

Summary
Allele loss and genetic alteration in chromosome 3p, particularly in 3p21.3 region, are the most frequent and the earliest genomic abnormalities found in lung cancer. Multiple 3p21.3 genes exhibit various degrees of tumour suppression activity suggesting that 3p21.3 genes may function as an integrated tumour suppressor region through their diverse biological activities. We have previously demonstrated growth inhibitory effects and tumour suppression mechanism of the H37/RBM5 gene which is one of the 19 genes residing in the 370kb minimal overlap region at 3p21.3. In the current study, in an attempt to find, if any, mutations in the H37 coding region in lung cancer cells, we compared nucleotide sequences of the entire H37 gene in tumour vs. adjacent normal tissues from 17 non-small cell lung cancer (NSCLC) patients. No mutations were detected, instead, we found the two silent single nucleotide polymorphisms (SNPs), C1138T and C2185T, within the coding region of the H37 gene. In addition, we found that specific allele types at these SNP positions are correlated with different histological subtypes of NSCLC; tumours containing heterozygous alleles (C+T) at these SNP positions are more likely to be associated with adenocarcinoma (AC) whereas homozygous alleles (either C or T) are associated with squamous cell carcinoma (SCC) (p=0.0098). We postulate that, these two silent polymorphisms may be in linkage disequilibrium (LD) with a disease causative allele in the 3p21.3 tumour suppressor region which is packed with a large number of important genes affecting lung cancer development. In addition, because of prevalent loss of heterozygosity (LOH) detected at 3p21.3 which precedes lung cancer initiation, these SNPs may be developed into a marker screening for the high risk individuals.
Keywords: lung cancer, H37/RBM5/Luca15, single nucleotide polymorphism, 3p21.3, tumour suppressor gene, linkage disequilibrium, loss of heterozygosity
1. Introduction
Lung cancer is the leading cause of cancer death in the United States resulting in nearly 1.2 million deaths worldwide and over 150,000 deaths in the United States each year [1, 2]. The lung cancer 5-year survival rate has remained 13 to 15% throughout the past 3 decades despite innovations in diagnostic testing, surgical technique, and development of new chemotherapeutic agents [3]. In contrast, the survival rates for other common cancers such as breast, prostate, and colon cancers, which are managed by similar principles of diagnosis, staging, resection, and chemotherapy, have improved dramatically. Although survival improvements in these cancers may be attributed to availability of effective early detection screening, it is possible that the poorer outcomes of lung cancer may be due to fundamental differences in its tumour biology; whereas breast, colon, and prostate malignancies are predominantly adenocarcinomas (ACs), lung cancer histology is heterogeneous. Small cell lung carcinomas (SCLCs) account for 20% of all lung cancers, and the rest 80% consists of non-small cell lung carcinomas (NSCLCs) which, in turn, is further sub-classified into 40% AC, 40% squamous cell carcinoma (SCC) and 20% large cell carcinoma [4]. Growing biological and epidemiological data suggest that different NSCLC histological subtypes, in particular the two most common, AC and SCC, have distinct etiologies, therefore, they should be treated differently. For example, SCC, originated in the tracheobronchial tree, tends to stay localized while growing slowly to large sizes and cavitating whereas AC, developed in the glandular tissue, metastasizes much earlier to the lymph nodes and brain [5, 6]. However, current clinical regimens are the same for both types of lung cancer because the presently available treatment options make no different outcomes in either type. Delineating genetic alterations specific to each subtype of lung cancer may be the most effective way of improving molecular markers for early detection and prediction of response to chemoprevention/chemotherapy as well as developing better individualized treatment.
Deletion and genetic alteration in chromosome 3p, particularly in 3p21.3 region, are the most frequent and the earliest genomic abnormalities found in lung cancer [7]. For instance, loss of heterozygosity (LOH, i.e. heterozygous deletion) in 3p21.3 region occurs in more than 65% of NSCLCs and 95% of SCLCs [8]. Genomic aberrations can be found as early as in preneoplastic lesions of smoker’s lung [9], suggesting one or more 3p21.3 genes may function as preventing tumour initiation. In addition, smokers whose peripheral blood lymphocytes showed greater damage in this 3p21.3 region after treatment with the benzo-a-pyrene diol epoxide carcinogen in vitro had an increased lung cancer risk, suggesting the potential for genetic polymorphisms in this region which predispose cells to lung cancer development [10]. Multiple 3p21.3 genes, in particular ~19 genes contained in the 370 kb smallest region of overlap, exhibit various characteristics of tumour suppressor activity including growth inhibition, apoptosis, and cell cycle arrest [8]. Therefore, it is believed that 3p21.3 genes may co-operate as an integrated ‘tumour suppressor region’ either to promote tumourigenesis through loss of expression, or to suppress tumour growth through activation of their tumour suppressing pathways. Since the genetic abnormalities in the 3p region occur in the earliest stage of lung cancer development, the 3p21.3 tumour suppressor genes (TSGs) hold particular promises to be developed as biomarkers for early cancer detection, screening and chemoprevention. In addition, their potent and multifunctional tumour suppressor functions as well as their direct protein-protein interactions with many important cellular target, confer these 3p21.3 genes great opportunities for gene replacement therapeutics in the future [8].
