

Abstract
Background
The pulmonary vasculopathy in pulmonary arterial hypertension (PAH) results in increased resistance to pulmonary blood flow, limiting the cardiac output required for the increased O2 demands of exercise.
Aims
We sought to determine the physiologic basis for clinical improvement in PAH patients receiving sildenafil, hypothesizing that the key mechanisms of improvement are improved blood flow and ventilatory efficiency, leading to improved exercise capacity and O2 pulse over time.
Methods
We studied 28 PAH patients with (n=14) and without (n=14) sildenafil treatment. All received warfarin and diuretic therapy, and 13/14 sildenafil-treated patients were already receiving specific PAH drugs. Cardiopulmonary exercise testing was performed before and after sildenafil.
Results
Peak V̇O2, peak O2 pulse, V̇E/V̇CO2 and PetCO2, were 0.84 ± 0.1 L/min, 6.1 ± 0.7 ml • beat−1, 49 ± 2 and 26 ± 1.5 mmHg, and improved after adding sildenafil to 0.91 ± 0.1 L/min, 6.8 ± 0.8 ml • beat−1, 43 ± 2, and 30 ± 1.9, respectively, whereas control patients worsened (p = 0.012, 0.008, 0.008 and 0.0002, treated vs. controls, respectively).
Conclusions
Sildenafil improves PetCO2, V̇E/V̇CO2, peak O2 pulse and peak V̇O2 during exercise compared to controls. A prospective, placebo-controlled study is needed to validate these findings.
Keywords: pulmonary arterial hypertension, cardiopulmonary exercise testing, ventilatory efficiency, pulmonary heart disease
Introduction
Pulmonary arterial hypertension (PAH) is a disorder of the pulmonary circulation that primarily strikes young women and leads to rapidly progressing right ventricular failure and death [1]. In PAH patients, despite increasing pulmonary artery pressure, pulmonary blood flow fails to increase appropriately with exercise. This leads to poor O2 delivery to the peripheral muscles and an increase in the resting ventilatory dead space fraction of the tidal volume (VD/VT), even during exercise, because the regions of ventilated lung that are underperfused at rest remain underperfused during exercise [2]. This results in dyspnoea and fatigue, which are prominent symptoms in these patients.
Recent advances have led to FDA approval of several new treatments for PAH, including the oral phosphodiesterase-5 inhibitor sildenafil in 2005. The benefits of most PAH therapies may not be due primarily to their ability to vasodilate the pulmonary vascular bed, but rather to their alteration of the vascular neurohormonal and cellular signals that lead to abnormal pulmonary vascular cellular proliferation, fibrosis, and thrombosis that are hallmarks of the PAH vasculopathy [3]. While sildenafil is known to mediate the effects of the nitric oxide pathway via cGMP, the precise pathophysiology of the clinical benefit with all PAH drugs has not been fully elucidated.
Because sildenafil has a direct action on the pulmonary circulation, we hypothesized that the predominant mechanism of clinical improvement in PAH patients would first be reflected by an improvement in pulmonary blood flow and more uniform ventilation-perfusion matching, thereby reducing the ventilatory requirement for exercise. This improvement in pulmonary blood flow would be reflected by improved ventilation (VE) relative to CO2 output (V̇CO2), and by improved end-tidal carbon dioxide tension (PetCO2), both measured at the anaerobic threshold (AT). This physiological improvement might precede the improvement in peak oxygen uptake and V̇O2 kinetics, which depend on an improved cardiac output response to exercise and gradually improved aerobic capacity.
Methods
We studied 28 patients with idiopathic PAH (n=22), PAH associated with connective tissue disease (n=4), and PAH associated with corrected congenital heart disease (n=2). All were stable and receiving either conventional therapy (n=2) or were on stable doses of prostanoids (n=12) or endothelin receptor antagonists (n=14). Relevant conventional therapy included diuretics, potassium, digoxin, and warfarin. None of the patients were either formally or informally undergoing exercise training. The diagnosis of PAH was based on clinical and laboratory data, always included right heart catheterization, and satisfied the diagnostic criteria described by the American College of Chest Physicians (ACCP) evidence-based clinical practice guidelines [4].
