

Abstract
Cockayne syndrome (CS) is characterized by increased photosensitivity, growth retardation, and neurological and skeletal abnormalities. The recovery of RNA synthesis is abnormally delayed in CS cells after exposure to UV radiation. Gene-specific repair studies have shown a defect in the transcription-coupled repair (TCR) of active genes in CS cells from genetic complementation groups A and B (CS-A and CS-B). We have analyzed transcription in vivo in intact and permeabilized CS-B cells. Uridine pulse labeling in intact CS-B fibroblasts and lymphoblasts shows a reduction of ≈50% compared with various normal cells and with cells from a patient with xeroderma pigmentosum (XP) group A. In permeabilized CS-B cells transcription in chromatin isolated under physiological conditions is reduced to about 50% of that in normal chromatin and there is a marked reduction in fluorescence intensity in transcription sites in interphase nuclei. Transcription in CS-B cells is sensitive to α-amanitin, suggesting that it is RNA polymerase II-dependent. The reduced transcription in CS-B cells is complemented in chromatin by the addition of normal cell extract, and in intact cells by transfection with the CSB gene. CS-B may be a primary transcription deficiency.
Cockayne syndrome (CS) is a rare inherited disorder belonging to the category of segmental progeriod conditions (1). Afflicted individuals suffer from severe growth retardation (2), cachectic dwarfism (3), mental retardation, deafness, optic atrophy, intracranial calcification, and a number of facial characteristics. Some features, such as severe photosensitivity and the premature aging phenotype, are shared with the human repair deficient disorder xeroderma pigmentosum (XP), but in contrast to XP patients, CS patients do not appear to have a significantly increased risk of cancer. Two complementation groups of CS have been identified, designated CS-A and CS-B, and the corresponding genes have been cloned (4, 5).
The cellular and molecular phenotype of CS includes a significantly increased sensitivity to a number of DNA-damaging agents including UV radiation (6–8). Studies in CS cells were initially confined to DNA repair in the general, overall genome, where no defect was found (reviewed in ref. 9). The observation that CS cells are deficient in the resumption of RNA synthesis after UV irradiation (8) led to the idea that these cells might be deficient in the repair of transcriptionally active genes. When assays for gene-specific repair became available (10), it was discovered that CS cells are indeed defective in the preferential repair of active genes and in the preferential repair of the transcribed strand of such genes (11, 12). This defect in transcription coupled repair (TCR) in CS is not only found after UV exposure but also after gamma radiation (13).
Recent developments have shown that a number of DNA repair proteins are also essential for RNA polymerase II (RNA pol II) transcription (14). The basal transcription factor TFIIH contains at least two known excision repair proteins (XPB and XPD) and possibly more. Other repair proteins may be associated with the TFIIH function in a less inclusive manner (15). The extent of overlap and linkage between DNA repair and transcription makes it difficult or impossible to clearly separate the two. Recently it has been shown that a mutant form of TFIIH can inhibit both DNA repair and transcription in normal cells in vitro (16). Hence, a primary transcription defect rather than a primary DNA repair defect may determine the complex clinical phenotypes of some of the clinical conditions that we have come to think of as DNA repair disorders. CS may be one such condition. Although the notion of “transcription syndromes” was formalized in the literature several years ago (17), there is still no experimental confirmation of this hypothesis.
Some data support the notion of a primary transcription defect rather than a primary DNA repair defect in CS cells. Gene-specific DNA repair studies in CS cells suggest that the defect in TCR cannot explain the hypersensitivity of these cells to DNA damage (18), and recent in vitro transcription experiments have shown reduced levels of RNA polymerase II, but not RNA polymerase I or III transcription in extracts of CS and XP-B/CS cells (G.L.D., J.-F. Houle, N. Iyer, V.A.B., and E.C.F., unpublished results). In the present study, we have explored transcription in CS-B cells in vivo using several approaches. We have measured uridine incorporation in intact CS-B lymphoblastoid cells and fibroblasts, and in addition, monitored transcription using immunological assays. We have also employed permeabilized cells in which transcription was studied in the chromatin fraction under quasi-physiological conditions. In all of these assays we observed a reduction of about 50% in the level of transcription in CS-B cells compared with normal human cells. As a control for transcription status we studied several normal cells lines as well as fibroblasts from a patient with xeroderma pigmentosum group A (XP-A), which is severely defective in DNA repair. XP-A cells show levels of transcription similar to that observed in the normal cell lines.