H37/RBM5 is among the 19 genes located within the 370 kb minimal overlapping deletion regions at 3p21.3. The growing literatures on H37 strongly suggest its involvement in apoptosis and cell cycle regulation, with all results converging on a role for H37 as a TSG. Using the lung cancer cell model, we showed that H37 clearly inhibits tumour growth both in vitro and in vivo with antitumour mechanisms involving cell cycle (G1) arrest and apoptosis [11]. H37 also mediates growth inhibition by eliciting reduced expression of cyclin A and pRB as well as by increased expression of pro-apoptotic protein Bax [11]. Consistently, H37 triggers mitochondrial apoptotic pathways downstream of Bax, in a p53-independent manner, which include breakdown in the mitochondrial membrane potential, cytochrome c release into cytosol, and enhanced caspase-9 and caspase-3 activities [11]. Further, as compared with adjacent normal tissue, our findings show reduced expression in primary lung cancers of H37 transcript and protein in 9 of 11 (82%) and 46 of 62 (73%) samples, respectively [12], although the H37/3p21.3 LOH genomic status for these samples was unknown. The involvement of H37 in malignancy has been also reported by other investigators for a variety of cancer types. For example, H37 was identified as a serum antigen reacting with autologous antibody in renal cancer [13]. H37 is downregulated in human schwannomas [14] and in ras-transformed Rat-1 embryonic fibroblasts [15]. Ectopic expression of H37 also suppresses growth of HT 1080 fibrosarcoma cells [15]. Alternatively-spliced transcripts from the H37 locus and antisense fragments also serve as apoptotic regulators in hematopoetic cells [16–20]. Most significantly, the H37/RBM5 gene was included in the 17 common gene signature associated with metastasis (one of the nine genes down-regulated in metastases) identified in multiple solid tumour types [21]. Various types of solid tumours including lung cancer carrying this gene expression signature had high rates of metastasis and poor clinical outcome.
In the current study, to further confirm H37’s direct functional role in lung tumour development, we compared the H37 coding DNA sequences in primary lung tumour vs. adjacent normal tissues. Our initial aim was to find, if any, mutations in the H37 coding region in tumour. No mutations were detected, instead what we found in the H37 coding region was the two silent single nucleotide polymorphisms (SNPs) which appear to be associated with different histological subtypes of NSCLCs. Given the important attributes of the 3p21.3 region for lung cancer development, we discuss potential value of the found H37 SNPs which may be in linkage disequilibrium (LD) with disease causative polymorphisms yet to be discovered in the vicinity of the H37 gene. Further, the H37 SNPs may be useful as a diagnostic tool to detect LOH at 3p21.3 which is suggestive of higher risk for developing lung cancer.
2. Materials and Methods
2.1. Patient samples
Primary NSCLC tumours and adjacent normal tissues used in RT-PCR analysis were archival specimens obtained from various hospitals during 1988–1989. After surgical removal, all of the samples were immediately snap-frozen in liquid nitrogen and stored at −80°C until total RNA was extracted by guanidinium/cesium chloride ultracentrifugation.
2.2. RT-PCR
Reverse transcription was performed using SuperScript ™ II reverse transcriptase (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. Briefly, 5 μg of total RNA was primed with 500 ng of oligo(dT)12–18 in a total of 12 μl reaction volume for 10 min. at 70°C followed by quick chill on ice. Subsequently, 4 μl 5x first strand buffer, 2 μl 0.1 M DTT, and 1 μl 10 mM dNTP mix were added and the reaction solution was incubated at 42°C for 2 min. before addition of 1 μl SuperScript II (200 U) and further incubation at 42°C for 50 min. Subsequently, the reverse transcriptase was inactivated by heating at 70°C for 15 min. followed by addition of 1 μl RNase H (2 U) at 37°C for 20 min. PCR reaction was performed using Elongase Amplification system (Invitrogen) according to the manufacturer’s suggestions. The sequences of the paired primers used were 5′-TACTGCTGCTTCGGTCTCTC- 3′ (forward) and 5′ –ACCAGGCAACCCACATAAG- 3′ (reverse) which span base positions −79 ~ −60 and 2613 ~ 2631, respectively, in reference to the H37 gene transcription start site. A 50 μl total of PCR reaction consisted of 2 μl 5X buffer A (5 mM MgSO4), 8 μl 5X buffer B (10 mM MgSO4), 2 μl template DNA (reverse- transcription reaction), 0.01 μl forward primer (1 mM), 0.01 μl reverse primer (1 mM), 2 μl elongase polymerase enzyme (1U/μl), 2 μl of 10 mM dNTP mix. PCR was performed in GeneAmp960 (Perkin-Elmer, Waltham, MA, USA) thermocycler for 25 cycles of denaturation (94°C, 30 sec.), annealing (58°C, 30 sec.) and extension (72°C, 3min.) respectively.
