

Abstract
During endochondral ossification, regulation of chondrocyte maturation governs the growth of the cartilage plate. The role of inorganic phosphate (Pi), whose levels strongly increase in the hypertrophic zone of the growth plate, on chondrocyte maturation has not yet been deciphered. Using the chondrogenic ATDC5 cells, we investigated the effects of Pi on cell maturation and on mineralization of the extracellular matrix. Pi induces matrix mineralization identical to that observed in growth plate cartilage and increases expression of the hypertrophic marker, collagen X. When calcium concentration is slightly increased (like in cartilage growth plate), Pi also stimulates apoptosis of differentiated ATDC5 cells, with a decrease in Bcl-2/Bax mRNA ratio, DNA fragmentation, characteristic morphological features and caspase-3 activation. All these effects are dependent on Pi entry into cells through sodium-dependent transporters. Finally, inhibition of apoptosis with ZVAD-fmk reduces Pi-induced mineralization. These findings suggest that Pi regulates chondrocyte maturation and apoptosis, which, in turn, controls skeletal development.
Keywords: Animals; Apoptosis; drug effects; Calcification, Physiologic; drug effects; Caspases; metabolism; Cell Differentiation; drug effects; Cell Line, Tumor; Chondrocytes; cytology; drug effects; enzymology; metabolism; Mice; Phosphates; pharmacology; RNA, Messenger; genetics; metabolism; Signal Transduction; drug effects
Introduction
Vertebrate long bones form through a process called endochondral ossification, in which a cartilage template is replaced by a bony matrix. This process begins with the condensation of undifferentiated mesenchyme, from the core of which cells differentiate into chondrocytes, whereas cells at the periphery form the perichondrium. After a phase of proliferation, chondrocytes differentiate expressing type II, type IX and type XI collagen and sulfated glycosaminoglycans. Then chondrocytes further differentiate and become hypertrophic, expressing mainly type X collagen. Finally, they release matrix vesicles (MV) that accumulate calcium and phosphorus and are likely involved in mineralization of the extracellular matrix (ECM). Changes in the composition of the cartilage matrix in the hypertrophic zone allow invasion by capillaries, degradation of the cartilage matrix and its replacement by the trabecular bone matrix secreted by invading osteoblasts. The fate of hypertrophic chondrocytes is still a matter of debate. The first hypothesis is that chondrocytes are converted into bone cells through a process called trans-differentiation.1,2 However, there is growing evidence that terminally differentiated chondrocyte undergo programmed cell death.3 In situ investigation of the cartilage growth plate revealed that hypertrophic chondrocytes are characterized by a loss of mitochondrial membrane potential,4 by a decrease in Bcl-2/Bax protein ratio,5 and are TUNEL positive.6–8 In addition, deletion of the gene encoding bcl-2 in mice leads to accelerated maturation of chondrocytes and shortening of long bones.5
The growth of a skeletal element depends on precise regulation of chondrocyte proliferation, differentiation and apoptosis. Among the factors modulating chondrocyte maturation, much attention has been paid to the parathyroid hormone-related peptide/indian hedgehog axis,9 bone morphogenic proteins,10 and oxygen supply.8 Surprisingly, whereas inorganic phosphate (Pi) levels strongly increase both in the ECM and in the cells from the proliferative to the hypertrophic region of the growth plate,11–14 the possible role of Pi on chondrocyte maturation and endochondral ossification has not been fully investigated. Disorders in Pi homeostasis lead to abnormal endochondral ossification. On the one hand, inefficient re-absorption of phosphate by kidney leading to hyphosphatemia is associated with defective mineralization of the skeleton. On the other hand, phosphate retention or hyperphosphatemia, accompanying chronic renal disease, is associated with soft tissue calcification and abnormal bone metabolism.15 Pi has been suggested to be rate-limiting for cartilage mineralization,16 and induces or stimulates mineralization in several models of chondrocyte culture.17,18 Pi has also recently been reported to modulate cell differentiation and apoptosis. Pi was shown to increase transcription and/or synthesis of type X collagen in chondrocytes,19,20 osteopontin in osteoblasts,21 and core binding factor αl (Cbfal) in smooth muscle cells.22 Pi also induces chondrocyte and osteoblast apoptosis,23 which involves loss of mitochondrial membrane permeability transition,24 and caspase activation.25 Moreover, Pi-induced apoptosis is dependent on Pi entry into cells,24,26 and is modulated by extracellular calcium (Ca) levels.25
Despite this large body of evidence indicating that Pi stimulates chondrocyte differentiation, mineralization and apoptosis, no study has precisely explored the relationships between these effects and the role of Pi during endochondral ossification remains unclear. In an elegant paper, Proudfoot et al. suggested that in hyperphosphatemic conditions, Pi induced mineralization of vascular smooth muscle cells through apoptotic-dependent mechanisms.27 Relationships between apoptosis and mineralization both in physiological and pathological situations have already been pointed out,28 and in growth plate cartilage, it was reported that matrix vesicles (MV) share several characteristics with apoptotic bodies.23,28,29 In this context, the aims of the present study were to investigate the role of Pi on chondrocyte maturation during endochondral ossification, and to elucidate the possible relationships between differentiation, apoptosis and mineralization using the ATDC5 cell line. This cell line, established by Atsumi et al. from the teratocarcinoma cells AT805,30 expresses the full range of events described for differentiation of epiphyseal chondrocytes and calcifies the ECM without addition of an exogenous phosphate source.31 This work is an effort to provide new insight in cartilage physiopathology and understand the role of apoptosis during organogenesis of mineralized tissues.
Results
As previously described,30 ATDC5 cells proliferate to reach confluence between day 5 and day 7. In the presence of insulin, they begin then to form nodules in which they differentiate,31 expressing type II collagen, link-protein and sulfated glycosaminoglycans. From day 12–15, cells in the nodules express type X collagen, become hypertrophic and from day 29 mineralize their ECM. In the present study effects of Pi were investigated on day 21 in differentiated cells, at a stage when mineralization has not yet started.
Pi induces mineralization in ATDC5 cultures
We first asked whether Pi could initiate mineralization by hypertrophic cells as early as on day 21. ATDC5 cells were grown for the first 21 days in DMEM/F12 (which contains 1 mM Ca and no ascorbic acid) and were treated on day 21 in α-MEM (which contains 2.4 mM Ca and ascorbate), as initially described for ATDC5 differentiation.31 Mineralization was quantified by measurement of Ca content by atomic absorption spectrometry and expressed as μg Ca per mg protein in cell layers. In these conditions, Pi dose-dependently stimulated Ca deposition in 7 days (Fig. 1a). The time course of Ca deposition indicated that mineralization with 4 mM Pi begins within 8 hours and strongly increases after 24 and 48 hours (Fig. 1b). Ca deposition induced by 4 mM Pi appeared to be dependent on Pi entry into cells since phosphonoformic acid (PFA), an inhibitor of sodium-dependent phosphate transporters (NaPiTs),32,33 blocked mineralization (Fig. 1c). Finally, we questioned whether Pi could initiate mineralization in DMEM/F12, which contains no ascorbate and only 1 mM Ca. In these conditions, while ascorbic acid was not necessary (data not shown), Pi-induced mineralization required 2.5 mM Ca, which comes close to Ca levels present in α-MEM (Fig. 2d).
Fig. 1. Pi-stimulated calcium deposition in cell layers.
