon protein folding indicates that reduced cytochrome c (cyt cII) is more stable toward unfolding than the oxidized protein (cyt cIII).
The folding energetics of two cytochromes c: horse (h-cyt c) and yeast (Saccharomyces cerevisiae, isoenzyme-1, Cys102Ser mutant; y-cyt c) have been investigated. Although the primary amino acid sequences of h-cyt c and y-cyt c are just 60% homologous (22), crystal-structure analyses show that the polypeptide backbones of the two proteins have quite similar folding patterns (23, 24). Unfolding either protein by addition of guanidinium chloride produces an increase in fluorescence, owing to the greater separation between Trp59 and the heme. The guanidinium chloride (GuHCl)-induced unfolding of oxidized and reduced h-cyt c at 22.5°C (pH 7) can be described by a two-state model, where ΔGf is a linear function of [GuHCl] (17) (Fig. 1). The value of Δ(ΔGf) obtained by extrapolation to [GuHCl] = 0 is in good agreement with the difference in reduction potentials of free heme and the folded protein (Δ(ΔGf) ∼ 0.35 eV ∼ −ΔEf). The additional finding that Δ(ΔGf) values are roughly the same for h-cyt c and y-cyt c confirms that the hydrophobic solvation of the heme is primarily responsible for the high potential of the protein (18).
The results for cytochrome c underscore the advantage of having a covalently attached heme group as the redox-active cofactor. Not only does the cofactor stay in the same position on the protein chain in the unfolded state, but the fixed tetragonal coordination geometry of the heme places limits on the number of variations that can be made in the active-site structure. As far as the unfolded reference point is concerned, these cofactor variations are restricted to the axial ligation of the tetragonal heme unit. And, in cytochrome c, since the axial histidine (His18) is firmly attached to the iron, only the sixth coordination site is available for ligand substitution. In aqueous GuHCl solution of Cyt c, only modest changes in axial ligation are possible, so the heme reduction potential in the unfolded protein should fall in a narrow range (±50 mV) around −100 mV vs. NHE. We conclude, then, that one of the key conditions for the operation of a two-state model (Fig. 1) is met by Cyt c.
If the redox-active cofactor is not in a prepackaged geometrical arrangement in the unfolded protein, then the number of possible metal-ligand structures is increased dramatically. As long as the metal ion is in the rack of the folded protein, it has no choice of ligands or geometrical arrangement. However, in the unfolded state, when it is released from the rack, it is free to migrate to the best binding site it can find, and it is likely that this site will differ for oxidized and reduced metal ions.
Azurin
Consider now the case of a blue copper protein, Pseudomonas aeruginosa azurin. Unlike Cyt c, all the ligands in the azurin binding site are supplied by the polypeptide chain; thus, when the fixed rack coordination is disrupted by unfolding, the metal ion is free to occupy the ligand site it prefers, which is expected to differ for the oxidized (CuII) and reduced (CuI) proteins. In the scheme shown in Fig. 2, which is based on many investigations of copper coordination chemistry (25, 26), the unfolded CuII protein prefers to occupy a tetragonal binding site, whereas CuI favors linear coordination. The predictions are clear. What do the experiments say?
Figure 2.

(Upper) Thermodynamic cycles and free-energy diagram for the oxidized (OX) and reduced (RED) states of unfolded (U) and folded (F) forms of azurin. The unfolded copper sites can have different conformations (U, U′) with different relative stabilities in the oxidized and reduced states. (Lower) GuHCl denaturation curves and unfolding isotherms for oxidized (CuII) and reduced (CuI) Pseudomonas aeruginosa azurin (22°C).