2.3. Sequencing
The sequencing reaction was performed using an automatic DNA sequencer (Applied Biosystems Model 373A, Foster City, CA, USA). The same primer set used for PCR as above was also used as initial sequencing primers. The entire template was sequenced both in forward and reverse directions by designing several internal primers.
3. Results
According to the Knudson’s two hit hypothesis, both alleles of a TSG have to be inactivated in order to form tumours [22]. For example, for the classical TSGs such as p53 and RB, the affected individuals are usually born with deletion of the first allele followed by somatic mutation of the second allele during their life-time. Considering that the 3p21.3 genes are heterozygously deleted in a majority of lung cancers, we wanted to test whether H37 coding region may contain genomic mutations on the second allele which are present only in the tumour of lung cancer patients but not in their adjacent normal lung epithelia, suggesting H37’s direct role in tumour development. Therefore, we set out to compare the H37 coding sequence in 17 lung cancer patients’ tumour vs. adjacent normal tissue to find, if any, DNA sequence discrepancies. Total RNAs extracted from respective tissues were reverse-transcribed and the resultant cDNA pool was used as a DNA template in PCR reaction to amplify the entire coding region of the H37 gene (Fig. 1). Subsequent genomic sequencing revealed that there are no differences in the H37 gene sequence between tumour and normal tissues in any of the 17 patients. This finding, at least, was consistent to others’ reports that somatic mutations are rarely found in the 3p21.3 TSGs, rather they are inactivated by alternative mechanisms such as gene silencing by promoter methylation and/or they exhibit haploinsufficiency (i.e. a single copy of the normal gene is incapable of providing sufficient protein production as to assure normal function) [8]. Nevertheless, during our sequence analysis, we, incidentally, found the two silent SNPs present in the H37 coding region; 1) C/T, mRNA position #1138 (a.a. position #330, coding for Alanine); 2) C/T, mRNA position #2185 (a.a. position #679, coding for Tyrosine) (Fig. 1). Table 1 shows C1138T and C2185T allele type status for each individual patient sample and their tumour characteristics/behaviors retrieved from pathology reports. Of a note, the frequent chromosomal deletion observed in the 3p21.3 region is known to be mostly heterozygous deletion (i.e. LOH) [7], therefore finding the H37 locus in all the 17 samples employed in our sequencing task (which used mRNA as the starting material) is not unexpected. In all the patients tested, the nucleotides at the two SNP positions, #1138–#2185, are always the same allele as in either C-C or T-T. This tight linkage is expected given the close proximity of these two nucleotide positions in the genome (only 8405 bp apart); the initial nucleotide configurations present in the ancestral gene pool were conserved due to rare cross-over events between the two positions. In the tumour tissue of the two patients (#s 1, 9), LOH of the H37 genomic region was apparent as shown by presence of the two sequence peaks (C and T; i.e. heterozygote) at both C1138T and C2185T positions in their normal tissue (Fig. 2A and B for the respective positions) whereas only a single peak (at both positions, the “C” allele for the patient #1 and “T” for the patient #9; i.e. homozygote) is present in their tumour counterpart (Fig. 2C for an example of the “C” homozygous allele at C2185T, as in patient #1). When both tumour and normal tissues contain homozygous alleles at these two polymorphic sites, it is difficult to determine whether the tumour underwent LOH in the H37 genomic region because the sequencing peak intensity is not quantitative to the amount of DNA template. Precise analysis by quantitative RT-PCR using the H37 gene- specific primers would be more helpful to discern LOH status in the tumour.
Fig. 1.

Map of the H37 gene coding region. The 3091 bp H37 cDNA is comprised of a total of 25 exons and translated into 816 amino acids. The C1138T and C2185T SNPs are located in exon 12 and 22 respectively. The forward and reverse primers used in RT-PCR and sequencing are from the 5′ and 3′ untranslated regions respectively. rbd, RNA binding domain; zf, zinc finger motifs; nls, nuclear localization signal.
Table 1.
17 primary lung tissue pairs used and their SNP status and tumor characteristics.