The experiments with permeabilized cells have an advantage because the cell extracts can be complemented with other extracts or components. When extracts from normal cells were added to permeabilized CS-B cells, transcription was restored to normal levels. Further, the transcription defect in CS-B was complemented by transfection with the c-DNA for the CSB gene. We have also addressed the question of which RNA polymerase was involved by using α-amanitin as an inhibitor. The residual transcription in CS-B cells is completely abolished by α-amanitin suggesting that the reduced transcription is primarily mediated by RNA polymerase II. The viability of these cell extracts was checked for in vitro DNA repair of a plasmid damaged with N-acetoxy-2-acetylaminofluorene (AAAF) and we found no difference between normal and CS-B extracts. We suggest that CS-B cells may be defective in a factor involved in the organization of an efficient transcription complex.
METHODS
Cell Lines and Culture Conditions.
The lymphoblastoid and fibroblast cell lines of normal and CS-B were obtained from Corriel Cell repository (New Jersey). The cell lines are identified in the figures or legends. Lymphoblastoid cell lines were routinely maintained in RPMI medium supplemented with 15% fetal bovine serum (FBS) and antibiotics. Fibroblast cell lines were maintained in minimal essential medium supplemented with 15% FBS, antibiotics, amino acids, and vitamins. The CS1AN cell lines transfected with plasmid pDR2 containing the CSB gene were maintained in the presence of hygromycin B (200 μg/ml).
RNA Synthesis in Intact Cells.
Before [3H]uridine incorporation in intact cells, exponentially growing cells were prelabeled with [14C]thymidine (0.02 μCi/ml; spec. act. 56 mCi/mmol; Amersham) for a day to uniformly label the DNA. RNA synthesis was measured by a 30- and 90-min pulse labeling with [3H]uridine (10 μCi/ml; 46 Ci/mmol; Amersham). Radioactivity incorporated into TCA-insoluble material was scintillation counted and expressed as cpm/106 cells. Pretreatment of cells with α-amanitin (1 μg/ml) alone or with α-amanitin (1 μg/ml) and actinomycin D (0.2 μg/ml) was for 16 hr.
Transcription in Permeabilized Cells. Encapsulation of cells.
The cells were prelabeled with [3H]thymidine (0.1 μCi/ml) to uniformly label the DNA. The procedure for encapsulation was essentially the same as described by Jackson and Cook (19). Briefly, 2.5% agarose (Sigma Type VII) in PBS was melted and cooled to 39°C. Cells (5 ml; 2–4 × 106 cells per ml) in complete medium were mixed with 1.25 ml of molten agarose in a conical flask at 39°C. After the addition of 15 ml of liquid paraffin oil (Fisher Scientific) to the cells in molten agarose, the mixture was vortexed for 30 sec and kept on ice with constant swirling of the flask for 2 min. Complete medium (15 ml) was added to the flask and the contents were transferred to a 50 ml centrifuge tube. The agarose beads were pelleted using a bench-top centrifuge (500 × g, 5 min). After the removal of paraffin and excess aqueous phase, the agarose beads were thoroughly washed thrice in PBS. These beads containing the cells were now ready for lysis.
Lysis of encapsulated cells.
The beads containing the cells were lysed either in 0.05% or 0.5% Triton X-100 in a modified physiological buffer (PB) for 15 min on ice. This buffer contained 10 mM Na2HPO4, 2.5 mM MgCl2, 65 mM KCl, 65 mM KC2 H3O2, 1 mM Na2ATP, 1 mM dithiothreitol, and 0.2 mM phenylmethylsulfonyl fluoride (PMSF). After lysis, cells were washed in PB and processed for in vitro reaction. The chromatin prepared by this way was found to replicate in vitro at 85% of the rate found in vivo in a cell cycle-specific manner (19).
In vitro transcription.
The reaction was started by the addition of 50 μl of 10× concentrated transcription mixture to 450 μl of beads. The transcription mixture contained a final concentration of 0.1 mM CTP, GTP, 5 μM cold UTP, [32P]UTP(50 μCi/ml; spec. act. 3,000 Ci/mmol; Amersham), 10 μM S-adenosylmethionine, 1 mM ATP, and 2.5 mM MgCl2 and RNasin (40 units per ml, Boehringer Mannheim). Initial transcription reactions were carried out both at 32°C and 37°C, and no difference in transcription rate was found at these incubation temperatures. The beads were incubated at 37°C and 100-μl beads were collected at 0, 5, 15, and 30 min. The beads were washed four times in ice-cold PB to remove the unincorporated nucleotides. Lysis was carried out with 0.1% SDS for 2 hr at 37°C. After lysis, an equal volume of 20% trichloroacetic acid (TCA) was added to the beads and the samples were spotted onto glass fiber discs (Whatmann). The filters were washed sequentially with 5% TCA, 70% ethanol, and acetone. The filters were dried under a red lamp, the radioactivity incorporated into acid-insoluble material was scintillation counted, and the incorporated radioactivity was expressed as pmol UMP incorporated per 106 cells. In some experiments, the lysed cells in the beads were incubated with α-amanitin (5 μg/ml) for 30 min on ice before the transcription reaction.
Labeling of Transcription Sites in Interphase Nuclei by Immunofluorescence.