When the unfolding of oxidized azurin by GuHCl is followed by changes in circular dichroism at 220 nm, there is an initial rapid change due to unfolding, followed by a slow change, which involves a redox reaction between the thiol ligand and the CuII ion (19). By subtracting the contribution from the slow process, it is possible to extract the unfolding curve (Fig. 2). An analysis of this curve yields a folding driving force of 0.54 eV. When the same experiment is done with the reduced protein under anaerobic conditions, there are no complications from redox reactions. The folding driving force extracted from the data is 0.41 eV (Fig. 2). Unlike the situation with cytochrome c, it is thus apparent that the driving force for folding the oxidized protein is higher than that for the reduced one. In terms of the thermodynamic cycle of Fig. 2, this means that the average reduction potential in the unfolded protein is ≈0.13 eV higher than the potential of native azurin. This is readily interpreted in terms of a model in which CuII prefers an unfolded conformation with tetragonal coordination, in which state the reduction potential is negative, whereas CuI favors linear or trigonal coordination. Such a CuI complex would have a high reduction potential, and consequently, the average reduction potential of the copper ions in the unfolded form would be higher than the reduction potential in the folded state (see Fig. 2). Electrochemical measurements on unfolded CuI azurin have shown that the average potential is ≈0.13 eV higher than in the the folded state, in excellent agreement with the prediction of the cycle (M. G. Hill, A. J. Di Bilio, and H.B.G., unpublished results).
A free-energy diagram that includes four unfolded states of azurin is shown in Fig. 2: the PU′RED state is favored in the case of the reduced protein, whereas the PUOX state is somewhat more stable for the oxidized form. The four-state model for the unfolded protein is analogous to the square scheme used to explain the redox reactions of copper model complexes (27). Thus, the only difference in the energy diagram compared with that for cytochrome c (Fig. 1) is that there are two high-lying states (URED, UOX′) that, because of their high energy, are not populated to any significant degree.
Functional Consequences
Protein folding can play a key role in active-site reactivity by adjusting the relative energies of the ground and transition states along a given reaction pathway. Consider, for example, the transfer of an electron between the active site of two molecules of the same protein; in this electron-exchange reaction (FOX + FRED ⇌ FRED + FOX), FOX and FRED must have identical configurations in the transition state (28). At this fixed point in nuclear configuration space, total energy is conserved when an electron transfers from donor to acceptor. This requirement for the transition-state configuration holds for exchange reactions of both folded and unfolded proteins. In addition to the folding free energies of the oxidized and reduced proteins (ΔGf,OX, ΔGf,RED), we can define a free-energy change associated with conversion of the electron-exchange-transition-state configuration of the unfolded protein ([UOX URED]‡) to the transition-state configuration of the folded protein ([FOX FRED]‡). This transition-state folding free-energy change (ΔGf,‡) differs from those for the folding around the equilibrium configuration in the sense that ΔGf,‡ must be the same for both the oxidized and reduced proteins.
A thermodynamic cycle connecting the folded and unfolded proteins and their electron-exchange transition states is shown in Fig. 3. This cycle leads to an expression for the difference in self-exchange reorganization energies for the folded and unfolded proteins (Δλf ≡ λF − λU) in terms of ΔGf,‡ and the mean of the folding free-energy changes for the oxidized and reduced proteins (〈ΔGf〉):
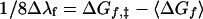 |
1 |
The hydrophobic interior of cytochrome c has been estimated to have a low dielectric constant (ɛ ∼ 2 − 3) (29, 30), which gives rise to both the high potential of the heme and a reduction in the outer-sphere (solvent) barrier to electron exchange (31). In this protein, folding stabilizes the electron-exchange transition-state configuration by lowering the polarizability (compared with water) of the medium surrounding the heme. The reorganization barrier to electron exchange in cytochrome c [λF = 0.7 eV (32, 33)] is 0.5 eV smaller than that for the Cyt c-heme octapeptide in aqueous solution (λU ∼ 1.2 eV).‖ Since the average folding free energy (〈ΔGf〉) for horse heart ferricytochrome c and ferrocytochrome c is −0.59 eV (18), it follows (Eq. 1) that the driving force for folding the protein around the transition state is −ΔGf,‡ ∼ 0.65 eV.
Figure 3.

Free-energy diagram for the oxidized (OX) and reduced (RED) states of unfolded (U) and folded (F) forms of a redox-active protein, and the corresponding transition states for electron exchange ([UOX|URED]‡, [FOX|FRED]‡).