| Patient/Tissues I.D.a | C1138T | C2185T | Age, gender, race | Tumor type | Tumor size (cm)/stage | Metastasis status/Commentsc |
|---|---|---|---|---|---|---|
| N1
T1 |
T+C
C |
T+C
C |
55, M, | SCC - Infiltrating, moderately differentiated, focally keratinizing | T2N0 (stage I) | Negative margin & nodes |
| N2
T2 |
C
C |
C
C |
53, F, White | AC - Moderate to well differentiated, mucin secreting | 2.5×2×1.5 | 2/9 node positive, margin negative |
| N3
T3 |
C
C |
C
C |
58, | Large cell poorly differentiated (anaplastic) | 5.5 | 1/8 positive, margin negative |
| N4
T4 |
T+C
T+C |
T+C
T+C |
34 | SCC - Moderately/poorly differentiated | 3.2×2.5×3 | Negative margin & nodes |
| N5
T5 |
C
C |
C
C |
59, M, | SCC - Moderate to poorly differentiated, invasive | Bronchial margin: mild squamous dysplasia, no tumor seen/node negative | |
| N6
T6 |
T+C
T+C |
T+C
T+C |
63,M, White | AC - Poorly differentiated | 2.5 | Node negative, Margin N/A |
| N7
T7 |
T+C
T+C |
T+C
T+C |
71, M, White | AC - Well differentiated | 4.8×3.5×2.5 | Margin negative, 1/8 node positive on frozen (no tumor on permanents) |
| N8
T8 |
T+C
T+C |
T+C
T+C |
50, M, White | AC - Moderately to poorly differentiated | 3.8×2.8×2.6 | Invasion into the chest wall and rib, margin & nodes negative |
| N9
T9 |
T+C
T |
T+C
T |
60, M, | SCC - Well differentiated, invasive | 3/8 node positive, negative margin | |
| N10
T10 |
C
C |
C
C |
77, M, | SCC - Poorly differentiated | Node negative, margin N/A | |
| N11
T11 |
T
T |
T
T |
64, M, | SCC - Poorly differentiated | 2/6 lymph node and bronchial margin positive for tumor | |
| N12
T12 |
C
C |
C
C |
75, F, White | SCC - Well to moderately differentiated | Tumor involves the visceral and parietal pleura, Negative margin & nodes | |
| N13
T13 |
T+C
T+C |
T+C
T+C |
51, M | AC - Moderately well differentiated | Node negative, margin N/A | |
| N14
T14 |
T+C
T+C |
T+C
T+C |
62, F, | AC (Bronchioalveolar cell carcinoma) | Node & margin negative | |
| N15
T15 |
T+C
T+C |
T+C
T+C |
66, F, White | AC - Poorly differentiated | T1N0 | Node & margin negative, no invasion of the pleura or blood vessels. |
| N16
T16 |
NA
C |
NA
C |
53, F, | SCC - Poorly differentiated | T3N1M0 (stage IIIA) | metastases to lymph nodes and tissues between superior vena cava and spine. |
| N17
T17 |
T
T |
T
T |
48, M, White | SCC - Infiltrating moderately differentiated, grade 2 | Margin & node negative |
N=adjacent normal tissue, T=lung tumor tissue
SCC=squamous cell carcinoma, AC=adenocarcinoma
margin N/A, margin status is not available in the pathology report.
Fig. 2.
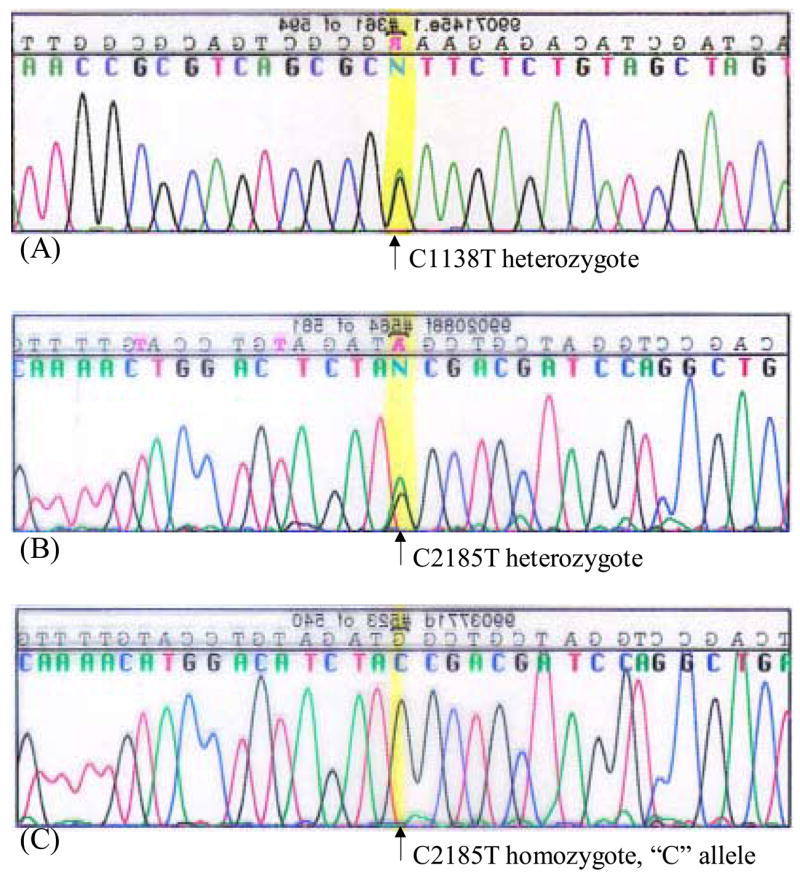
Sequencing peak profiles of the C1138T/C2185T heterozygote vs. homozygote. (A and B) Representative sequence peak diagrams for the C1138T and C2185T heterozygotes respectively, as in the patient #s 1 and 9’s normal tissue. Shown here is the sequencing of the anti-sense strand and the yellow highlighted peaks show overlap of the green colored “A” (anti-parallel of “T”) and the black colored “G” (anti-parallel of “C”) nucleotides. (C) A representative sequence peak diagram for the homozygous allele in the C1138T/C2185T SNP position, as in the patient #s 1 and 9’s tumour tissue. Illustrated here is a specific example for the homozygous allele “G” (anti-parallel complementary of “C”) in the C2185T SNP position, i.e. patient #1, the peak highlighted in yellow color.