The fibroblast cells of normal (WI 38) and CS-B (GM1098) were used for this assay. The procedure was very similar to the one described above. For immunolabeling, 0.1 mM 5-bromouridine 5′-triphosphate (Br-UTP) was used instead of [32P]UTP. After the reaction, beads were washed several times in PB. The nuclei were permeabilized in ice-cold PB containing 0.5% Triton X-100 for 10 min and washed three times in PB. The beads were subsequently washed in PBS supplemented with 0.05% Tween 20. Transcription sites were indirectly immunolabeled using a primary antibody raised against bromodeoxyuridine-BSA conjugate [Anti-BrdU mouse monoclonal IgG (1:1,000 dilution in PB, 4 hr at 4°C; Boehringer Mannheim), which reacts only with single-stranded Br-RNA. After incubation with primary antibody, beads were washed extensively with PB plus 0.05% Tween 20 and incubated with secondary antibody for 16 hr at 4°C (Sheep anti-mouse IgG Texas red conjugated; 1:500 dilution; Amersham). The beads were then washed thoroughly in PB and PBS. Vectashield (25 μl; Vector Laboratories) mounting medium containing 0.1 μg/ml of 4′,6-diamidino-2-phenylindole (DAPI) was mixed with 10 μl of beads and the mixture was placed on a glass slide and covered with a coverslip. Photographs were taken with a Zeiss Axiovert microscope using Kodak color 400 ASA film (100× oil immersion objective).
Measurement of Fluorescence Intensity of Transcription Foci.
Fluorescent images of 25 individual nuclei from normal and Cockayne cells were captured using a CCD camera (JVC) and analyzed using the image proplus software program (Media Cybernetics, Silver Spring, MD). The transcription foci were measured within the intensity range of 10–255 pixels.
In Vitro Complementation Studies.
Cell extracts were prepared by the method of Manley et al. (20). The extracts were dialyzed overnight at 4°C against 25 mM Hepes⋅KOH (pH7.9), 1 mM DTT, 1 mM EDTA, 17% glycerol, 12 mM MgCl2 containing 0.1 M KCl, aliquoted, and stored at −80°C. The same extracts from normal and CS-B cells were used for in vitro transcription and in vitro repair (see below). The transcription procedure was essentially the same for in vitro complementation studies except that the chromatin prepared from normal and CS-B cells was preincubated with 100 μg of whole cell extracts (WCEs) and the transcription was measured using [3H]UTP.
In Vitro Nucleotide Excision Repair (NER) Assay.
Plasmids pRS and pUC 19 used in NER assay were purified by cesium chloride and sucrose gradient centrifugation (21). The pUC 19 DNA was treated with 3 μM N-acetoxy-2-acetylaminofluorene in 10 mM Tris⋅HCl (pH 7.5) and 10% ethanol for 3 hr at 37°C prior to sucrose gradient purification. The NER assay was performed essentially as described by Wood et al. (22).
RESULTS
Transcription in Intact Normal and Cockayne Cells.
Fig. 1 A and B show in vivo transcription rates in intact normal and CS-B human cell lines measured by total incorporation of [3H]uridine after pulse labeling for 30 and 90 min. The use of short pulses of [3H]uridine predominantly measures RNA pol II transcription (23). RNA synthesis in CS-B lymphoblastoid cells was reduced to 42% of that in normal lymphoblasts (Fig. 1A). Fig. 1B shows transcription in intact CS-B fibroblasts derived from three different patients. All three cell lines showed approximately 50% reduction in the rate of transcription, which was more pronounced at 90 min than at 30 min. We observed normal rates of transcription in several other normal human cell lines used as controls. In addition, we measured transcription by [3H]uridine labeling of intact cells from the excision repair-deficient syndrome, XP-A (GM2250E). There was no defect in XP-A cells compared with normal lymphoblastoid cells.
Figure 1.