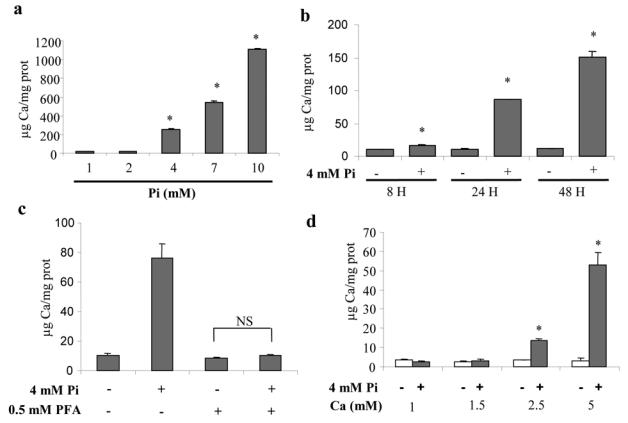
Cells were cultured in DMEM/F12 (1 mM Pi, 1 mM Ca) for 21 days before treatment. Calcium deposition in cell layers was determined by spectrometry, as detailed in the materials and methods section, (a) Calcium deposition in cell layers after 7 days of treatment with indicated Pi concentrations in α-MEM (1 mM Pi, 2.4 mM Ca). (b) Time course of calcium deposition after treatment with 4 mM Pi in α-MEM. (c) Effects of 0.5 mM PFA on calcium deposition in cells treated with 4 mM Pi for 24 hours in α-MEM. PFA was added 30 minutes before Pi treatment, (d) Effects of calcium concentration in DMEM/F12 on calcium deposition. *p<0.05 compared with control without Pi and Ca supplementation. **p<0.05 compared with 4 mM Pi treated cells. NS: not significant.
Fig. 2. Pi-stimulated ECM mineralization.
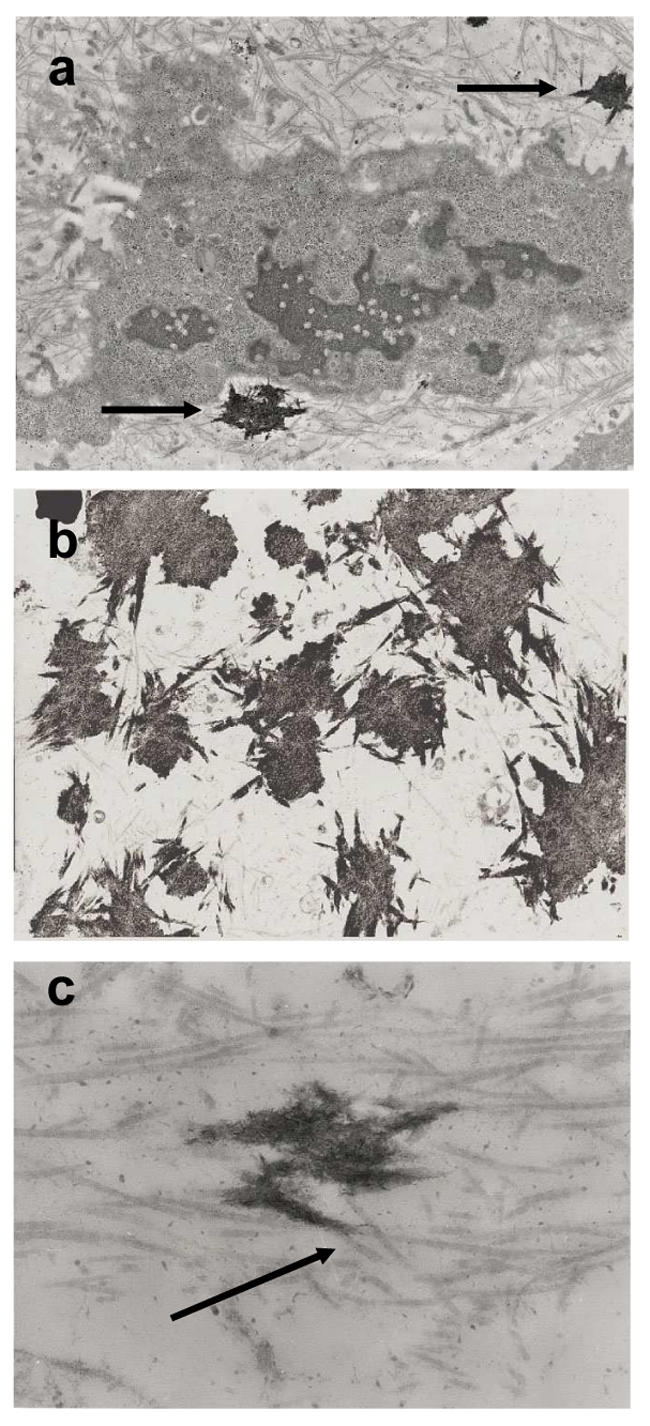
ATDC5 cells were cultured for 21 days in DMEM/F12 and treated for 7 days with 4 mM Pi in α-MEM (2.4 mM Ca). Calcified nodule sections were observed by TEM, as detailed in the materials and methods section, (a; × 5,000) Note the localization of crystals in ECM (arrows), (b; × 6,000) Mineralized areas join together along collagen fibrils, (c; × 25,000) Crystals are associated with collagen fibrils (arrow).
Pi-induced mineralization is identical to that in cartilage growth plate
Mineralization at the different Pi concentrations was first analyzed by transmission electron microscopy (TEM) after 8 days treatment in α-MEM (Fig. 2). With 4 mM Pi, crystals were deposited in the ECM (Fig. 2a), in association with collagen fibrils (Fig. 2b). TEM micrographs showed thin and relatively long crystals associated parallel to the long axis of collagen fibrils, consistent with physiological mineralization (Fig. 2c). In contrast, with higher Pi concentrations, non-physiologic crystal deposition was observed, probably due to a mere physicochemical precipitation (data not shown). Thus crystal composition and maturation in vitro with 4 mM Pi (Fig. 3a) were investigated by Fourier transform infrared microspectroscopy (FTIR-M) and compared with those of crystals in murine growth plate cartilage ex vivo (Fig. 3b). After deconvolution between 1200 and 900 cm−1 of the ν1ν3 PO4 domain (which gives information on crystal composition and organization), the high intensity of the bands at 1145, 1125, and 1112 cm−1 indicated that crystals both in vitro and ex vivo contain great amounts of HPO42− ions and non-apatitic PO43− ions (Figs 3c and 3d). This suggests that crystals formed in vitro and ex vivo are poorly crystalline and may contain weakly organized regions. These crystals have also a very poor if not nil carbonate content as revealed by the weak intensity of the ν2CO3 domain (data not shown). During maturation, crystals in vitro and ex vivo evolved similarly toward a greater crystallinity with a loss of HPO42− (1145 cm−1) and non-apatitic PO43− ions (1125 and 1112 cm−1).
Fig. 3. Crystal composition and maturation in vitro and ex vivo,
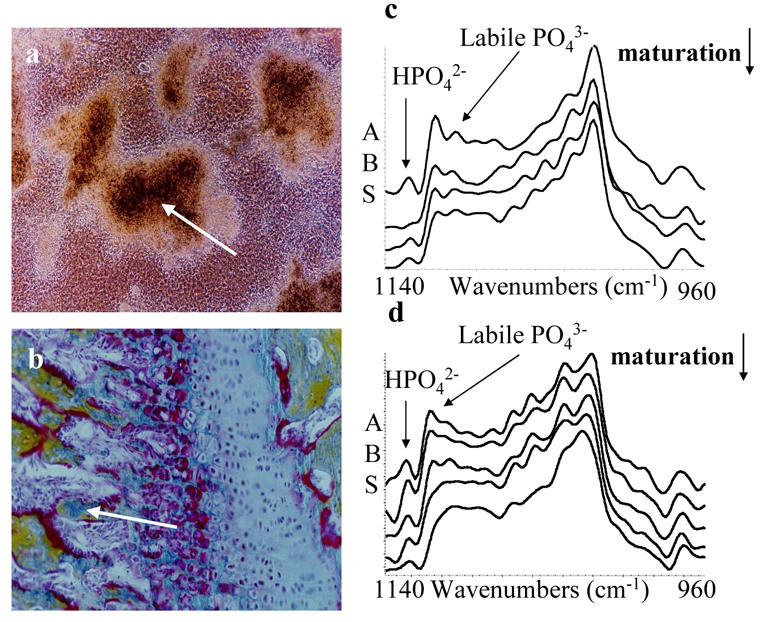
(a) Nodules in ATDC5 cell culture grown for 21 days in DMEM/F12 before treatement with 4 mM Pi in α-MEM for 7 days, (b) Murine cartilage growth plate stained with Movat’s pentachrome. Crystal maturation, i.e. evolution of crystal composition during aging, from the periphery to the center of a mineralizing nodule in culture (a; arrrow) and from the mineralizing septae to the junction with newly formed bone in unstained cartilage growth plate (b; arrow), was monitored by FTIR microspectroscopy (respectively c and d). Spectra in the ν1ν3PO4 domain between 1200 and 900 cm−1 were deconvoluted as described in the materials and methods.