There was no formal study protocol dictating the use of background therapy or the addition of sildenafil. For 13 of the 26 patients receiving background PAH therapy, the decision to add sildenafil was based on lack of an adequate clinical response to treatment. This was based on criteria which included, but were not limited to WHO/NYHA functional class, 6-minute walk (6MW) distance, symptoms, findings on physical examination, and laboratory testing. None of the patients in this study were treated with sildenafil based on CPET findings and there was no a priori definition of clinical response. Sildenafil was neither given to nor withheld from patients with the intent of studying their CPET responses. One patient was given sildenafil as first-line treatment. The control group consisted of 14 patients not given sildenafil, retrospectively selected during a similar time period.
Both sildenafil-treated and control patients underwent cardiopulmonary exercise testing (CPET) several months before being entered into the study, at baseline, and 4 ± 2 months later. At our institution, all PAH patients have CPET performed at 3–6 month intervals as part of their routine clinical care. All patients were exercised using a progressively increasing work rate test to maximum tolerance on an electromagnetically braked cycle ergometer [2]. Gas exchange was measured using MedGraphics (St. Paul, Minnesota) equipment with recordings of oxyhaemoglobin saturation (SpO2), heart rate (HR), ventilation (VE), CO2 output (V̇CO2), O2 uptake (V̇O2), work rate (WR), end-tidal carbon dioxide tension (PetCO2) and other variables, breath-by-breath [5]. From these data, peak work rate, peak V̇O2, AT, peak O2 pulse (peak V̇O2 / peak HR), and the ventilatory equivalent for CO2 (V̇E/V̇CO2 @AT or nadir) and end-tidal CO2 (PetCO2 @AT or at a later high point) were analyzed using standard techniques [5–6].
Statistical Analysis
We retrospectively analyzed before and after changes using paired t-tests. For data with non-normal distribution, the Wilcoxon signed-rank test for nonparametric data was used. Between group comparisons of changes (deltas) were performed using 2-tailed, unpaired t-tests. Data are presented as mean ± SEM. A p-value <0.05 was considered statistically significant.
This study was conducted in accordance with Good Clinical Practices, the principles outlined in the Declaration of Helsinki, and with local institutional regulations. The local ethics review committee (Human Subjects Committee) approved the protocol; written informed consent was obtained for all patients.
Results
Table 1 shows the patient’s demographics and baseline characteristics. The sildenafil-treated baseline mean peak V̇O2, peak O2 pulse, V̇E/V̇CO2 @AT, and PetCO2 @AT were 0.84±0.11 L/min, 6.1±0.7 ml/beat, 49±2, and 26±1.5 mmHg respectively (Table 2), reflecting severe pulmonary vascular disease despite their prior treatment. The demographics of the control group did not differ significantly from the sildenafil-treated group. The baseline resting pulmonary haemodynamics obtained at cardiac catheterization were also consistent with severe PAH in both groups. Following treatment with sildenafil, peak V̇O2 trended towards an increase, peak O2 pulse increased, V̇E/V̇CO2 decreased, and PetCO2 increased significantly (Figure 1). The increase in peak WR was borderline significant; there were no significant changes in AT, peak HR, and lowest SpO2 (Table 2). However, since the control patients either did not significantly improve, or worsened, between group comparisons of the changes in peak V̇O2, peak O2 pulse, V̇E/V̇CO2 @AT, and PetCO2 @AT for the sildenafil-treated compared to the control group were all highly significant (p = 0.012, 0.008, 0.008 and 0.0002, respectively). The course of change from the first test to the baseline test was similar in both the control and sildenafil-treated groups.
TABLE 1.