(A) RNA synthesis in intact normal (▪) and CS-B (•) lymphoblastoid cells. [14C]Thymidine-prelabeled cells were pulse labeled with [3H]uridine for 30 and 90 min. Radioactivity incorporated into acid-insoluble material was measured by scintillation counting. The absolute transcription rate was expressed as [3H]uridine incorporation per 106 cells based on the specific activity of [14C]thymidine per cell. (B) RNA synthesis in intact normal (▪) and three different CS-B fibroblast cell lines (•, ▴, ▾). [14C]Prelabeled cells were given a pulse with [3H]uridine for 30 and 90 min and the radioactivity incorporated into acid-insoluble material was measured. The transcription rate was expressed as [3H]uridine incorporation per 106 cells based on the specific activity of [14C]thymidine per cell. (C and D). Visualization of transcription sites in permeabilized normal (C) and CS-B (D) fibroblast cells. Cells were encapsulated in 0.5% agarose beads, lysed with 0.05% Triton X-100. The transcription reaction was carried out for 30 min using triphosphates and 0.1 mM Br-UTP at 37°C. After the reaction, the beads were washed in PB and the nuclei were permeabilized with 0.5% Triton X-100 for 10 min. Transcription sites were indirectly immunolabeled using a primary antibody against bromodeoxyuridine-BSA conjugate, which reacts only with single-stranded Br-RNA (Anti-BrdU mouse monoclonal IgG). After washing, the beads were incubated with Texas red conjugated secondary antibody (Sheep anti-mouse IgG). (E) Transcription in normal and CS-B chromatin in permeabilized cells. [3H]Thymidine-labeled normal (▪) and CS-B (•) lymphoblastoid cells were encapsulated in 0.5% agarose beads and lysed with 0.05% Triton X-100. The transcription was carried out as described. The beads were incubated at 37°C and 100-μl aliquots were collected at 0, 15, and 30 min to determine the rate of incorporation. The beads were washed thrice in ice-cold PB and lysed with 0.1% SDS for 2 hr at 37°C. The radioactivity incorporated into acid-insoluble material was measured and the transcription was expressed as pmol UMP incorporated per 106 cells. (F) In vitro transcription using different concentrations of Triton X-100. [3H]Thymidine-labeled normal (GM1712A, ▪, ▴) and CSB (•, ▾) lymphoblastoid cells were encapsulated and lysed with 0.05% and 0.5% Triton X-100. The beads were incubated at 37°C in transcription mixture, 100-μl aliquots were collected, and the incorporated radioactivity was determined by scintillation counting. The radioactivity incorporated into acid-insoluble material was measured and the transcription was expressed as pmol UMP incorporated per 106 cells.
We examined the spatial distribution of transcription sites in interphase nuclei of permeabilized cells by incorporation of the UTP analogue Br-UTP into nascent RNA chains. Incorporated Br-UTP was visualized by indirect immunofluorescence microscopy. In normal cells, bright, punctuated labeling of RNA pol II transcription was observed (Fig. 1C), whereas in CS-B cells, the fluorescence intensity of transcription foci appeared much less (Fig. 1D). Using this approach, we again detected about 40% less transcription in CS-B than in normal cells, Table 1.
Table 1.
Fluorescence intensity of transcription foci
| Cell line | Fluorescence intensity |
|---|---|
| Normal (WI 38) | 6,997 ± 1,020 |
| CS-B (GM1098) | 4,298 ± 1,423 |
Relative fluorescence intensities of transcription foci were measured using a Texas red isothiocyanate (TRITC) filter within the intensity range of 10–255 pixels. Values were derived from the analysis of 25 interphase nuclei randomly chosen from each cell line. Numbers in parentheses indicate the cell line used.
Chromatin Transcription in Permeabilized Normal and Cockayne Cells.
We prepared permeabilized cells that were depleted of soluble proteins using an assay that protects chromatin from shearing and preserves it in its native state in isotonic buffers (24). This chromatin fraction has been shown to carry out most of the replicative and transcriptional activities characteristic of intact living cells (19, 25). An advantage of this system is that the chromatin is accessible to a variety of proteins and is suitable for in vitro complementation. Transcription rates in permeabilized normal and CS-B cells are presented in Fig. 1E, and the transcription rate in CS-B cells was reduced by about 50%, similar to the results of our studies with intact cells (Fig. 1E).
Effects of Triton X-100 on Chromatin Transcription.
We determined the effects of lysis with different concentrations of Triton X-100 on transcription rates in chromatin from normal and CS-B lymphoblasts. Increasing the Triton concentration appeared to disrupt chromatin structure more in CS-B cells than in normal cells. In normal cells, transcription rates expressed as pmol UMP incorporation per 106 cells remained essentially the same after lysis with 0.05% and 0.5% Triton X-100 (Fig. 1F). At a concentration of 0.5%, Triton X-100 released 60% of the proteins in normal cells and 82% in CS-B cells. This suggests that the transcription complex is tightly associated with chromatin even after the removal of more than 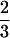 of the total proteins. In contrast, the transcription rate in CS-B cells was significantly reduced (by 75%) when the Triton concentration was increased to 0.5% (Fig. 1F). It is possible that CS-B cells have a defective chromatin organization that could account for the reduced transcription.
of the total proteins. In contrast, the transcription rate in CS-B cells was significantly reduced (by 75%) when the Triton concentration was increased to 0.5% (Fig. 1F). It is possible that CS-B cells have a defective chromatin organization that could account for the reduced transcription.
Effect of α-Amanitin.