Consequently, since TEM and FTIR-M experiments indicate that a 4 mM Pi concentration reproduces in vitro the cartilage mineralization observed in cartilage growth plate, subsequent experiments were conducted using this concentration.
Pi stimulates chondrocyte terminal differentiation
The effects of Pi on ATDC5 differentiation were investigated by RT-PCR (Fig. 4). Cells were incubated on day 20 in DMEM/F12 without insulin containing 0.5% FCS and treated on day 21 with 4 mM Pi. Results show that Pi increased type X collagen mRNA levels whereas it does not increase mRNA levels for osteopontin (OPN) (Fig. 4a). The effect of Pi on collagen X expression was inhibited by 0.5 mM PFA suggesting that Pi enters cells via NaPiTs to increase type X collagen mRNA levels (Fig. 4b). To investigate whether Pi-stimulated collagen X production was involved in mediating the effect of Pi on mineralization, we measured Ca deposition after treatment with Pi for 48 hours in the presence of the translation inhibitor cycloheximide. In these conditions, cycloheximide not only failed to inhibit mineralization, but even enhanced Ca deposition induced by 4 mM Pi and 2.4 mM Ca (Fig. 4c). Since cycloheximide is also an apoptogen, we next investigated the effects of Pi on apoptosis.
Fig. 4. Pi-stimulated collagen X mRNA expression and effect of cycloheximide on ECM mineralization.
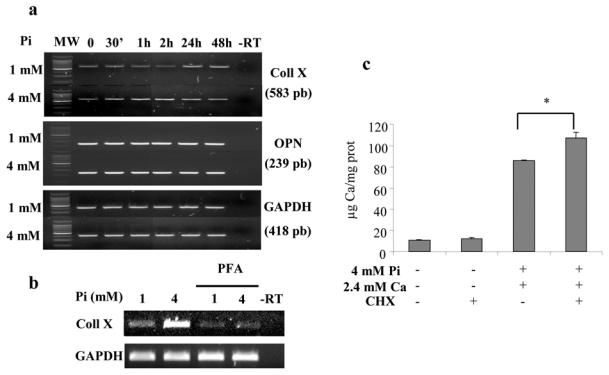
ATDC5 cells were cultured for 21 days in DMEM/F12 before treatment. (a) The effect of 4 mM Pi on collagen X (Coll X) and osteopontin (OPN) mRNA levels was assessed by RT-PCR, as described in materials and methods. A representative agarose gel is shown. (b) The effect of the Pi transport inhibitor PEA (0.5 mM) on collagen X mRNA expression induced by 4 mM Pi was assessed by RT-PCR. PEA was added 30 minutes before treatment with Pi for 2 hours. A representative agarose gel is shown. MW: molecular weight marker; −RT: control without reverse transcriptase. (c) Effects of the protein translation inhibitor, cycloheximide (CHX, 5 μM) on Pi-induced mineralization. CHX was added 30 minutes before treatment with Pi and Ca for 48 hours. Calcium deposition in cell layers was determined by spectrometry as detailed in the materials and methods section. *p<0.05 compared with ion pair treated cells.
Pi triggers cell death of differentiated chondrocytes
The effect of Pi on cell viability was assessed by measuring the mitochondrial conversion of MTS into formazan. Figure 5 shows that, if Pi alone had no effect on cell viability on day 21, it dramatically reduced MTS activity when added together with 2.4 mM Ca (Fig. 5a). Cycloheximide used alone only slightly decreased cell viability, but increased the effects of Pi and Ca on ATDC5 cell death. Inhibition of cellular Pi uptake with 0.5 mM PFA blocked cell death induced by Pi and Ca, suggesting that cell death is dependent on Pi entry into ATDC5 cells (Fig. 5b). Similar experiments realized with undifferentiated ATDC5 cells, cultured for only 7 days, indicated that in the absence of nodules, Pi and Ca fail to stimulate cell death (data not shown). Confocal microscopy was used to localize living and dead cells within the nodules after 48 hours treatment with 4 mM Pi and 2.4 mM Ca. Cultures were treated with CTG and EthD-1, which respectively stain living cells in green and dead cells in red, and pictures were collected from the top to the bottom of the nodules (Fig. 5c). In both treated and untreated cultures, dead cells were found essentially in the center of the nodule (differentiated cells), whereas viable cells were mostly observed in the nodule periphery (undifferentiated cells). The proportion of dead cells in nodules was markedly increased in cells treated with 4 mM Pi as compared to untreated cells (data not shown).
Fig. 5. Pi effects on ATDC5 cell viability.
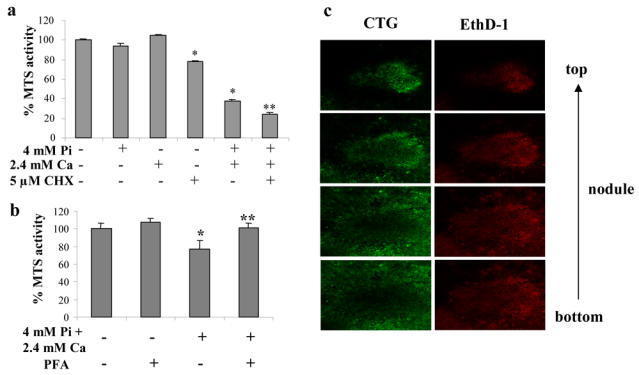
ATDC5 cells were cultured for 21 days in DMEM/F12 before treatment. (a) ATDC5 cell viability was assessed by MTS activity after 48 hours treatment with either 4 mM Pi, 2.4 mM Ca or both, in the presence or absence of 5 μM cycloheximide (CHX). (b) Effect of the Pi transport inhibitor PEA (0.5 mM) on MTS activity after treatment with 4 mM Pi and 2.4 mM Ca. PEA was added 30 minutes before treatment with 4 mM Pi and 2.4 mM Ca for 48 hours, (c; x160) Confocal microscopic localization of viable cells, stained with CTG, and dead cells, stained with EthD-1, in a nodule after treatment with 4 mM Pi and 2.4 mM Ca for 48 hours *p<0.05 compared with control. **p<0.05 compared with ion pair treated cells.
ATDC5 cell death is apoptotic
To examine whether cell death in response to Pi occurs through an apoptotic process, biochemical and morphological features of apoptosis were investigated. Treatment of ATDC5 cells for 24 hours with Pi and/or Ca affected the expression levels of mRNAs encoding for bcl-2 and bax, which are respectively anti- and pro-apopotic proteins. Densitometric analysis of agarose bands indicated that the value of bcl-2/bax ratio is reduced after treatment with both Pi and Ca (Fig. 6). On the other hand, treatment with Pi or Ca alone had weak or no effects on the bcl-2/bax ratio.
Fig. 6. Apoptotic features of ion pair induced-cell death.