Baseline patient characteristics
| Demographics | sildenafil | controls | P value |
|---|---|---|---|
| Age (yrs) | 41.4 ± 3.4 | 45.4 ± 2.4 | 0.4 |
| Sex (F/M) | 13/1 | 13/1 | 0.5 |
| Height (cm) | 163 ± 2.6 | 165 ± 2.4 | 0.9 |
| Weight (kg) | 73 ± 5 | 72 ± 11 | 0.7 |
| Race | 0.5 | ||
| White (%) | 7 (50) | 5 (37) | |
| Hispanic (%) | 4 (29) | 3 (21) | |
| Asian (%) | 2 (14) | 6 (42) | |
| Black (%) | 1 (7) | 0 | |
| NYHA/WHO Class | 0.5 | ||
| II | 1 | 4 | |
| III | 11 | 9 | |
| IV | 2 | 1 | |
| Cause of pulmonary hypertension | 0.5 | ||
| Idiopathic (n) | 11 | 11 | |
| Associated with CTD (n) | 2 | 2 | |
| Corrected CHD (n) | 1 | 1 | |
| Resting haemodynamics | |||
| mRAP, mmHg | 9.1 ± 1.0 | 8.1 ± 0.9 | 0.4 |
| mPAP, mmHg | 50 ± 3.3 | 54 ± 4.3 | 0.4 |
| mPWP, mmHg | 10 ± 1.3 | 11 ± 1.1 | 0.9 |
| CO, L • min−1 | 3.9 ± 0.4 | 4.0 ± 0.4 | 1.0 |
| Cardiac Index, L • min−1• m² | 2.2 ± 0.2 | 2.2 ± 0.2 | 1.0 |
| PVR dyne•s•cm−5 | 901 ± 127 | 1048 ± 175 | 0.4 |
| mBP, mmHg | 91 ± 3 | 92 ± 4 | 0.7 |
| Background PAH therapy | 0.5 | ||
| prostacyclin analogue (n) | 6 | 7 | |
| endothelin antagonist (n) | 7 | 6 | |
| none (n) | 1 | 1 |
CTD = connective tissue disease; CHD = congenital heart disease
TABLE 2.
Responses of sildenafil-treated patients (n=14) and control group (n=14)*
| Variable | Baseline control group | Baseline sildenafil group | 4 ± 2 months after sildenafil | Δ% after sildenafil | P value¶ |
|---|---|---|---|---|---|
| peak V̇O2, L • min−1 | 0.79 ± 0.18 | 0.84 ± 0.11 | 0.91 ± 0.10 | 8.8 | 0.074 |
| peak V̇O2, ml •min−1•kg−1 | 11.34 ± 0.57 | 11.41 ± 1.15 | 12.38 ± 0.94 | 8.8 | 0.050 |
| AT, L • min−1 | 0.61 ± 0.04 | 0.61 ± 0.06 | 0.67 ± 0.05 | 4.1 | NS |
| V̇E/V̇CO2 | 50 ± 2.9 | 49 ± 2.3 | 43 ± 2.2 | −14.9 | 0.009 |
| peak WR, watts | 57 ± 5.4 | 60 ± 10 | 68 ± 8.9 | 13.7 | 0.050 |
| PetCO2, mmHg | 27 ± 1.5 | 26 ± 1.5 | 30 ± 1.9 | 10.4 | 0.006 |
| rest HR | 81 ± 4.4 | 88 ± 1.9 | 87 ± 2.3 | −1.2 | NS |
| peak HR, bpm | 136 ± 5.1 | 139 ± 5.0 | 139 ± 4.4 | 0.1 | NS |
| peak O2 pulse, ml•min−1•beat−1 | 6.2 ± 0.4 | 6.1 ± 0.7 | 6.8 ± 0.8 | 10.0 | 0.014 |
| Lowest SaO2, % | 92 ± 1.5 | 90 ± 2.3 | 90 ± 2.3 | 0.4 | NS |
| peak RER | 1.16 ± 0.03 | 1.18 ± 0.03 | 1.21 ± 0.03 | 2.4 | NS |
values are ± SEM
post-sildenafil compared to baseline
Figure 1.