Next, we wanted to assess whether there are any correlations between the different allele types in individual patients and their tumour characteristics. According to the statistical analysis (Exact Chi-square test), the only significant correlation found was: the tumours containing both C and T alleles (heterozygotes) at the two SNP spots are mostly AC (6 of 7 samples), whereas the tumors containing either only C allele or only T allele (homozygotes) are mostly SCC (8 of 9 samples), p=0.0098. Of the sample population used in this study, only 1 of 7 AC patients was homozygous and only 1 of 9 SCC patients was heterozygous in the two SNP positions.
Lastly, we surveyed the SNP public database (dbSNP, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=snp) to integrate C1138T and C2185T to the previously reported SNP records. Intriguingly, the H37 SNPs, registered as rs1061474 and rs1138536 respectively, are the two of the most extensively reported SNPs in the 3p21.3 region in terms of a total number of individuals as well as different (ethnic) populations studied, extent of population diversity, and a total number of different submitters, etc., although these public database SNPs are mostly analysed in the general (normal) population samples. Of an interesting note, in the White population, “C” and “T” alleles are evenly distributed (C ratio 0.502/0.548 for C1138T/C2185T respectively) whereas either Asian or Black populations have dominantly “C” allele (C ratio 0.875/0.87 and 0.808/0.776 for Asian and the Black populations respectively). Fig. 3 and Table 2 illustrate, respectively, the genomic distribution and population diversity of other major SNPs within the coding region of the 19 genes at 3p21.3 to compare with the two H37 SNPs.
Fig. 3.

Genomic distribution of the 19 genes and other major SNPs in the 3p21.3 tumour suppressor region. Included here are the SNPs only from the coding region of the genes for which more than 200 individuals were analysed with outcome of significant difference in population diversity (Table 2).
Table 2.
Other major SNPs present in the 3p21.3 gene coding region.
| dbSNP no.a | individual #b | allelesc | amino acids | allele frequencyd | ||
|---|---|---|---|---|---|---|
| Asian | Black | White | ||||
| rs2023953 | 938 | G/A | Gln [Q] | 0.923 | 0.945 | 0.864 |
| rs7061 | 596 | T/C | Tyr [Y] | 0.881 | 0.793 | 0.605 |
| rs1061474 | 1258 | C/T | Ala[A] | 0.875 | 0.808 | 0.502 |
| rs1138536 | 607 | C/T | Tyr[Y] | 0.87 | 0.776 | 0.548 |
| rs1046956 | 361 | A/T | Met[M]/Leu[L] | 1 | 0.685 | 0.649 |
| rs928946 | 240 | G/A | Gly[G] | 0.944 | 0.611 | 1 |
| rs13100173 | 344 | C/T | His[H]/Tyr[Y] | 0.863 | 0.886 | 0.543 |
| rs709210 | 487 | G/T | Ala[A]/Ser[S] | 0.783 | 0.96 | 0.639 |
A total number of individuals analyzed.
The first nucleotide listed is the more frequently occurring allele.
Frequency of the first allele.
4. Discussion
Finding genetic association in population samples hold the promise in uncovering the susceptibility genes linked to development of complex or common disease. Most association studies rely on the use of surrogate markers including SNPs, restriction fragment length polymorphisms (RFLP) and microsatellite markers. SNPs are the most frequent form of DNA variation present in the human genome, occurring approximately every 1200 bp [23, 24], and thus far more than 2 million of them have been identified in the public database [25]. Because of their abundance, even spacing, and stability across the genome, SNPs offer significant advantage in diagnostic potential for complex human diseases including cancers, compared with the conventional polymorphisms such as RFLP and microsatellite markers. In addition, scoring of SNPs can be easily automated as validated in highdensity oligonucleotide arrays which are currently being used for large-scale highthroughput SNP analysis [26]. Given the large number of markers and individuals that must be genotyped for studies of complex traits, SNPs may become a key component in future studies.
For lung cancer, in particular, only ~11% of heavy cigarette smokers ultimately develop the malignancy [27], suggesting that there may be genetic factors predisposing to lung cancer risk. In support of this notion, genetic epidemiologists found that there is a 2.5-fold risk of lung cancer from family history after controlling for cigarette smoking [27]. Certain genetic polymorphisms frequent in the population may significantly contribute to the majority of inherited lung cancer risk.