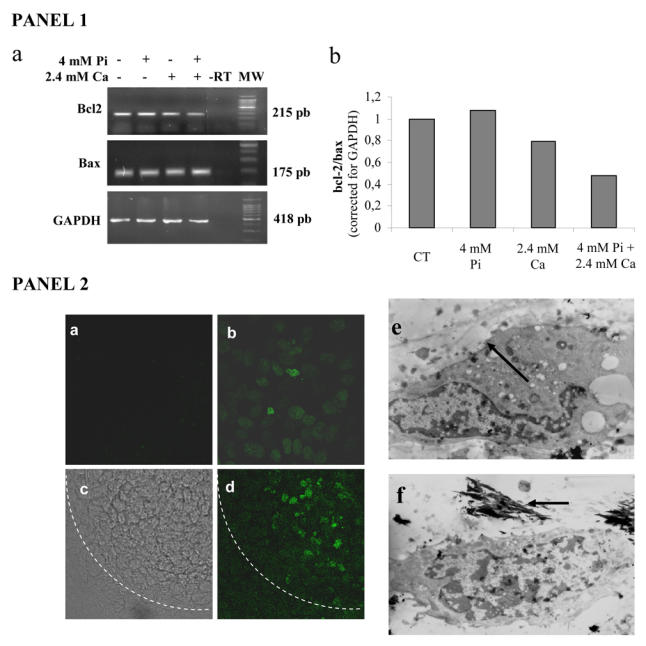
ATDC5 cells were cultured for 21 days in DMEM/F12 before treatment. PANEL 1: (a) The effects of 24 hours treatment with 4 mM Pi and/or 2.4 mM Ca on bcl-2 and bax mRNA expression were investigated by RT-PCR, as described in materials and methods. A representative agarose gel is shown, (b) Densitometric representation after normalisation to GAPDH levels (MW: molecular weight; −RT: control without reverse transcriptase). PANEL 2: (a–d) Effects of treatment with 4 mM Pi and 2.4 mM Ca for 48 hours on ATDC5 DNA fragmentation as determined by TUNEL. (a) Negative control (without enzyme; × 630). (b) Positive control (DNAse treatment; × 630). Nodule observation in light microscopy (c) and fluorescence (d; × 160). The dotted line indicates the nodule periphery, (e–f) ATDC5 cells were treated with 4 mM Pi for 7 days in a-MEM (2.4 mM Ca). The TEM micrographs shows chromatin condensation (e; arrow) and mineralized collagen fibrils (f; arrow).
DNA fragmentation in Pi and Ca treated cells was investigated by the TUNEL method. Figure 6 shows that only Pi and Ca treated cells in the center of the nodules were stained. The negative control without TUNEL enzyme was only faintly stained and cells treated with DNase (positive control) stained positively, independently of their localization. Cell morphology was then assessed by TEM after 7 days of treatment with 4 mM Pi in α-MEM (Fig. 6). Only individual cells, surrounded by dense collagen framework or located in proximity of mineralized areas exhibited typical apoptotic morphology with chromatin condensation at the periphery of the nucleus. Moreover, the cells exhibited a loss of organelle structure and vacuolation.
Implication of caspases in Pi stimulated apoptotis and mineralization
Finally, implication of caspases in Pi induced cell death was investigated by staining ATDC5 cells with an anti active caspase-3 antibody (Fig. 7). Positive staining was observed specifically within the nodules. Cells that stained positively for active caspase-3 (Fig. 7a) also stained positive for EthD-1 (Fig. 7b), indicating that they had already lost part of their viability. Merging of the images shown in figures 7a and 7b showed condensed chromatin stained with EthD-1 and cytoplasmic localization of active caspase-3 (Fig. 7c). The negative control showed only a barely noticeable green staining (Figure 7d). Involvement of caspases in Pi induced cell death is further suggested by the fact that ZVAD-fmk, a broad range caspase inhibitor, blocked the decrease in cell viability induced by Pi and Ca (Fig. 7e). Finally, we investigated whether modulating apoptosis by caspase inhibition might affect mineralization. Inhibition of apoptosis with ZVAD-fmk significantly decreased mineralization induced by Pi and Ca (Fig. 7f), suggesting that mineralization is at least partially dependent on caspase activity.
Fig. 7. Implication of caspases in ATDC5 cell apoptosis and ECM mineralization.
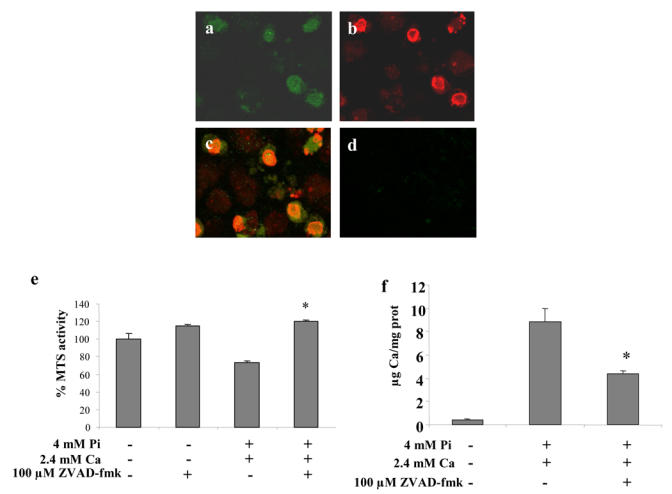
ATDC5 cells were cultured for 21 days in DMEM/F12 before 24 hours treatment with 4 mM Pi and 2.4 mM Ca. Confocal observation (× 630) of (a) anti-active caspase-3 immunostaining and (b) EThD-1 staining. (c) Merging of (a) and (b). (d) Negative control. (e–f) Effect of 100 μM ZVAD-fmk on (e) MTS activity and (f) mineralization. ZVAD-fmk was added 30 minutes before treatment with ions. *p<0.05 compared with ion pair treated cells.
Discussion
In the cartilage growth plate, Pi levels increase considerably from the proliferative to the hypertrophic region, both in cells and in the ECM.13,14 Whether increase in intracellular Pi concentration results from an active Pi transport from the ECM or from intracellular Pi generation is not yet fully understood. On the one hand, chondrocytes highly express NaPiTs that could account for high intracellular Pi levels;26,34 on the other hand, this high Pi content and the low ATP/Pi ratio reported in vivo,13 could result from hypoxic injury as it was reported in other cell types.35 In the poorly vascularized growth plate, the most hypoxic chondrocytes are localized in the hypertrophic zone.8 In the present study, we sought to explore the relationship between chondrocyte maturation and cartilage mineralization induced by Pi using the ATDC5 in vitro model of endochondral ossification.31 Among the reported models of chondrocyte differentiation, ATDC5 presently remains the only cell line expressing the full range of events described for differentiation of epiphyseal chondrocytes and the ability to mineralize spontaneously. Moreover, these cells are a convenient tool to study Pi effects on chondrocyte biology because like chondrocytes in vivo,34 they express type III NaPiTs, including glvr-1 and/or ram, both in the cells and in matrix vesicles.36 Finally, results of the present study demonstrate that mineralization in ATDC5 culture is identical to that in murine growth plate cartilage, indicating that this cell line is also suitable to study the mineralization process in vitro.
In in vitro models of chondrocyte cultures, an exogenous phosphate source, consisting of Pi20 or β-glycerophosphate (β-GP) salts,18,19 is frequently added to induce or stimulate mineralization. Although some studies have precisely investigated the composition of the mineral phase formed with varying concentrations of Pi or β-GP,37 Pi or β-GP are often used at concentrations far beyond the physiological range. In the present study for instance, if Pi dose and time-dependently increased Ca deposition in ATDC5 cell culture from 1 to 10 mM, concentrations above 4 mM led to non physiologic calcifications (data not shown). Ultra-structural and spectroscopic analyses with the different Pi concentrations demonstrated that a Pi level of 4 mM in the ATDC5 culture medium induces a mineralization identical to that in murine growth plate ex vivo. The crystals in vitro are deposited in the ECM. They are long-shaped,38 deposited parallel to the long axis of collagen fibrils. Moreover, this crystal formation is not merely due to Pi and Ca from the medium precipitating on collagen fibrils because inhibition of NaPiTs with PEA blocks Ca deposition, suggesting that Pi needs to enter cells or matrix vesicles.39 Crystal composition was analyzed by FTIR-M that associates FTIR spectroscopy to microscopy and allows crystal composition40 to be studied as a function of location.37 Both ex vivo and in vitro crystals consist in poorly organized apatite, contain large amounts of HPO42− and very few carbonate ions and evolve toward a greater crystallinity with a loss of acidic phosphate groups. These findings are consistent with previously published data on cartilage mineralization38,40 and suggest that 4 mM is an adequate concentration to study the in vitro effects of Pi. The same Pi concentration was reported to mimic in primary chondrocyte culture the in vivo mineralization.37,41
Pi reportedly induces or up-regulates expression of genes exclusively expressed in mineralizing cell types.19–21 For instance, Pi stimulates osteopontin gene transcription in osteoblastic MC3T3-E1 cells.21 Moreover, it was recently shown that Pi induced the osteoblastic phenotype in culture of smooth muscle cells, with induction of Cbfal expression.22,32 In ATDC5 cells, Pi specifically up-regulates mRNA levels for the late chondrogenic marker collagen X. Since collagen X is the only matrix protein expressed exclusively by hypertrophic chondrocytes, these results suggest that Pi accelerates terminal chondrocyte differentiation.19,20 In cartilage growth plate, the increase in Pi levels could thus play a key role during chondrocyte maturation. On the other hand, Pi does not seem to stimulate osteopontin transcription in ATDC5 cells as it was reported in osteoblastic MC3T3-E1 cells,21 suggesting that Pi stimulation of collagen X mRNA levels are specific and not merely due to stimulation of the basal transcriptional activity of hypertrophic chondrocytes. The precise molecular mechanisms involved by Pi in gene expression remain to be elucidated. These effects are at least dependent on Pi entry into cells because they are blocked by PFA,21,32 which was demonstrated to inhibit Pi uptake in the concentration range used in the present study.32,33 Moreover, these effects were dependent on the presence of sodium in culture medium, further arguing for intracellular uptake through NaPiTs.21 Today, specific signaling pathways potentially activated by Pi transport or by increasing intracellular Pi levels have not yet been reported and are under intense investigation.