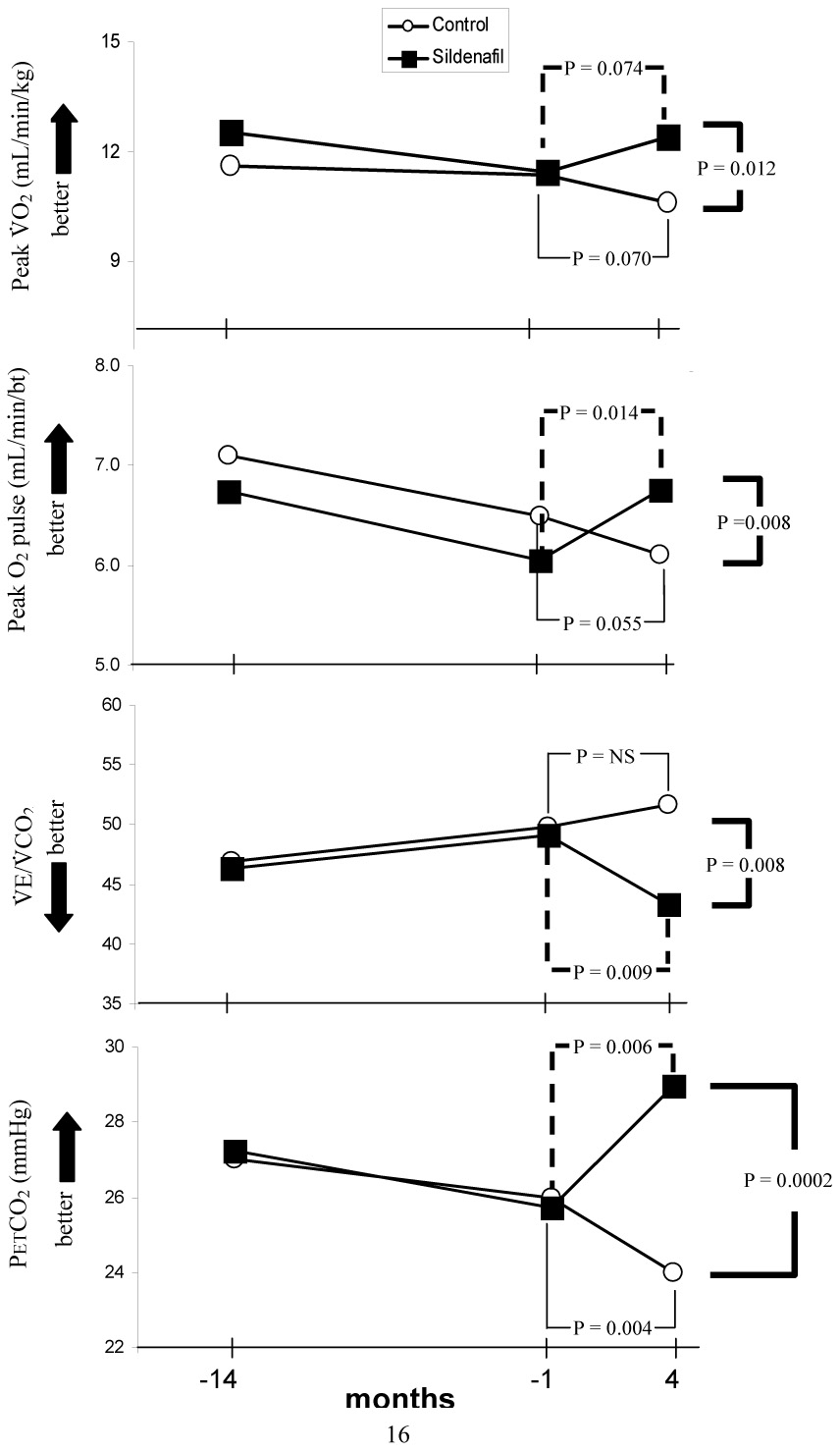
Serial measurements in peak V̇O2, peak O2 pulse, V̇E/V̇CO2, and PetCO2 made at 14 ± 3.2 months and 1.4 ± 0.6 months prior to, and 4 ± 2 months after the start of sildenafil treatment (squares); circles represent control PAH patients treated over a similar time period, but without the addition of sildenafil.