In the current study, we reported the two silent SNPs found in the H37 lung cancer TSG located at chromosome 3p21.3, which is one of the ‘hottest’ spots in the genome implicated in lung cancer development. In addition, we found that tumors containing the heterozygote alleles at the SNP positions (C+T) are associated with being AC while the homozygous alleles are associated with being SCC. Although our study employed a relatively small sample size (n=17), the observed association is statistically significant (p=0.0098) and is a novel finding, therefore, warrants expanded evaluations with a larger number of samples. Traditionally, silent SNPs have largely been assumed to exert no direct functional effects on phenotype difference because they don’t result in different protein sequence. However, as recently as in the past months, a landmark study was published demonstrating that naturally occurring silent SNPs can, by altering translation kinetics of mRNA, affect in vivo protein folding and, subsequently, function [28]. It is anticipated that this pioneering study may open up a new venue in SNP research proving that silent SNPs as well might contribute to development and progression of certain diseases. Also, it has been demonstrated that silent SNPs can still have an important effect on the stability and expression of the gene; for example, a synonymous (silent) SNP in the human dopamine receptor D2, DRD2, gene functionally alters mRNA stability, leading to measurable differences in gene expression [29].
However, given the potential significance of the 19 genes residing in the 370kb 3p21.3 lung cancer tumor suppressor region, it is equally tempting to postulate that the H37 SNPs C1138T and C2185T may be in LD with some distant polymorphic regulatory element of the H37 gene and/or associated with other disease causative variants within the 3p21.3 tumor suppressor region. In LD, some combinations of alleles or genetic markers occur more frequently than would be expected by chance alone, therefore one allele can serve as a surrogate marker for the mapping of the other; non-random associations between any two markers are measured by the degree of LD. Therefore, disease risk alleles can be mapped by taking advantage of LD relationships to neighboring alleles of genetic markers, and, this way, multiple silent polymorphisms have been found to be associated with human diseases [30]. For example, a link has been found between the silent SNP 102C/T of the 5-HT2A receptor gene in the temporal cortex and the pathogenesis of schizophrenia [31]. Jones and colleagues have shown that silent polymorphisms of the angiotensin-2 receptor increase cardiovascular risk in hypertension [32], while Arriba-Mendez and colleagues have found that when the C allele is present in the silent SNP 927T/C, male children may be predisposed to asthma and atopic dermatitis [33]. Similarly, a silent SNP, 988C→T of the Fas gene has been shown to have significant implications in the development of papillary thyroid carcinoma [33].
It is well known that the development of SCC is strongly associated with smoking compared to AC [30] indicating that carcinogenic processes are different among the histological subtypes of lung cancers. Recently, arduous efforts have been made to differentiate these two types of lung cancers at a molecular level. For example, it was found that somatic p53 mutations occur more frequently in SCC than in AC [34]. In addition, individuals with AC and p53 somatic mutations have a shorter survival compared with SCC [30], and both LOH and somatic mutation patterns of the P53 gene among AC and SCC patients are distinctly different [35]. In another study, comparing gene expression profiles by microarray analysis demonstrated that different subtypes of NSCLCs exhibit substantially different molecular signatures which recapitulate distinctive lung developmental pathways known to be associated with respective subtypes [3]. Taken together, it can be hypothesized that inheritance of certain polymorphic gene alleles may not only predispose individuals to develop lung cancer but also contribute to the development of specific subtypes of cancer. However, still very little is known about genetic polymorphisms related to development of specific subtypes of lung cancers. The identification of disparate genetic pathways leading to specific lung tumour histological classes is critical for future approaches of developing improved diagnostic biomarkers and chemoprevention as well as individualized therapeutics customized to reflect histological subtype-specific genetic alterations.
Because of the considerable benefits in developing SNP as a diagnostic screening tool over the conventional polymorphic markers, lung cancer researchers have recently validated that SNP array can discern LOH patterns with a high degree of concordance to previous microsatellite analyses in the same cancer samples [36]. In addition, relevant to the current study, SNP arrays are able to cluster lung cancer samples to different subtypes based on their shared regions of LOH [37]. Further, high-density SNP arrays can be used to infer chromosomal copy number changes (i.e. gains or losses) in tumour cells with a high degree of accuracy [26]. Although in our study based on sequencing technique there were only two patient samples (out of 17) that we could convincingly conclude occurrence of LOH in the tumor cells, H37- C1138T and C2185T have good potential to serve as a LOH marker for the 3p21.3 region by the SNP array strategy and/or quantitative RT-PCR. Early detection of 3p21.3 LOH will be crucial as it is prevalent at the earliest stage of lung cancer development and renders poorer prognosis.
5. Conclusion
Taken together, the H37 SNP C1138T and C2185T from the 3p21.3 lung cancer tumour suppressor region have good potential to be associated with genetic elements causing different subtypes of NSCLC and may be useful for future strategy of utilizing the SNP array for detection of LOH at 3p21.3. Given the significant importance of the 3p21.3 region for implication of lung cancer and for future development of cancer diagnostics/therapeutics, further studies using a lager number of samples are warranted to validate true biologic correlation between the C1138T/C2185T allele status and the NSCLC subtypes.
Acknowledgments
We thank Drs. Yan Cui and He-Jing Wang for their generous help with statistical analysis in this study.