Pi significantly stimulates apoptosis in the presence of 2.4 mM Ca, whereas separately Pi and Ca had no effect. Several studies reported that Pi induces chondrocyte or osteoblast apoptosis,4,23 but a recent report indicated that Pi-stimulated cell death was dramatically modulated by Ca levels.25 Pi-induced apoptosis in chondrocytes is dependent on Pi entry into cells, probably through type III and/or type II NaPiTs.26,34,36 On the other hand, it was reported that inhibitors of Ca transport fail to rescue cells from ion-pair treated cultures,25 suggesting an implication of Ca sensing receptors in chondrocyte maturation.42 It is unlikely that Pi and Ca induced cell death results from direct cellular toxicity, at the mitochondrial level for instance,43 because it is maturation stage dependent.23 ATDC5 apoptosis in our study was restricted to differentiated cells (within the nodules), and when Pi and Ca were added in the proliferation phase before the formation of nodules, there was no induction of apoptosis. The maturation stage dependency of Pi-induced apoptosis may be related to the fact that only hypertrophic chondrocytes contain large amounts of intracellular Ca,11,12,44 particularly within their mitochondria.45 An alternative possibility is that terminally differentiated chondrocytes undergo a maturation-dependent loss of mitochondrial function4 independent of Pi levels that, together with a decrease in levels of the cell death inhibitor bcl-25 would render cells more susceptible to Pi toxicity.4 In the cartilage growth plate, there is a decrease in the Bcl-2/Bax protein ratio from the proliferative to the hypertrophic zone,5 coinciding with the reported increase in Pi13,14 and Ca11,12,44 levels. In the present study, treatment with Pi and Ca led to a decrease in the Bcl-2/Bax ratio, which is believed to disrupt the mitochondrial membrane and promote release of mitochondrial components, irreversibly engaging the cell toward apoptosis. Among the mitochondrial components released during the apoptotic process, cytochrome c leads to subsequent activation of caspases. Accordingly, Pi and Ca treatment led to caspase-3 activation in differentiated ATDC5 cells. Co-localization of cytoplasmic active caspase-3 and nuclear EthD-1 staining indicated that active caspase-3 positive cells were no more viable. Activation of caspases by Pi and Ca ions was further reinforced by the fact that a broad-range caspase inhibitor (ZVAD-fmk) completely rescued cells from death. Caspase-3 is known to cleave the inhibitor of caspase-3 activated DNase (CAD) leading to CAD release that is responsible for oligonucleosome fragmentation (DNA laddering). In the present study, confocal microscopic observations of ion pair treated cells revealed that only the differentiated cells in nodules exhibited DNA fragmentation as determined by TUNEL, which was further confirmed by TEM observations that showed cells with characteristic apoptotic features, particularly chromatin condensation.
In the present ATDC5 in vitro model, Pi accelerates chondrocyte terminal differentiation, and together with Ca induces apoptosis and ECM mineralization. Mineralization does not seem to require de novo protein synthesis, since blocking protein translation with cycloheximide did not prevent Ca deposition. In contrast, results of the present study highlight the relationships between apoptosis and mineralization: Ca deposition is significantly enhanced by the apoptogen cycloheximide and reduced by the apoptosis inhibitor ZVAD-fmk. Moreover, TEM observation showed that apoptotic cells were almost always located in the vicinity of mineralized collagen matrix. Although a relationship between apoptosis and mineralization had already observed in other tissues in pathological conditions, such as vascular calcifications,27 this study is, to our knowledge, the first one to report dependence of mineralization on apoptosis in a model of endochondral ossification. Precise mechanisms of chondrocyte apoptosis induced by Pi and Ca and leading to mineralization remain largely unknown. It is likely that matrix vesicles (MV) play an important role in epiphyseal calcification46 and some studies have implied that apoptotic bodies, similarly to MV, could initiate pathological or physiological calcification.27–29,46 Moreover, it was reported that mitochondrial phosphate and Ca released during chondrocyte apoptosis are included into nascent MV and could participate to matrix mineralization.11,45,47,48 ZVAD-fmk could thus inhibit mineralization by blocking the release of apoptotic bodies from cells.49 Finally, it was reported that if Pi levels regularly increase from proliferative to hypertrophic regions of the growth plate,14 Ca levels are relatively constant in all zones of the plate, increasing only in hypertrophic zones, both in extracellular50 and intracellular compartments.11 Our data indicating that Pi is able to accelerate terminal chondrocyte differentiation on its own, but requires Ca to induce apoptosis-dependent mineralization might thus be of physiological relevance.
In conclusion, results presented in this study suggest an important role of Pi during endochondral ossification. Increased Pi levels from proliferative to hypertrophic regions may accelerate chondrogenic differentiation, and together with increased Ca levels in the hypertrophic zone induce apoptosis-dependent mineralization. Moreover, if apoptosis is believed to play an important role during organogenesis, this study suggests that programmed cell death is an essential process regulating skeletal growth.
Materials and Methods
Materials
Cell culture plasticware was purchased from Falcon (Becton-Dickinson, Franklin Lakes, NJ) and Corning-Costar (Integra Biosciences, Wallisellen, Switzerland). Fetal calf serum (FCS) was obtained from Dominique Dutscher (Brumath, France). α-MEM, glutamine, antibiotics, trypsin/ethylene-diamine tetraacetic acid (EDTA) were obtained from Life Technologies Ltd. (Paisley, UK). A 1:1 mixture of DMEM and Ham’s F12 medium (DMEM/F12) was provided by ICN Biochemicals (Orsay, France). PFA, cycloheximide, ascorbic acid, calcium chloride, dimethyl sulfoxide (DMSO), bovine insulin, transferrin and sodium selenite were purchased from Sigma (St Louis, MO). ZVAD-fmk was obtained from R&D Systems (Abingdon, UK). Cycloheximide and ZVAD-fmk were dissolved as concentrated solutions in DMSO.
DNase I and Taq DNA polymerase were obtained from Life Technologies. Avian myeloblastosis virus-reverse transcriptase (AMV-RT), random hexamers and recombinant ribonuclease inhibitor were purchased from Promega (Madison, WI). Trizol reagent was from Life technologies.
Cell tracker green (CTG) and ethidium homodimer 1 (EthD-1) were purchased from Molecular Probes (Leiden, The Netherlands). The in situ cell death detection kit, based on the TdT-mediated dUTP nick end labeling (TUNEL) was obtained from Roche Molecular Biochemicals (Meylan, France). Antibodies (BD PharMingen) were kindly provided by Dr. Lisa Valette. All other chemicals were from standard laboratory suppliers and were of the highest purity available.