Pretreatment of intact cells with α-amanitin, an inhibitor of RNA pol II, reduced transcription to 22% in normal cells and 8% in CS-B lymphoblastoid cells (Fig. 2A). Treatment of normal and CS cells with both α-amanitin and actinomycin D completely abolished total RNA synthesis (Fig. 2A), indicating that this assay measures legitimate transcription. Transcription in chromatin was also sensitive to α-amanitin. Transcription in the presence of α-amanitin was reduced by 75% in normal fibroblasts and was completely abolished in CS-B fibroblasts (Fig. 2B). We conclude from these and the above experiments that the transcription we measured in cells and chromatin is mainly due to RNA pol II activity.
Figure 2.

(A) Effect of α-amanitin and actinomycin D on transcription in intact normal and CS-B cells. 14C-labeled cells were pretreated with α-amanitin (2 μg/ml) alone or α-amanitin and actinomycin D (0.2 μg/ml) for 16 hr. The cells were then labeled with [3H]uridine for 30 and 90 min. The absolute transcription rate was expressed as [3H]uridine incorporation per 106 cells based on the specific activity of [14C]thymidine per cell. Normal lymphoblastoid cells (▪, •, ▾), CS-B (▾, ♦, +). (B) Effect of α-amanitin on in vitro transcription in normal and CS-B chromatin. 14C-prelabeled cells were encapsulated in 0.5% agarose beads and lysed with 0.05% Triton X-100. Prior to the transcription reaction, a fraction of the beads were incubated with the inhibitor of RNA pol II, α-amanitin (5 μg/ml, 30 min on ice), and the transcription was carried out with triphosphates and [32P]UTP using the conditions described in Fig. 3. Aliquot of beads (100 μl) was collected at 0, 5, and 30 min. After washing the beads in PB to remove the unincorporated nucleotides, the beads were lysed in 0.2% SDS for 2 hr and the acid insoluble radioactivity was measured by scintillation counting. The transcription rate was expressed as pmol UMP incorporated per 106 cells. Normal (▴, untreated; ▾, α-amanitin treated) and CS-B (▪, untreated; •, α-amanitin treated).
Transcription Complementation.
We utilized the chromatin depleted of soluble proteins for in vitro complementation studies. Three different transcription reactions were carried out using the CS-B chromatin supplemented either with CS-B or normal WCE. The transcription measured in the chromatin without any extract served as a control for the determination of the effect of extract on chromatin transcription. Addition of WCE from CS-B lymphoblastoid cells to CS-B chromatin resulted in a minor elevation (18%) of transcription compared with the transcription in chromatin without extract (Fig. 3A). Addition of normal cell extract to CS-B chromatin enhanced the transcription rate substantially and made it marginally higher than the control, normal chromatin supplemented with normal cell extract (Fig. 3A). The addition of CS-B extract to chromatin from normal cells caused a slight inhibition of transcription (13%) compared with transcription without extract (data not shown).
Figure 3.

(A) Complementation of transcription in CS-B chromatin by WCEs. [14C]Thymidine-prelabeled lymphoblastoid cells were encapsulated and lysed in 0.5% Triton X-100 in PB. Before the transcription reaction, the beads containing the protein depleted chromatin were incubated either with 100 μg of whole cell CS-B (•) or normal (▴) extract for 30 min on ice. The chromatin not supplemented with either of the WCE (▪) served as control. The transcription reaction was carried out using triphosphates and [3H]UTP and the radioactivity incorporated into acid-insoluble material was measured. The transcription rate was expressed as pmol UMP incorporated per 106 cells. (B and C) Transcription in complemented cell lines. Transcription in intact (B) SV40-transformed normal fibroblasts (GM00637D) (▪) and in CS-B cells transfected with the CSB c-DNA (CS1AN/CSB/pDR2) (•). CS-B cells transfected with the plasmid alone (CS1AN/pDR2) (▴) were used for comparison. Transcription measured in chromatin (C) SV40-transformed normal fibroblasts (▴), CS-B cells transfected with CSB cDNA (•), and CS-B transfected with plasmid alone (▪).
Complementation of the reduced transcription with the CSB gene was next examined both in vivo (intact) and in vitro (permeabilized cells) using simian virus 40 (SV40)-transformed CSB fibroblasts (CS1AN) transfected with the CSB cDNA (Figs. 3 B and C). CS1AN cells transfected only with plasmid pDR2 served as a control. SV40-transformed normal human fibroblast (GM00637D) were used for comparison with transcription in CS-B cells. The results clearly demonstrate that CSB gene completely restores the transcription deficiency in CS-B cells both in intact cells (Fig. 3B) and in permeabilized cells (Fig. 3C). The introduction of the CSB c-DNA restored transcription to normal levels in both of these assays, supporting the conclusion that the transcription deficiency is due to dysfunction of the CSB gene. This corroborates the results of the in vitro complementation studies where addition of normal cell extract considerably enhanced the rate of transcription.
In Vitro DNA Repair.