P values are shown for within-group and between group comparisons.
We compared the baseline peak V̇O2, peak O2 pulse, V̇E/V̇CO2 and PetCO2 values in the sildenafil-treated patients to those obtained 14 ± 3 months before adding sildenafil, and also compared the serial changes in V̇O2, peak O2 pulse, V̇E/V̇CO2, and PetCO2 to the changes in the control group over a similar time period (Figure 1). It is evident that both the sildenafil-treated and control patients tended to worsen prior to baseline, while thereafter the control patients continued to worsen and the sildenafil-treated patients improved. By the end of 4 months of sildenafil treatment, the changes in peak V̇O2, peak O2 pulse, V̇E/V̇CO2 and PetCO2 were all significantly improved compared to controls.
There was no apparent influence of background PAH therapy upon the change in CPET performance in either the sildenafil-treated group or the control group. However, this may have been due to the small sample size and/or the heterogeneous distribution of background therapy.
Discussion
Several uncontrolled studies of sildenafil in PAH have reported improvements in several clinical measurements such as functional class, exercise capacity, and haemodynamics [15], right ventricular dilation and hypertrophy [16], and quality of life [18]. In a large, multicenter trial of sildenafil vs. placebo for PAH patients, Galié and colleagues demonstrated improvements in exercise capacity as measured by the 6MW distance, functional class and, to a minor degree, resting haemodynamics with sildenafil [8]. However, the physiologic mechanisms of these improvements were not explored, and the haemodynamic assessments in this study were performed at rest, rather than during exercise, when PAH symptoms predominantly occur. Resting haemodynamic measurements do not directly address the ability of the cardiovascular system to respond to exercise.
Mathai, et al also studied sildenafil added to bosentan and found salutary changes in patients with IPAH and more modest changes in patients with connective tissues disease associated PAH [17]. Hoeper, et al. studied 9 patients with PAH who were given sildenafil added to background therapy consisting of the endothelin antagonist bosentan [9]. They demonstrated significant improvements in 6MW distance and peak V̇O2 after 2–3 months of sildenafil, but did not explore mechanisms. Lunze K, et al measured V̇E/V̇CO2 in 11 patients with pulmonary hypertension treated with sildenafil however did not report the changes after treatment [19].
In the present study, we demonstrated that brief treatment with sildenafil resulted in improvement of ventilatory efficiency (decrease in V̇E/V̇CO2 and increase in PetCO2). This improvement in ventilatory efficiency seen in our study serves to explain the primary mechanism of clinical improvement with sildenafil in addition to acting as a marker of improvement. The utility of PetCO2 in particular to measure ventilatory efficiency during exercise was previously shown in the PAH patient population [7], however this is the first study to demonstrate changes in PetCO2 over time. PetCO2 is a simple measurement that can easily be obtained online during exercise and does not require sophisticated means to interpret the results.
Compatible with the improvement in pulmonary perfusion, both peak work rate and peak O2 pulse improved significantly with sildenafil. In PAH, the low cardiac output response to exercise results in a high arterial-mixed venous O2 difference [C(a-v̄)O2], and low stroke volume (SV) at a low work rate. Because C(a-v̄)O2 at peak V̇O2 is likely unchanged from one study to the next,[5], the low peak O2 pulse {SV × [C(a-v̄)O2]} likely reflects a low stroke volume (SV) at peak exercise. Thus, the increase in peak O2 pulse seen in our treated patients likely reflects an improvement in stroke volume and cardiac output at peak exercise with sildenafil, although an increase in [C(a-v̄)O2] cannot be absolutely excluded. These haemodynamic changes during exercise have not been previously reported.