Cell and culture conditions
ATDC5 cells were routinely grown in a maintenance medium consisting of DMEM/F12 (1:1) containing 5% FCS, 10 μg/mL human transferrin (T), 3 × 10−8 mol/L sodium selenite (S), 1% antibiotics, 1% glutamine. Cells were subcultured once a week using trypsin/EDTA, and maintained at 37°C in a humidified atmosphere of 5% CO2 in air. To induce chondrogenesis and cartilage nodule formation, ATDC5 cells (1.5 × 104/cm2) were seeded in a differentiation medium consisting of maintenance medium supplemented with 10 μ/mL of bovine insulin (I) for the first three weeks. When indicated, DMEM/F12 was replaced on day 21 by α-MEM containing 5% FCS and the ITS supplement at 37°C in a humidified atmosphere of 3% CO2 in air. Medium was replaced every second day. In these conditions, mineralization begins at day 29. DMEM/F12 and α-MEM contain 0.9 and 1 mM Pi respectively. Pi concentrations of 2, 4, 7 and 10 mM were obtained by addition of a mixture of NaH2PO4 and Na2HPO4 pH 7.3. DMEM/F12 and α-MEM contain 1 and 2.4 mM Ca respectively; higher Ca concentrations were obtained by adding calcium chloride. To reduce the non-specific effects of agonists present in FCS, the cells were incubated in low-serum (0.5 %) medium for 24 hours before stimulation with Pi (in all experiments excepted those in which cells were cultured for 7 days). All inhibitors were added 30 minutes before treatments with agonists.
RNA isolation
Cells were seeded in 25 cm2 flasks for RNA isolation. After indicated times, media were removed, cell layers rinsed with RNase free PBS and stored at −80°C until total RNA was extracted using the Trizol reagent according to the manufacturer’s instructions. Briefly, lysis of the cells in Trizol was followed by centrifugation at 10,000 g, 4°C, for 15 min in the presence of chloroform. The upper aqueous phase was collected and the RNA was precipitated by addition of isopropanol and centrifugation at 7,500 g, 4°C, for 5 minutes. RNA pellets were washed with cold 75% ethanol, dried, reconstituted with sterile water, and quantified by spectrometry.
Reverse transcription and polymerase chain reaction (RT-PCR) analysis
After DNase I digestion, RNA samples (2 μg) were reverse transcribed using AMV-RT and random hexamer primers in a total volume of 30 μL. Template cDNAs (5 μl) were then amplified in a typical 50 μL PCR reaction containing 20 mM Tris-HCl (pH 8.4), 50 mM KC1, 1 μM of the respective primers, 200 μM dNTP and 2.5 units of Taq DNA polymerase. The magnesium chloride concentration was 1.5 mM. The absence of DNA contamination in RNA preparations was tested by including RNA samples which had not been reverse-transcribed. Primers sequences were as follows: GAPDH, forward 5′ CAC CAT GGA GAA GGC CGG GG3′, reverse 5′ GAC GGA CAC ATT GGG GGT AG 3′ (55°C, 418 bp);51 type X collagen, forward 5′ CCA CCT GGG TTA GAT GGA AAA 3′, reverse 5′ AAT CTC ATC AAA TGG GAT GGG 3′ (56°C, 583 bp);52 osteopontin, forward 5′ ACA CTT TCA CTC CAA TCG TCC 3′, reverse 5′ TGC CCT TTC CGT TGT TGT CC 3′ (55°C, 239 bp);51 bcl-2, forward 5′ TGT GTG TGG AGA GCG TCA ACA 3′, reverse 5′ CCA GGT ATG CAC CCA GAG TGA T 3′ (60°C, 215 bp);53 bax, forward 5′ AGC TGC AGA GGA TGA TTG CT 3′, reverse 5′ GAT CAG CTC GGG CAC TTT AG 3′ (60°C, 175 bp).53 Amplifications were carried out in an Eppendorf master cycler (Dr. Vaudaux AG, Schonenbuch, Switzerland) under the following conditions: denaturation for 3 minutes at 94°C followed by cycles of 30 s denaturation at 94° C, 30 s annealing at the primer specific temperature and 45 s elongation at 72°C. All PCR results show amplification products obtained in the linear range of amplification. Semi-quantitative analysis of RNA levels and normalization to GAPDH levels were realized by densitometry (Leica Q500).
Confocal microscopy
Imaging was performed using a confocal laser-scanning inverted microscope (Leica TCS SP1, Heidelberg, Germany) equipped with an argon/krypton laser. Cells were visualized using a X 63/1.4 APO objective lens. The data were collected with a simultaneous dual channel detector and visualized with a 24-bit imaging system including Leica TCS NT software.
Briefly, for cell tracker green (CTG) and ethidium homodimer 1 (EthD-1) staining, cells were treated with 5 μM CTG for 30 minutes at 37°C, then for 30 minutes in FCS containing medium and finally for 30 minutes in 1 μM EthD-1 at room temperature. The fluorescence of CTG incorporated into living cells was detected using an isothiocyanate (FITC)-fluorescent set: λex = 488 nm; λem collected = 490–560 nm. The red EthD-1 emission was viewed using the confocal microscope (λex = 568 nm; λem collected = 570–700 nm).
For anti-active caspase-3 immunostaining, cells were fixed in 4% paraformaldehyde for 30 minutes at room temperature, permeabilized for 5 minutes on ice and treated with a rabbit anti-active caspase 3 antibody for 30 minutes at room temperature. After washing the cells, a anti-rabbit FITC-conjugated antibody was added for 30 minutes in obscurity. The fluorescence was detected using an isothiocyanate (FITC)-fluorescent set: λex = 488 nm; λem collected = 490–560 nm. Before incubation with antibodies, some coverslips were stained with EthD-1 as described above.
For DNA fragmentation staining, cells were fixed in 4% paraformaldehyde for 15 minutes at room temperature, permeabilized for 5 minutes on ice and treated by the TUNEL reagent for 1 hour at 37°C. All coverslips were mounted with Antifade kit (Molecular Probes). The green TUNEL emission was analyzed using an isothiocyanate (FITC)-fluorescent set as described above.
Fourier transform infrared microspectroscopy
ATDC5 nodules (6 multiwell plates) were fixed in 70 % ethanol and placed on a barium fluoride (BaF2) disk. The crystallinity was analyzed from the periphery to the center of the nodules to study the maturation of the mineral phase. In parallel, growth plate cartilage was studied as a control. Briefly, 1 week old mice were sacrificed, their tibiae were fixed in 70 % ethanol, dehydrated and embedded in glycolmethylmethacrylate. Sections (2 μm-thick) were cut with a Supercut 2050 microtome (Reichert-Jung, Heidelberg, Germany) and placed on a BaF2 disk. Spectra were recorded with a Magna-IR 550 spectrometer (Nicolet, Trappes, France) equipped with an IR-plan Advantage microscope (Spectra-Tech, Shelton, CT; X15 Reflachromat lens) fitted with a high-sensitivity mercury cadmium tellurite (MCT) detector. Sample positioning was realized with a motorized x-y stage under computer control. FTIR data were acquired with the spatial resolution provided by a 20 × 20 μm2 aperture in order to prevent diffraction artifacts and maximize the signal/noise ratio. Infrared spectra were recorded at 4 cm−1 resolution with 512 or 1024 interferograms co-added and Happ-Genzel apodization. Omnic software (Nicolet, Trappes, France) was used for data analysis. Residual H2O and CO2 absorptions were automatically subtracted. Before mineral analysis by deconvolution of the ν1ν3 PO4 domain (k=2.3 and σ=22.5 cm−1), collagen and proteoglycan absorptions were minimized by subtracting the spectrum non-mineralized regions.
Transmission electron microscopy
ATDC5 nodules (6 multiwell plates) were fixed in cacodylate buffered 4% glutaraldehyde for 15 minutes at 4°C, washed and post-fixed in cacodylate buffered 2% osmium tetroxide for 20 minutes at 4°C. Nodules were dehydrated in successive dilutions of ethanol and embedded overnight in Epon at 37°C and for two other days at 55°C. Sections (80 nm-thick) were cut with an Ultracut E ultramicrotome (Reichert-Jung, Heidelberg, Germany), mounted on copper grids, stained with uranyle acetate and observed on a Jeol 1010 electron microscope at a voltage of 100 kV.