To verify that the CS-B extracts were not nonspecifically inactivated, an in vitro DNA repair experiment was performed. Plasmid DNA was damaged with AAAF, and NER was monitored by in vitro repair synthesis. The specificity of DNA synthesis was determined by the use of an undamaged plasmid of a slightly larger size. The results of such an experiment are shown in Fig. 4. The relative repair is similar in the damaged plasmids supplemented with normal (lane 1) and CS-B (lane 2) extracts. This result indicates that the CS-B extract used for the in vitro complementation studies is active, and it also shows that overall genome repair of this DNA lesion in CS-B cells is as efficient as in normal human cells.
Figure 4.

In vitro NER by normal and CS-B cell free extract. Reactions contained 0.3 μg of each of N-acetoxy-2-acetylaminofluorene damaged and undamaged plasmid supplemented with 100 μg protein of either normal (lane 1) or CS-B (lane 2) cell free extract. Following incubation at 30°C for 1 hr, plasmid DNA was linearized and electrophoresed on 1% agarose gel. (A) Ethidium bromide-stained agarose gel showing equal loading of both damaged and undamaged plasmids. (B) Autoradiograph of the same gel showing repair incorporation.
DISCUSSION
The rate of transcription in several CS-B cells measured in vivo and in vitro using multiple different experimental approaches is reduced to about 50% of normal. The in vitro chromatin transcription assay used in the present study is analogous to nuclear run-on transcription and basically measures only elongation of those transcription sites that are initiated in vivo (19, 25). We conclude that the transcription elongation rate is substantially reduced in CS-B cells. It remains to be determined whether there is also a defect in transcription initiation. In support of our results, a previous study found that expression of a metallo protease was reduced to 50% in CS cells (26), and very recently it was reported that the CSB protein affects transcription in a purified-component in vitro assay (27).
An advantage of the permeabilized system is that the chromatin is readily accessible to a variety of proteins and is highly suitable for in vitro complementation assays on intact chromatin. Furthermore, activities can be modulated by differential extraction of proteins using varying concentrations of Triton X-100. In normal cells, the transcription rate remained essentially the same after the removal of up to 85% of the soluble proteins from the chromatin, while in CS-B cells there was a drastic reduction in transcription with increased removal of proteins. In normal cells, the factors involved in the organization of the transcription complex are apparently tightly associated with chromatin and hence cannot be easily solubilized with Triton X-100. This confirms earlier observations that transcription complexes are firmly attached to a nucleoskeleton (28, 29). The same may not be true for CS-B cells in which the increased removal of proteins resulted in a decrease in transcription rate. The association between the transcription complex and chromatin may be disrupted in CS-B cells, and their chromatin organization appears to be more “loose” than that in normal cells.
The transcription defect in CS-B chromatin can be complemented to wild-type levels with extracts from normal cells. Addition of CS-B extract to CS-B chromatin or to chromatin from normal cells did not have any detectable effect. This indicates that some of the factors required for the formation and maintenance of an efficient transcription complex may be lacking in CS-B cells rather than the presence of an inhibitory factor in CS-B. Transcription was also complemented in transfectants carrying the CSB c-DNA, further documenting that mutations in this gene are directly related to the defect in transcription.
It is of interest to clarify whether CS cells are defective in the processing of endogenous damage such as oxidative DNA lesions generated by oxidative stress. Oxidative DNA damage is the result of endogenous processes, and an accumulation of these lesions could contribute to the CS phenotype. It is unclear whether repair of oxidative damage can be transcription coupled, and recent work from this laboratory (Laboratory of Molecular Genetics) has shown that the repair of a major lesion introduced by oxidative stress, 8-oxo-guanine, is repaired in a nonpreferential manner without obvious strand bias (30). That suggests that this lesion is not repaired by TCR. CS cells, which are thought to be deficient only in TCR, would then be expected to be proficient in the repair of oxidative damage. However, one study reported a deficiency in the repair of damage introduced by ionizing irradiation in CS cells (13).
Henning et al. (5) showed that in vitro translated CSA protein interacts with in vitro translated CSB protein, and with the p44 subunit of TFIIH. The CSB protein does not appear to be an integral component of the TFIIH transcription complex, and it is not established whether these interactions occur in vivo. However, mutations in CSA or CSB gene products could indirectly modulate the activity of TFIIH via the p44 subunit or other associations. The CSB protein also interacts with the XPG protein (31), which has been shown to incise damaged DNA at the 3′ side of lesions during NER (32). There has been speculation that the XPG protein may play a role in TCR, and its interaction with CSB may be an important link to transcription.