Utility of Non-invasive Monitoring
In PAH, ventilation of relatively underperfused alveoli during exercise results in an increase in dead space ventilation, shown as an increase in V̇E relative to the V̇CO2, best measured at the AT or nadir of the V̇E/V̇CO2 ratio (1). The breathlessness that occurs with exercise in PAH patients may be primarily due to the mismatching of ventilation to perfusion, as patients with PAH nearly always have a high breathing reserve at peak exercise. Their elevated V̇E/V̇CO2, measured during CPET, is accompanied by reductions in PetCO2 [7] and elevations in physiologic dead-space-to-tidal-volume ratio [13]. Thus these non-invasive measurements provide important documentation and explanation of a therapeutic effect. In this study, a relatively rapid improvement in pulmonary perfusion was demonstrated using CPET, without invasive testing, supporting the utility of CPET in evaluating responses to PAH therapies. Further, these findings explain the mechanism of the clinical improvements seen with sildenafil.
Comparison of ventilatory efficiency to aerobic capacity
The pulmonary vasculopathy of PAH can be likened to a relatively “fixed stenosis” of the pulmonary circulation, limiting the increase in pulmonary blood flow during exercise. The required increase in cardiac output during exercise in PAH is blunted by the inability of the pulmonary vascular bed to accept an adequate increase in blood flow, despite increased force of contraction by the right ventricle. This results in a limited or relatively “fixed” level of blood flow that can perfuse the lungs at peak exercise. This limitation in cardiac output to adequately increase during exercise, results in decreased aerobic capacity and deconditioning of the peripheral muscles.
With sildenafil treatment, while pulmonary perfusion is improved over the short term, improvements in aerobic capacity (i.e. peak V̇O2 and/or AT) may be delayed. A similar situation is seen after the surgical correction of severe mitral stenosis [10]. In mitral stenosis, although symptomatic improvement occurs rapidly after mitral commisurotomy, functional improvement is not immediate. Rather, it occurs over several months, despite the immediate increase in pulmonary blood flow afforded by the commisurotomy [11–12]. Thus, in PAH, patients with chronically impaired pulmonary blood flow may not manifest significant gains in aerobic capacity until vascular remodelling and/or aerobic training allows the skeletal musculature to use the greater cardiac output afforded by the improved pulmonary blood flow. This may in part account for the findings in another PAH clinical trial in which peak V̇O2 did not appreciably increase after the administration of sitaxsentan for 3 months [14]. In our study, compared to the control group, the change in aerobic capacity (peak V̇O2 and peak O2 pulse) was less in magnitude and statistical significance than the change in ventilatory inefficiency (PetCO2 @AT and V̇E/V̇CO2 @AT). Had we performed CPET more frequently after baseline, a significant delay in aerobic capacity improvement relative to ventilatory efficiency might have been appreciated. Further study is required to determine the time course of ventilatory and aerobic improvements more precisely in PAH patients treated with sildenafil.
Limitations
The addition of sildenafil to our patients’ treatment regimen was not part of a formal study protocol. There was no blinded control group to assure that the findings were not related to a placebo effect. In addition, no attempt was made to control for background PAH therapy or self-driven exercise training at home. Nevertheless, nearly all sildenafil-treated patients experienced an improvement in ventilatory efficiency, in contrast to the control group, and this improvement did not appear to be influenced by their background therapy. Finally, the resting haemodynamic effects of sildenafil were not studied because repeat catheterization was not clinically indicated.
In summary, the improvements in V̇E/V̇CO2, PetCO2, peak O2 pulse and peak V̇O2, in PAH patients treated with sildenafil provides an objective physiologic explanation for the improved exercise performance that is seen in patients. A prospective, placebo-controlled study is needed to validate these findings.