Grant support: Developmental Research Program Award from the University of California at Los Angeles Lung Cancer Specialized Programs of Research Excellence grant P50C A90388 (J.J. Oh), American Lung Association of California Research Grant (J.J. Oh), and the Wolfen Family Lung Cancer Clinical/Translational Research Program at University of California at Los Angeles Jonsson Comprehensive Cancer Center.
Footnotes
Conflict of interest statement
None of the authors in this study has any financial or personal relationships with other people or organizations that could inappropriately influence their work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Landis SH, Murray T, Bolden S, Wingo PA. Cancer statistics, 1999. CA Cancer J Clin. 1999;49(1):8–31. 1. doi: 10.3322/canjclin.49.1.8. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.Borczuk AC, Gorenstein L, Walter KL, Assaad AA, Wang L, Powell CA. Non-small-cell lung cancer molecular signatures recapitulate lung developmental pathways. Am J Pathol. 2003;163(5):1949–60. doi: 10.1016/S0002-9440(10)63553-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Travis WDCT, Corrin B, Shimosato Y, Brambilla E. World Health Organization International Histological Classification of Tumours. New York: Springer-Verlag; 1999. Histological typing of lung and pleural tumors. [Google Scholar]
- 5.Rehm S, Lijinsky W. Squamous metaplasia of bronchiolar cell-derived adenocarcinoma induced by N-nitrosomethyl-n-heptylamine in Syrian hamsters. Vet Pathol. 1994;31(5):561–71. doi: 10.1177/030098589403100508. [DOI] [PubMed] [Google Scholar]
- 6.Tatsuro Okamoto RM, Suemitsu Ryuichi, Aoki Yoshiro, Wataya Hiroshi, Kojo Miyako, Ichinose Yukito. Prognostic value of the histological subtype in completely resected non-small cell lung cancer. Interact CardioVasc Thorac Surg. 2006;5:362–6. doi: 10.1510/icvts.2005.125989. [DOI] [PubMed] [Google Scholar]
- 7.Lerman MI, Minna JD. The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: identification and evaluation of the resident candidate tumor suppressor genes. The International Lung Cancer Chromosome 3p21.3 Tumor Suppressor Gene Consortium. Cancer Res. 2000;60(21):6116–33. [PubMed] [Google Scholar]
- 8.Ji L, Minna JD, Roth JA. 3p21.3 tumor suppressor cluster: prospects for translational applications. Future Oncol. 2005;1(1):79–92. doi: 10.1517/14796694.1.1.79. [DOI] [PubMed] [Google Scholar]
- 9.Wistuba II, Lam S, Behrens C, et al. Molecular damage in the bronchial epithelium of current and former smokers. J Natl Cancer Inst. 1997;89(18):1366–73. doi: 10.1093/jnci/89.18.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X, Zhao Y, Honn SE, et al. Benzo[a]pyrene diol epoxide-induced 3p21.3 aberrations and genetic predisposition to lung cancer. Cancer Res. 1998;58(8):1605–8. [PubMed] [Google Scholar]
- 11.Oh JJ, Razfar A, Delgado I, et al. 3p21.3 tumor suppressor gene H37/Luca15/RBM5 inhibits growth of human lung cancer cells through cell cycle arrest and apoptosis. Cancer Res. 2006;66(7):3419–27. doi: 10.1158/0008-5472.CAN-05-1667. [DOI] [PubMed] [Google Scholar]
- 12.Oh JJ, West AR, Fishbein MC, Slamon DJ. A candidate tumor suppressor gene, H37, from the human lung cancer tumor suppressor locus 3p21.3. Cancer Res. 2002;62(11):3207–13. [PubMed] [Google Scholar]
- 13.Scanlan MJ, Gordan JD, Williamson B, et al. Antigens recognized by autologous antibody in patients with renal-cell carcinoma. Int J Cancer. 1999;83(4):456–64. doi: 10.1002/(sici)1097-0215(19991112)83:4<456::aid-ijc4>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 14.Welling DB, Lasak JM, Akhmametyeva E, Ghaheri B, Chang LS. cDNA microarray analysis of vestibular schwannomas. Otol Neurotol. 2002;23(5):736–48. doi: 10.1097/00129492-200209000-00022. [DOI] [PubMed] [Google Scholar]
- 15.Edamatsu H, Kaziro Y, Itoh H. LUCA15, a putative tumour suppressor gene encoding an RNA-binding nuclear protein, is down-regulated in ras-transformed Rat-1 cells. Genes Cells. 2000;5(10):849–58. doi: 10.1046/j.1365-2443.2000.00370.x. [DOI] [PubMed] [Google Scholar]
- 16.Mourtada-Maarabouni M, Sutherland LC, Meredith JM, Williams GT. Simultaneous acceleration of the cell cycle and suppression of apoptosis by splice variant delta-6 of the candidate tumour suppressor LUCA-15/RBM5. Genes Cells. 2003;8(2):109–19. doi: 10.1046/j.1365-2443.2003.00619.x. [DOI] [PubMed] [Google Scholar]