Analytical methods
ECM mineralization was measured as the amount (μg) of total Ca present in cell layers (12 multiwell plates) was determined by atomic absorption spectrometry at 422.7 nm (Unicam 989 AA spectrometer, SOLAR) after extraction of the cells with 4 M HCl containing 1 % LaCl3. Protein content (mg) was determined with the Pierce Coomassie Plus assay reagent (Pierce, Rockford, IL). Cell survival (12 multiwell plates) was determined by measuring at 490 nm the formation of formazan from MTS tetrazolium with the CellTiter 96 Aqueus Non-radioactive cell proliferation assay (Promega, Wallisellen, Switzerland). Results are expressed as relative MTS activity. Each experiment was repeated at least once with similar results. Results are expressed as mean ± SEM of triplicate experiments. Comparative studies of means were performed using one way analysis of variance followed by a post-hoc test (Fisher’s projected least significant difference) with a statistical significance at p<0.05.
References
- 1.Cancedda R, Descalzi Cancedda F, Castagnola P. Chondrocyte differentiation. Int Rev Cytol. 1995;159:265–358. doi: 10.1016/s0074-7696(08)62109-9. [DOI] [PubMed] [Google Scholar]
- 2.Roach HI. New aspects of endochondral ossification in the chick: chondrocyte apoptosis, bone formation by former chondrocytes, and acid phosphatase activity in the endochondral bone matrix. J Bone Miner Res. 1997;12:795–805. doi: 10.1359/jbmr.1997.12.5.795. [DOI] [PubMed] [Google Scholar]
- 3.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajpurohit R, Mansfield K, Ohyama K, Ewert D, Shapiro IM. Chondrocyte death is linked to development of a mitochondrial membrane permeability transition in the growth plate. J Cell Physiol. 1999;179:287–296. doi: 10.1002/(SICI)1097-4652(199906)179:3<287::AID-JCP6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 5.Amling M, Neff L, Tanaka S, Inoue D, Kuida K, Weir E, Philbrick WM, Broadus AE, Baron R. Bcl-2 lies downstream of parathyroid hormone-related peptide in a signaling pathway that regulates chondrocyte maturation during skeletal development. J Cell Biol. 1997;136:205–213. doi: 10.1083/jcb.136.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gibson GJ, Kohler WJ, Schaffler MB. Chondrocyte apoptosis in endochondral ossification of chick sterna. Dev Dyn. 1995;203:468–476. doi: 10.1002/aja.1002030409. [DOI] [PubMed] [Google Scholar]
- 7.Hatori M, Klatte KJ, Teixeira CC, Shapiro IM. End labeling studies of fragmented DNA in the avian growth plate: evidence of apoptosis in terminally differentiated chondrocytes. J Bone Miner Res. 1995;10:1960–1968. doi: 10.1002/jbmr.5650101216. [DOI] [PubMed] [Google Scholar]
- 8.Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–2876. doi: 10.1101/gad.934301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 10.Enomoto-Iwamoto M, Iwamoto M, Mukudai Y, Kawakami Y, Nohno T, Higuchi Y, Takemoto S, Ohuchi H, Noji S, Kurisu K. Bone morphogenetic protein signaling is required for maintenance of differentiated phenotype, control of proliferation, and hypertrophy in chondrocytes. J Cell Biol. 1998;140:409–418. doi: 10.1083/jcb.140.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyde A, Shapiro IM. Energy dispersive X-ray elemental analysis of isolated epiphyseal growth plate chondrocyte fragments. Histochemistry. 1980;69:85–94. doi: 10.1007/BF00508369. [DOI] [PubMed] [Google Scholar]
- 12.Shapiro IM, Boyde A. Microdissection-elemental analysis of the mineralizing growth cartilage of the normal and rachitic chick. Metab Bone Dis Relat Res. 1984;5:317–326. doi: 10.1016/0221-8747(84)90019-5. [DOI] [PubMed] [Google Scholar]
- 13.Wuthier RE. Involvement of cellular metabolism of calcium and phosphate in calcification of avian growth plate cartilage. J Nutr. 1993;123:301–309. doi: 10.1093/jn/123.suppl_2.301. [DOI] [PubMed] [Google Scholar]
- 14.Mwale F, Tchetina E, Wu CW, Poole AR. The assembly and remodeling of the extracellular matrix in the growth plate in relationship to mineral deposition and cellular hypertrophy: an in situ study of collagens II and IX and proteoglycan. J Bone Miner Res. 2002;17:275–283. doi: 10.1359/jbmr.2002.17.2.275. [DOI] [PubMed] [Google Scholar]
- 15.Malluche HH, Monier-Faugere MC. Hyperphosphatemia: pharmacologic intervention yesterday, today and tomorrow. Clin Nephrol. 2000;54:309–317. [PubMed] [Google Scholar]
- 16.Hunter GK, Holmyard DP, Pritzker KP. Calcification of chick vertebral chondrocytes grown in agarose gels: a biochemical and ultrastructural study. J Cell Sci. 1993;104:1031–1038. doi: 10.1242/jcs.104.4.1031. [DOI] [PubMed] [Google Scholar]
- 17.Thomas JT, Boot-Handford RP, Grant ME. Modulation of type X collagen gene expression by calcium beta-glycerophosphate and levamisole: implications for a possible role for type X collagen in endochondral bone formation. J Cell Sci. 1990;95:639–648. doi: 10.1242/jcs.95.4.639. [DOI] [PubMed] [Google Scholar]
- 18.Bellows CG, Aubin JE, Heersche JN. Initiation and progression of mineralization of bone nodules formed in vitro: the role of alkaline phosphatase and organic phosphate. Bone Miner. 1991;14:27–40. doi: 10.1016/0169-6009(91)90100-e. [DOI] [PubMed] [Google Scholar]
- 19.Coe MR, Summers TA, Parsons SJ, Boskey AL, Balian G. Matrix mineralization in hypertrophic chondrocyte cultures. Beta glycerophosphate increases type X collagen messenger RNA and the specific activity of pp60c-src kinase. Bone Miner. 1992;18:91–106. doi: 10.1016/0169-6009(92)90850-d. [DOI] [PubMed] [Google Scholar]
- 20.Fujita T, Meguro T, Izumo N, Yasutomi C, Fukuyama R, Nakamuta H, Koida M. Phosphate stimulates differentiation and mineralization of the chondroprogenitor clone ATDC5. Jpn J Pharmacol. 2001;85:278–281. doi: 10.1254/jjp.85.278. [DOI] [PubMed] [Google Scholar]
- 21.Beck GR, Jr, Zerler B, Moran E. Phosphate is a specific signal for induction of osteopontin gene expression. Proc Natl Acad Sci U S A. 2000;97:8352–8357. doi: 10.1073/pnas.140021997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- 23.Mansfield K, Rajpurohit R, Shapiro IM. Extracellular phosphate ions cause apoptosis of terminally differentiated epiphyseal chondrocytes. J Cell Physiol. 1999;179:276–286. doi: 10.1002/(SICI)1097-4652(199906)179:3<276::AID-JCP5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 24.Meleti Z, Shapiro IM, Adams CS. Inorganic phosphate induces apoptosis of osteoblast-like cells in culture. Bone. 2000;27:359–366. doi: 10.1016/s8756-3282(00)00346-x. [DOI] [PubMed] [Google Scholar]
- 25.Adams CS, Mansfield K, Perlot RL, Shapiro IM. Matrix regulation of skeletal cell apoptosis. Role of calcium and phosphate ions. J Biol Chem. 2001;276:20316–20322. doi: 10.1074/jbc.M006492200. [DOI] [PubMed] [Google Scholar]
- 26.Mansfield K, Teixeira CC, Adams CS, Shapiro IM. Phosphate ions mediate chondrocyte apoptosis through a plasma membrane transporter mechanism. Bone. 2001;28:1–8. doi: 10.1016/s8756-3282(00)00409-9. [DOI] [PubMed] [Google Scholar]