The situation is different in the case of azurin, for which the self-exchange reorganization energy has been estimated to be similar to that of cytochrome c (λF ∼ 0.70 eV) (A. J. Di Bilio, M. G. Hill, L. K. Skov, N. Bonander, B. G. Karlsson, J.R.W., B.G.M., and H.B.G., unpublished results). Self-exchange reactions of CuII/I model complexes tend to have large inner-sphere reorganization barriers, owing to the different geometric preferences of the two oxidation states (26). The rate constant for aqueous Cu(phen)22+/+ electron exchange [50 M−1s−1 (35)] is much smaller than that for Ru(bpy)33+/2+ [2 × 109 M−1s−1 (28)]. The relative rates of these two exchange reactions imply that the reorganization energy for Cu(phen)22+/+ exchange is 1.8 eV greater than that for Ru(bpy)33+/2+ in aqueous solution. The reorganization energy for Ru(bpy)33+/2+ exchange [0.6 eV (28)] arises primarily from solvent reorientation; the 1.8-eV increase for Cu(phen)22+/+ reflects the inner-sphere contributions and suggests a value of λU ∼ 2.4 eV for blue copper proteins. For azurin, then, Δλf ∼ −1.70 eV, 〈ΔGf〉 = −0.47 eV, and the driving force for protein folding around the electron-exchange transition-state configuration is −ΔGf,‡ ∼ 0.68 eV. Virtually all of this stabilization arises from the elimination of the large inner-sphere barriers for CuII/I electron exchange [a spectroscopic estimate of the inner-sphere reorganization associated with thiolate-to-CuII charge transfer in azurin is 0.2 eV (36)]. Clearly, the folded polypeptide around the copper site greatly facilitates electron flow through the protein.
Acknowledgments
We thank N. Sutin and E. I. Solomon for helpful comments. This work was supported by the National Science Foundation (MCB9630465), National Institutes of Health (DK19038), and Natural Science Research Council (Sweden).
ABBREVIATIONS
- phen
1,10-phenanthroline
- bpy
2,2′-bipyridine
- GuHCl
guanidinium chloride
- NHE
normal hydrogen electrode
- Cyt c
cytochrome c
Footnotes
References
- 1.Lumry R, Eyring H. J Phys Chem. 1954;58:110–120. [Google Scholar]
- 2.Eyring H, Lumry R, Spikes J D. In: The Mechanism of Enzyme Action. McElroy W D, Glass B, editors. Baltimore: The Johns Hopkins Press; 1954. pp. 123–136. [Google Scholar]
- 3.Lumry R, Biltonen R. In: Structure and Stability of Biological Macromolecules. Timasheff S N, Fasman G D, editors. New York: Marcel Dekker; 1969. pp. 65–212. [Google Scholar]
- 4.Vallee B L, Williams R J P. Proc Natl Acad Sci USA. 1968;59:498–505. doi: 10.1073/pnas.59.2.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malmström B G, Vänngård T. J Mol Biol. 1960;2:118–124. doi: 10.1016/s0022-2836(62)80074-6. [DOI] [PubMed] [Google Scholar]
- 6.Malmström B G. In: Oxidases and Related Redox Systems. King T E, Mason H S, Morrison M, editors. New York: Wiley; 1964. pp. 207–216. [Google Scholar]
- 7.Gray H B, Malmström B G. Comments Inorg Chem. 1983;2:202–209. [Google Scholar]
- 8.Brill A S. Mol Phys. 1995;85:727–734. [Google Scholar]
- 9.Solomon E I, Penfield K W, Gewirth A A, Lowery M D, Shadle S E, Guckert J A, LaCroix L B. Inorg Chim Acta. 1996;243:67–78. [Google Scholar]