- 17.Mourtada-Maarabouni M, Sutherland LC, Williams GT. Candidate tumour suppressor LUCA-15 can regulate multiple apoptotic pathways. Apoptosis. 2002;7(5):421–32. doi: 10.1023/a:1020083008017. [DOI] [PubMed] [Google Scholar]
- 18.Rintala-Maki ND, Sutherland LC. LUCA-15/RBM5, a putative tumour suppressor, enhances multiple receptor-initiated death signals. Apoptosis. 2004;9(4):475–84. doi: 10.1023/B:APPT.0000031455.79352.57. [DOI] [PubMed] [Google Scholar]
- 19.Sutherland LC, Edwards SE, Cable HC, et al. LUCA-15-encoded sequence variants regulate CD95-mediated apoptosis. Oncogene. 2000;19(33):3774–81. doi: 10.1038/sj.onc.1203720. [DOI] [PubMed] [Google Scholar]
- 20.Sutherland LC, Lerman M, Williams GT, Miller BA. LUCA-15 suppresses CD95-mediated apoptosis in Jurkat T cells. Oncogene. 2001;20(21):2713–9. doi: 10.1038/sj.onc.1204371. [DOI] [PubMed] [Google Scholar]
- 21.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33(1):49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 22.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reich DE, Gabriel SB, Altshuler D. Quality and completeness of SNP databases. Nat Genet. 2003;33(4):457–8. doi: 10.1038/ng1133. [DOI] [PubMed] [Google Scholar]
- 24.Sachidanandam R, Weissman D, Schmidt SC, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409(6822):928–33. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- 25.Solinas-Toldo S, Lampel S, Stilgenbauer S, et al. Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer. 1997;20(4):399–407. [PubMed] [Google Scholar]
- 26.Zhao X, Li C, Paez JG, et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 2004;64(9):3060–71. doi: 10.1158/0008-5472.can-03-3308. [DOI] [PubMed] [Google Scholar]
- 27.Amos CI, Xu W, Spitz MR. Is there a genetic basis for lung cancer susceptibility? Recent Results Cancer Res. 1999;151:3–12. doi: 10.1007/978-3-642-59945-3_1. [DOI] [PubMed] [Google Scholar]
- 28.Kimchi-Sarfaty C, Oh JM, Kim IW, et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315(5811):525–8. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 29.Malhotra AK. Candidate gene studies of antipsychotic drug efficacy and drug-induced weight gain. Neurotox Res. 2004;6(1):51–6. doi: 10.1007/BF03033296. [DOI] [PubMed] [Google Scholar]
- 30.Basolo F, Giannini R, Faviana P, et al. Thyroid papillary carcinoma: preliminary evidence for a germ-line single nucleotide polymorphism in the Fas gene. J Endocrinol. 2004;182(3):479–84. doi: 10.1677/joe.0.1820479. [DOI] [PubMed] [Google Scholar]
- 31.Polesskaya OO, Sokolov BP. Differential expression of the “C” and “T” alleles of the 5-HT2A receptor gene in the temporal cortex of normal individuals and schizophrenics. J Neurosci Res. 2002;67(6):812–22. doi: 10.1002/jnr.10173. [DOI] [PubMed] [Google Scholar]
- 32.Jones A, Dhamrait SS, Payne JR, et al. Genetic variants of angiotensin II receptors and cardiovascular risk in hypertension. Hypertension. 2003;42(4):500–6. doi: 10.1161/01.HYP.0000088853.27673.D0. [DOI] [PubMed] [Google Scholar]
- 33.Arriba-Mendez S, Sanz C, Isidoro-Garcia M, et al. 927T>C polymorphism of the cysteinyl-leukotriene type-1 receptor (CYSLTR1) gene in children with asthma and atopic dermatitis. Pediatr Allergy Immunol. 2006;17(5):323–8. doi: 10.1111/j.1399-3038.2006.00416.x. [DOI] [PubMed] [Google Scholar]
- 34.Liu G, Miller DP, Zhou W, et al. Differential association of the codon 72 p53 and GSTM1 polymorphisms on histological subtype of non-small cell lung carcinoma. Cancer Res. 2001;61(24):8718–22. [PubMed] [Google Scholar]
- 35.Marchetti A, Pellegrini S, Sozzi G, et al. Genetic analysis of lung tumours of non-smoking subjects: p53 gene mutations are constantly associated with loss of heterozygosity at the FHIT locus. Br J Cancer. 1998;78(1):73–8. doi: 10.1038/bjc.1998.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindblad-Toh K, Tanenbaum DM, Daly MJ, et al. Loss-of-heterozygosity analysis of small-cell lung carcinomas using single-nucleotide polymorphism arrays. Nat Biotechnol. 2000;18(9):1001–5. doi: 10.1038/79269. [DOI] [PubMed] [Google Scholar]
- 37.Janne PA, Li C, Zhao X, et al. High-resolution single-nucleotide polymorphism array and clustering analysis of loss of heterozygosity in human lung cancer cell lines. Oncogene. 2004;23(15):2716–26. doi: 10.1038/sj.onc.1207329. [DOI] [PubMed] [Google Scholar]