Mutations in the gene encoding the CSB protein appear to perturb the elongation of RNA pol II, as we have shown in several experiments both in vivo and in vitro. In a recent model (33), it was suggested that mutational inactivation of either CSA or CSB proteins could affect their interactions with XPB, XPD, and XPG proteins, resulting in a reduced efficiency of displacement of RNA polymerase II from damaged sites or natural transcription pause sites. This might explain the delayed recovery of RNA synthesis after UV irradiation of CS-B cell lines. Hence, a deficiency in the rate of elongation of RNA pol II arising from the mutational inactivation of either of the genes CSA, CSB, XPD or XPB might possibly be the basis for the CS phenotype. In addition, mutations may in other ways perturb or alter the transcription complex via some of the above-mentioned interactions. This might explain our observations with Triton-X treatments in which the CS chromatin appears to have a “looser” configuration.
The predicted amino acid sequence of the CSB protein shows homology with the ATPase subunit of the yeast SWI/SNF transcription activation complex (34), which is composed of at least 12 different subunits (35). In fact, a yeast homologue of CSB, Rad26, has been shown to exhibit DNA-dependent ATPase activity in vitro (36). The SWI/SNF complex activates transcription by facilitating access of the transcriptional machinery to the promoter regions of active genes. Recent work showed that the SWI/SNF complex may be an integral component of the RNA pol II holoenzyme (37). The homology of CSB to one of the subunits of the SWI/SNF complex suggests a role for CSB protein in transcription. Hence, mutations in the CSB gene could affect the overall efficiency of transcription. If the CSB protein is involved in chromatin remodeling, mutational inactivation might impair this event not only during transcription, but also during NER.
The presence of photoproducts in the transcribed strand of active genes blocks elongation by RNA pol II (38). If the elongation process is deficient, it may also be more sensitive to DNA damage or possibly other modifications of DNA than in normal cells. This might explain the delay in RNA synthesis resumption after UV exposure that has been observed in CS cells (8), and would suggest that a primary transcription defect is in fact responsible for the impairment of a specific NER pathway in CS cells. Thus, CS may be a “transcription syndrome” rather than a “repair syndrome.”
Acknowledgments
We thank the Danish center for Molecular Gerontology for constructive interactions, and Drs. David Orren and Robert Brosh (Laboratory of Molecular Genetics) for critical comments.
ABBREVIATIONS
- CS
Cockayne syndrome
- XP
xeroderma pigmentosum
- TCR
transcription-coupled repair
- WCE
whole cell extract
- NER
nucleotide excision repair
- TFIIH
basal transcription factor
- PB
physiological buffer
- SV40
simian virus 40
- RNA pol II
RNA polymerase II
References
- 1.Beauregard S, Gilchrest B. Dermatol Clin. 1987;5:109–121. [PubMed] [Google Scholar]
- 2.Cockayne E A. Arch Dis Child. 1936;11:1–8. doi: 10.1136/adc.11.61.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nance M, Berry S. Am J Med Genet. 1992;42:68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- 4.Troelstra C, van Gool A, De Wit J, Vermeulen W, Bootsma D, Hoeijmakers H J. Cell. 1992;71:939–953. doi: 10.1016/0092-8674(92)90390-x. [DOI] [PubMed] [Google Scholar]
- 5.Henning K, Li L, Legerski R, Iyer N, McDaniel L, Schultz R, Stefanini M, Lehmann A, Mayne L, Friedberg E. Cell. 1995;82:555–566. doi: 10.1016/0092-8674(95)90028-4. [DOI] [PubMed] [Google Scholar]
- 6.Schmickel R, Chu E, Trosko J, Chang C. Pediatrics. 1979;60:135–139. [PubMed] [Google Scholar]
- 7.Andrews A D, Barett S F, Robbins J H. Proc Natl Acad Sci USA. 1978;75:1984–1988. doi: 10.1073/pnas.75.4.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehmann A R, Mayne L V. In: Chromosome Damage and Repair. Seeberg E, Kleppe K, editors. New York: Plenum; 1981. pp. 367–371. [Google Scholar]
- 9.Friedberg E C, Walker G C, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 10.Bohr V A, Smith C A, Okumoto D S, Hanawalt P C. Cell. 1985;40:359–369. doi: 10.1016/0092-8674(85)90150-3. [DOI] [PubMed] [Google Scholar]