- 10.Ryde U, Olsson M H M, Peierloot K, Roos B O. J Mol Biol. 1996;261:586–596. doi: 10.1006/jmbi.1996.0484. [DOI] [PubMed] [Google Scholar]
- 11.Malmström B G. Eur J Biochem. 1994;223:711–718. doi: 10.1111/j.1432-1033.1994.tb19044.x. [DOI] [PubMed] [Google Scholar]
- 12.Williams R J P. Eur J Biochem. 1995;234:363–381. doi: 10.1111/j.1432-1033.1995.363_b.x. [DOI] [PubMed] [Google Scholar]
- 13.Bushnell A M, Brayer G D. J Mol Biol. 1992;214:585–595. doi: 10.1016/0022-2836(90)90200-6. [DOI] [PubMed] [Google Scholar]
- 14.Nar H, Messerschmidt A, Huber R, van de Kamp M, Canters G W. J Mol Biol. 1991;218:427–447. doi: 10.1016/0022-2836(91)90723-j. [DOI] [PubMed] [Google Scholar]
- 15.Wuttke D S, Gray H B. Curr Opin Struct Biol. 1993;3:555–563. [Google Scholar]
- 16.Rafferty S P, Guillemette J G, Berghuis A M, Smith M, Brayer G D, Mauk A G. Biochemistry. 1996;35:10784–10792. doi: 10.1021/bi960430v. [DOI] [PubMed] [Google Scholar]
- 17.Pascher T, Chesick J P, Winkler J R, Gray H B. Science. 1996;271:1558–1560. doi: 10.1126/science.271.5255.1558. [DOI] [PubMed] [Google Scholar]
- 18.Mines G A, Pascher T, Lee S C, Winkler J R, Gray H B. Chem Biol. 1996;3:491–497. doi: 10.1016/s1074-5521(96)90097-6. [DOI] [PubMed] [Google Scholar]
- 19.Leckner, J., Wittung, P., Bonander, N., Karlsson, B. G. & Malmström, B. G. (1997) J. Biol. Inorg. Chem., in press.
- 20.Bryngelson J D, Onuchic J N, Wolynes P G. Proteins Struct Funct Genet. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 21.Onuchic J N, Wolynes P G, Luthey-Schultzen Z, Socci N D. Proc Natl Acad Sci USA. 1995;92:3626–3630. doi: 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore G R, Pettigrew G W. Cytochromes c: Evolutionary, Structural, and Physicochemical Aspects. New York: Springer; 1990. [Google Scholar]
- 23.Bushnell G W, Louie G V, Brayer G D. J Mol Biol. 1990;214:585–595. doi: 10.1016/0022-2836(90)90200-6. [DOI] [PubMed] [Google Scholar]
- 24.Berghuis A M, Brayer G D. J Mol Biol. 1992;223:959–976. doi: 10.1016/0022-2836(92)90255-i. [DOI] [PubMed] [Google Scholar]
- 25.Kitajima N, Fujisawa K, Moro-oka Y. J Am Chem Soc. 1990;112:3210–3212. [Google Scholar]
- 26.Jameson G B, Ibers J A. In: Bioinorganic Chemistry. Bertini I, Gray H B, Lippard S J, Valentine J S, editors. Mill Valley, CA: University Science Books; 1994. pp. 167–252. [Google Scholar]
- 27.Bernardo M M, Robandt P V, Schroeder R R, Rorabacher D B. J Am Chem Soc. 1989;111:1224–1231. [Google Scholar]
- 28.Marcus R A, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 29.Simonson T, Perahia D. J Am Chem Soc. 1995;117:7987–8000. [Google Scholar]
- 30.Simonson T, Brooks C L., III J Am Chem Soc. 1996;118:8452–8458. [Google Scholar]
- 31.Churg A, Warshel A. Biochemistry. 1986;25:1675–1681. doi: 10.1021/bi00355a035. [DOI] [PubMed] [Google Scholar]
- 32.Dixon D W, Hong X, Woehler S E, Mauk A G, Sishta B P. J Am Chem Soc. 1990;112:1082–1088. [Google Scholar]
- 33.Andrew S M, Rhomasson K A, Northrup S H. J Am Chem Soc. 1993;115:5516–5521. [Google Scholar]
- 34.McLendon G, Smith M. Inorg Chem. 1982;21:847–850. [Google Scholar]
- 35.Augustin M A, Yandell J K. Inorg Chem. 1979;18:577–583. [Google Scholar]
- 36.Fraga E, Webb M A, Loppnow G R. J Phys Chem. 1996;100:3278–3287. [Google Scholar]