- 27.Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;87:1055–1062. doi: 10.1161/01.res.87.11.1055. [DOI] [PubMed] [Google Scholar]
- 28.Kim KM. Apoptosis and calcification. Scanning Microsc. 1995;9:1137–1175. discussion 1175–1138. [PubMed] [Google Scholar]
- 29.Hashimoto S, Ochs RL, Rosen F, Quach J, McCabe G, Solan J, Seegmiller JE, Terkeltaub R, Lotz M. Chondrocyte-derived apoptotic bodies and calcification of articular cartilage. Proc Natl Acad Sci U S A. 1998;95:3094–3099. doi: 10.1073/pnas.95.6.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atsumi T, Miwa Y, Kimata K, Ikawa Y. A chondrogenic cell line derived from a differentiating culture of AT805 teratocarcinoma cells. Cell Differ Dev. 1990;30:109–116. doi: 10.1016/0922-3371(90)90079-c. [DOI] [PubMed] [Google Scholar]
- 31.Shukunami C, Ishizeki K, Atsumi T, Ohta Y, Suzuki F, Hiraki Y. Cellular hypertrophy and calcification of embryonal carcinoma-derived chondrogenic cell line ATDC5 in vitro. J Bone Miner Res. 1997;12:1174–1188. doi: 10.1359/jbmr.1997.12.8.1174. [DOI] [PubMed] [Google Scholar]
- 32.Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli CM. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:E10–17. doi: 10.1161/01.res.87.7.e10. [DOI] [PubMed] [Google Scholar]
- 33.Montessuit C, Caverzasio J, Bonjour JP. Characterization of a Pi transport system in cartilage matrix vesicles. Potential role in the calcification process. J Biol Chem. 1991;266:17791–17797. [PubMed] [Google Scholar]
- 34.Palmer G, Zhao J, Bonjour J, Hofstetter W, Caverzasio J. In vivo expression of transcripts encoding the Glvr-1 phosphate transporter/retrovirus receptor during bone development. Bone. 1999;24:1–7. doi: 10.1016/s8756-3282(98)00151-3. [DOI] [PubMed] [Google Scholar]
- 35.Marcussen M. Induction of cell surface blebbing by increased cellular Pi concentration. Biochem J. 1996;318:955–958. doi: 10.1042/bj3180955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guicheux J, Palmer G, Shukunami C, Hiraki Y, Bonjour JP, Caverzasio J. A novel in vitro culture system for analysis of functional role of phosphate transport in endochondral ossification. Bone. 2000;27:69–74. doi: 10.1016/s8756-3282(00)00302-1. [DOI] [PubMed] [Google Scholar]
- 37.Boskey AL, Guidon P, Doty SB, Stiner D, Leboy P, Binderman I. The mechanism of beta-glycerophosphate action in mineralizing chick limb-bud mesenchymal cell cultures. J Bone Miner Res. 1996;11:1694–1702. doi: 10.1002/jbmr.5650111113. [DOI] [PubMed] [Google Scholar]
- 38.Kim H, Rey C, Glimcher MJ. X-ray diffraction, electron microscopy, and Fourier transform infrared spectroscopy of apatite crystals isolated from chicken and bovine calcified cartilage. Calcif Tissue Int. 1996;59:58–63. doi: 10.1007/s002239900086. [DOI] [PubMed] [Google Scholar]
- 39.Wu LN, Ishikawa Y, Sauer GR, Genge BR, Mwale F, Mishima H, Wuthier RE. Morphological and biochemical characterization of mineralizing primary cultures of avian growth plate chondrocytes: evidence for cellular processing of Ca2+ and Pi prior to matrix mineralization. J Cell Biochem. 1995;57:218–237. doi: 10.1002/jcb.240570206. [DOI] [PubMed] [Google Scholar]
- 40.Rey C, Shimizu M, Collins B, Glimcher MJ. Resolution-enhanced Fourier transform infrared spectroscopy study of the environment of phosphate ion in the early deposits of a solid phase of calcium phosphate in bone and enamel and their evolution with age: 2. Investigations in the nu3PO4 domain. Calcif Tissue Int. 1991;49:383–388. doi: 10.1007/BF02555847. [DOI] [PubMed] [Google Scholar]
- 41.Boskey AL, Camacho NP, Mendelsohn R, Doty SB, Binderman I. FT-IR microscopic mappings of early mineralization in chick limb bud mesenchymal cell cultures. Calcif Tissue Int. 1992;51:443–448. doi: 10.1007/BF00296678. [DOI] [PubMed] [Google Scholar]
- 42.Chang W, Tu C, Pratt S, Chen TH, Shoback D. Extracellular Ca(2+)-Sensing Receptors Modulate Matrix Production and Mineralization in Chondrogenic RCJ3.1C5.18 Cells. Endocrinology. 2002;143:1467–1474. doi: 10.1210/endo.143.4.8709. [DOI] [PubMed] [Google Scholar]
- 43.Kowaltowski AJ, Castilho RF, Grijalba MT, Bechara EJ, Vercesi AE. Effect of inorganic phosphate concentration on the nature of inner mitochondrial membrane alterations mediated by Ca2+ ions. A proposed model for phosphate-stimulated lipid peroxidation. J Biol Chem. 1996;271:2929–2934. doi: 10.1074/jbc.271.6.2929. [DOI] [PubMed] [Google Scholar]
- 44.Gunter TE, Zuscik MJ, Puzas JE, Gunter KK, Rosier RN. Cytosolic free calcium concentrations in avian growth plate chondrocytes. Cell Calcium. 1990;11:445–457. doi: 10.1016/0143-4160(90)90077-8. [DOI] [PubMed] [Google Scholar]
- 45.Hargest TE, Gay CV, Schraer H, Wasserman AJ. Vertical distribution of elements in cells and matrix of epiphyseal growth plate cartilage determined by quantitative electron probe analysis. J Histochem Cytochem. 1985;33:275–286. doi: 10.1177/33.4.3980981. [DOI] [PubMed] [Google Scholar]
- 46.Anderson HC. Molecular biology of matrix vesicles. Clin Orthop. 1995:266–280. [PubMed] [Google Scholar]
- 47.Iannotti JP, Naidu S, Noguchi Y, Hunt RM, Brighton CT. Growth plate matrix vesicle biogenesis. The role of intracellular calcium. Clin Orthop. 1994:222–229. [PubMed] [Google Scholar]
- 48.Wu LN, Wuthier MG, Genge BR, Wuthier RE. In situ levels of intracellular Ca2+ and pH in avian growth plate cartilage. Clin Orthop. 1997:310–324. [PubMed] [Google Scholar]
- 49.Zhang J, Reedy MC, Hannun YA, Obeid LM. Inhibition of caspases inhibits the release of apoptotic bodies: Bcl-2 inhibits the initiation of formation of apoptotic bodies in chemotherapeutic agent-induced apoptosis. J Cell Biol. 1999;145:99–108. doi: 10.1083/jcb.145.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Althoff J, Quint P, Krefting ER, Hohling HJ. Morphological studies on the epiphyseal growth plate combined with biochemical and X-ray microprobe analyses. Histochemistry. 1982;74:541–552. doi: 10.1007/BF00496668. [DOI] [PubMed] [Google Scholar]
- 51.Qu Q, Perala-Heape M, Kapanen A, Dahllund J, Salo J, Vaananen HK, Harkonen P. Estrogen enhances differentiation of osteoblasts in mouse bone marrow culture. Bone. 1998;22:201–209. doi: 10.1016/s8756-3282(97)00276-7. [DOI] [PubMed] [Google Scholar]
- 52.Valcourt U, Ronziere MC, Winkler P, Rosen V, Herbage D, Mallein-Gerin F. Different effects of bone morphogenetic proteins 2, 4, 12, and 13 on the expression of cartilage and bone markers in the MC615 chondrocyte cell line. Exp Cell Res. 1999;251:264–274. doi: 10.1006/excr.1999.4584. [DOI] [PubMed] [Google Scholar]
- 53.Newman B, Gigout LI, Sudre L, Grant ME, Wallis GA. Coordinated expression of matrix Gla protein is required during endochondral ossification for chondrocyte survival. J Cell Biol. 2001;154:659–666. doi: 10.1083/jcb.200106040. [DOI] [PMC free article] [PubMed] [Google Scholar]