- 11.Venema J, Mullenders L H F, Natarajan A T, van Zeeland A A, Mayne L V. Proc Natl Acad Sci USA. 1990;87:4707–4711. doi: 10.1073/pnas.87.12.4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans M K, Bohr V A. Mutat Res. 1994;314:221–231. doi: 10.1016/0921-8777(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 13.Leadon S A, Cooper P K. Proc Natl Acad Sci USA. 1993;90:10499–10503. doi: 10.1073/pnas.90.22.10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaeffer L, Roy R, Humbert S, Moncollin V, Vermeulen W, Hoeijmakers J H J, Chambon P, Egly J M. Science. 1993;260:58–63. doi: 10.1126/science.8465201. [DOI] [PubMed] [Google Scholar]
- 15.Friedberg E C. Annu Rev Biochem. 1996;65:15–42. doi: 10.1146/annurev.bi.65.070196.000311. [DOI] [PubMed] [Google Scholar]
- 16.Hwang J R, Moncollin V, Vermeulen W, Seroz T, Van Vuuren H, Hoeijmakers J H J, Egly J M. J Biol Chem. 1996;271:15898–15904. doi: 10.1074/jbc.271.27.15898. [DOI] [PubMed] [Google Scholar]
- 17.Bootsma D, Hoeijmakers J H. Nature (London) 1993;363:114–115. doi: 10.1038/363114a0. [DOI] [PubMed] [Google Scholar]
- 18.Oosterwijk M F, Versteeg A, Filon R, van Zeeland A A, Mullenders L H F. Mol Cell Biol. 1996;16:4436–4444. doi: 10.1128/mcb.16.8.4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson D A, Cook P R. J Cell Sci. 1988;90:365–378. doi: 10.1242/jcs.90.3.365. [DOI] [PubMed] [Google Scholar]
- 20.Manley J L, Fire A, Cano A, Sharp P A, Gefter M L. Proc Natl Acad Sci USA. 1980;77:3855–3859. doi: 10.1073/pnas.77.7.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biggerstaff M, Robins P, Coverly D, Wood R D. Mutat Res. 1991;254:217–224. doi: 10.1016/0921-8777(91)90059-x. [DOI] [PubMed] [Google Scholar]
- 22.Wood R D, Robins P, Lindahl T. Cell. 1988;53:97–106. doi: 10.1016/0092-8674(88)90491-6. [DOI] [PubMed] [Google Scholar]
- 23.Wansink D G, Schul W, van der Kraan I, van Steensel B, van Driel R, de Jong L. J Cell Biol. 1993;122:283–293. doi: 10.1083/jcb.122.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cook P R. EMBO J. 1984;3:1837–1842. doi: 10.1002/j.1460-2075.1984.tb02056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson D A, Cook P R. J Mol Biol. 1986;192:77–86. doi: 10.1016/0022-2836(86)90465-1. [DOI] [PubMed] [Google Scholar]
- 26.Millis A J T, Hoyle M, McCue H M, Martini H. Exp Cell Res. 1992;201:373–379. doi: 10.1016/0014-4827(92)90286-h. [DOI] [PubMed] [Google Scholar]
- 27.Selby C P, Sancar A. J Biol Chem. 1997;272:1885–1897. doi: 10.1074/jbc.272.3.1885. [DOI] [PubMed] [Google Scholar]
- 28.Pardoll D M, Vogelstein B, Coffey D S. Cell. 1980;19:527–536. doi: 10.1016/0092-8674(80)90527-9. [DOI] [PubMed] [Google Scholar]
- 29.Robinson S H, Nelkin B D, Vogelstein B. Cell. 1982;28:99–106. doi: 10.1016/0092-8674(82)90379-8. [DOI] [PubMed] [Google Scholar]
- 30.Taffe B, Larminat F, Laval J, Croteau D, Anson R M, Bohr V A. Mutat Res. 1996;364:183–192. doi: 10.1016/s0921-8777(96)00031-6. [DOI] [PubMed] [Google Scholar]
- 31.Iyer N, Reagan M S, Wu K-J, Canagarajah B, Friedberg E C. Biochemistry. 1996;35:2157–2167. doi: 10.1021/bi9524124. [DOI] [PubMed] [Google Scholar]
- 32.Sancar A. J Biol Chem. 1995;270:15915–15918. doi: 10.1074/jbc.270.27.15915. [DOI] [PubMed] [Google Scholar]
- 33.Habraken Y, Sung P, Prakash S, Prakash L. Proc Natl Acad Sci USA. 1996;93:10718–10722. doi: 10.1073/pnas.93.20.10718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richmond E, Peterson C L. Nucleic Acids Res. 1996;24:3685–3692. doi: 10.1093/nar/24.19.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cairns B R, Kim Y, Sayre M H, Laurent B C, Kornberg R D. Proc Natl Acad Sci USA. 1994;91:1950–1954. doi: 10.1073/pnas.91.5.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guzder S N, Sung P, Prakash L, Prakash S. J Biol Chem. 1996;271:8903–8910. doi: 10.1074/jbc.271.15.8903. [DOI] [PubMed] [Google Scholar]
- 37.Wilson C J, Chao D M, Imbalzano A N, Schnitzler G R, Kingston R E, Young R A. Cell. 1996;84:235–244. doi: 10.1016/s0092-8674(00)80978-2. [DOI] [PubMed] [Google Scholar]
- 38.van Hoffen A, Venema J, Meschini R, Van Zeeland A, Mullenders L N F. EMBO J. 1995;14:360–367. doi: 10.1002/j.1460-2075.1995.tb07010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]