Acknowledgments
This research was supported by NIH grant # 1 K23 RR17596-01 and General Clinical Research Centers grant # MO1 RR 425.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–1436. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- 2.Sun XG, Hansen JE, Oudiz RJ, Wasserman K. Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation. 2001;104:429–435. doi: 10.1161/hc2901.093198. [DOI] [PubMed] [Google Scholar]
- 3.Farber HW, Loscalzo J. Mechanisms of Disease: Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 4.McGoon M, Gutterman D, Steen V, et al. American College of Chest Physicians. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126:14S–34S. doi: 10.1378/chest.126.1_suppl.14S. [DOI] [PubMed] [Google Scholar]
- 5.Wasserman K, Hansen JE, Sue DY, Stringer WW, Whipp BJ. Principles of Exercise Testing and Interpretation. 4th Ed. Philadelphia: Lippincott, Williams & Wilkins; 2005. 2005. [Google Scholar]
- 6.Beaver WL, Wasserman K, Whipp BJ. A new method for detecting the anaerobic threshold by gas exchange. J Appl Physiol. 1986;60:2020–2027. doi: 10.1152/jappl.1986.60.6.2020. [DOI] [PubMed] [Google Scholar]
- 7.Yasunobu Y, Oudiz RJ, Sun XG, Hansen JE, Wasserman K. End-tidal PCO2 abnormality and exercise limitation in patients with primary pulmonary hypertension. Chest. 2005;127:1637–1646. doi: 10.1378/chest.127.5.1637. [DOI] [PubMed] [Google Scholar]
- 8.Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 9.Hoeper MM, Faulenbach C, Golpon H, Winkler J, Welte T, Niedermeyer J. Combination therapy with bosentan and sildenafil in idiopathic pulmonary arterial hypertension. Eur Respir J. 2004;24:1007–1010. doi: 10.1183/09031936.04.00051104. [DOI] [PubMed] [Google Scholar]
- 10.Herrmann HC. Acute and chronic efficacy of percutaneous transvenous mitral commissurotomy: implications for patient selection. Cathet Cardiovasc Diagn. Suppl 2:61–68. [PubMed] [Google Scholar]
- 11.Donald KW, Bishop JM, Wade OL, Wormald PN. Cardio-respiratory function two years after mitral valvotomy. Clin Sci. 1957;16:325–350. [PubMed] [Google Scholar]
- 12.Wade OL, Bishop JM, Donald KW. The effect of mitral valvotomy on cardiorespiratory function. Clin Sci. 1954;13:511–533. [PubMed] [Google Scholar]
- 13.Wasserman K, Whipp BJ. Exercise physiology in health and disease. Am Rev Respir Dis. 1975;112:219–249. doi: 10.1164/arrd.1975.112.2.219. [DOI] [PubMed] [Google Scholar]
- 14.Barst RJ, Langleben D, Frost A, et al. STRIDE-1 Study Group. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;169:441–447. doi: 10.1164/rccm.200307-957OC. [DOI] [PubMed] [Google Scholar]
- 15.Garg N, Sharma MK, Sinha N. Role of oral sildenafil in severe pulmonary arterial hypertension: Clinical efficacy and dose response relationship. Int J Cardiol. 2006 Dec 14; doi: 10.1016/j.ijcard.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 16.van Wolferen SA, Boonstra A, Marcus JT, et al. Right ventricular reverse remodelling after sildenafil in pulmonary arterial hypertension. Heart. 2006;92:1860–1861. doi: 10.1136/hrt.2005.085118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathai SC, Girgis RE, Fisher MR, et al. Addition of sildenafil to bosentan monotherapy in pulmonary arterial hypertension. Eur Respir J. 2007;29:469–475. doi: 10.1183/09031936.00081706. [DOI] [PubMed] [Google Scholar]
- 18.Wong RC, Koh GM, Choong PH, Yip WL. Oral sildenafil therapy improves health-related quality of life and functional status in pulmonary arterial hypertension. Int J Cardiol. 2006 Oct 23; doi: 10.1016/j.ijcard.2006.07.170. [DOI] [PubMed] [Google Scholar]
- 19.Lunze K, Gilbert N, Mebus S, et al. First experience with an oral combination therapy using bosentan and sildenafil for pulmonary arterial hypertension. Eur J Clin Invest. 2006;36 Suppl 3:32–38. doi: 10.1111/j.1365-2362.2006.01692.x. [DOI] [PubMed] [Google Scholar]