

Abstract
The structural properties of the influenza A virus M2 transmembrane channel in dimyristoylphosphatidylcholine bilayer for each of the four protonation states of the proton-gating His-37 tetrad and their effects on proton transport for this low-pH activated, highly proton-selective channel are studied by classical molecular dynamics with the multistate empirical valence-bond (MS-EVB) methodology. The excess proton permeation free energy profile and maximum ion conductance calculated from the MS-EVB simulation data combined with the Poisson-Nernst-Planck theory indicates that the triply protonated His-37 state is the most likely open state via a significant side-chain conformational change of the His-37 tetrad. This proposed open state of M2 has a calculated proton permeation free energy barrier of 7 kcal/mol and a maximum conductance of 53 pS compared to the experimental value of 6 pS. By contrast, the maximum conductance for Na+ is calculated to be four orders of magnitude lower, in reasonable agreement with the experimentally observed proton selectivity. The pH value to activate the channel opening is estimated to be 5.5 from dielectric continuum theory, which is also consistent with experimental results. This study further reveals that the Ala-29 residue region is the primary binding site for the antiflu drug amantadine (AMT), probably because that domain is relatively spacious and hydrophobic. The presence of AMT is calculated to reduce the proton conductance by 99.8% due to a significant dehydration penalty of the excess proton in the vicinity of the channel-bound AMT.
INTRODUCTION
The influenza A virus M2 protein contains an integral transmembrane (TM) four-helix channel. This channel is believed to play a key role in the viral life cycle by allowing proton flux into the virion, which activates the uncoating of the viral nucleic acid in endosomes, which is essential for them to enter into the host cells (1,2). In addition, the M2 channel can stabilize the accumulation of hemagglutinin glycoprotein in its native form from an otherwise premature acidic conformation by increasing vesicular pH of the trans-Golgi network in some influenza virus strains (3–5). The M2 channel has been shown to function as a proton channel by measurements of its proton permeation current when expressed in mouse erythroleukemia cells (6) or by measuring the intracellular pH changes of oocytes of Xenopus laevis in low pH medium (7).
The activity of the M2 channel can be specifically abated with the binding of the hydrophobic antiinfluenza drug amantadine (AMT), which resides in the outside region between Val-27 and Ser-31 as revealed by neutron diffraction (8). However, the precise proton blockage mechanism is not yet clear, i.e., whether the AMT deforms the M2 channel (9,10) or instead acts as a intrachannel steric blocker (8).
The uniqueness of the M2 channel is further exemplified by its high proton selectivity over other monovalent cations, as demonstrated by physiological experiments in both oocytes and mammalian cells (11,12). This selectivity can be notably reduced by replacing the His-37 residues with Ala, Gly, or Glu (13). Because the M2 channel is of great relevance to virology and drug design, considerable effort has been invested to characterize its TM backbone structure by solid-state NMR (14,15) and site-specific infrared dichroism (16,17) and to investigate its proton conductance mechanism by site-directed mutagenesis (18), ultraviolet resonance Raman spectroscopy (19), and molecular modeling (20–25).
The M2 channel is a 97-amino acid nonglycosylated polypeptide that consists of a 54-amino acid intracellular terminus, a 19-amino acid TM domain, and a 24-amino acid extracellular region (26). The TM domain is of primary interest because it has been demonstrated to contain a channel pore that is essential to the M2 channel ion conductance activity (12,27,28). The backbone structure of the TM domain has been determined to be a right-handed α-helix homotetramer (15,29,30), with each monomer consisting of hydrophobic residues except the highly conserved hydrophilic “fingerprint” of Ser-31, His-37, and Trp-41. The formation of tetramer is believed to be stabilized by the disulfide bonds between the cysteine residues located at the N-terminus (29,31).
The truncated and isolated membrane-spanning portion of M2, namely the M2 TM polypeptide (M2-TMP), has been observed to have ion activity and AMT sensitivity in planar lipid bilayers similar to that of intact M2 (27). As a result, a 25-residue peptide—namely the Udorn Influenza A strain (Ace-Ser-Ser-Asp-Pro-Leu-Val-Val-Ala-Ala-Ser-Ile-Ile-Gly-Ile-Leu-His-Leu-Ile-Leu-Trp-Ile-Leu-Asp-Arg-Leu-NH2 (32)), which corresponds to the residues 22–46—has been selected for this study. This choice is motivated by the observation that this particular sequence is more susceptible to proton blockage by AMT than the Rostock and Weybridge strains (33). A helical tilt angle of 38°, with respect to the bilayer normal, has been reported for M2-TMP (34) when incorporated into dimyristoylphosphatidylcholine (DMPC) vesicles, which is found to be slightly larger than that of 20° for intact M2 (15). This difference is suggested to be related to the interhelical interaction ascribed to the disulfide bonding (35), which can be examined by measuring the peptide association energy as a function of the bilayer thickness (36). Nevertheless, the M2-TMP is still a reasonably good model for investigating the structure and proton transport mechanism of the M2 channel in phospholipid bilayer.
Unlike most other proton channels which are voltage gated (37), the proton conductivity of the M2 channel is activated by elevating the proton concentration of the extracellular bathing medium (i.e., it is pH gated). Despite the slightly different threshold activation pH values of 5.8 and 4.0 reported by two research groups (13,38), a saturation proton current has been consistently observed (6,38,39) and can be explained by a three-site quantitative mechanism (40). The lumen of the M2 channel is found to be large enough to form a hydrogen-bonded water chain with the only interruption at His-37 (41). Its conformational change upon protonation and its interaction with the proximate pore-lining Trp-41 are believed to be essential for gating the proton transport (18–20,25,41).
Two proton conductance mechanisms, namely the “shuttling” mechanism (41) and the “shutter” mechanism (20), have been proposed from mutagenesis studies (41), infrared spectroscopy (19), and molecular modeling (20,23). The shuttling mechanism postulates that a His-37 imidazole nitrogen atom, which faces the exterior of the virus, is protonated by the entering proton before the other imidazole nitrogen atom at the same His-37 residue releases its proton to the interior of the virus. Then, the sequence is sustained by a ring flip or tautomerization. Since the His-37 acts as a proton relay conductor by this hypothesis, only one of the His-37 tetrad is necessarily protonated. Alternatively, the shutter mechanism suggests that the pore opening is ascribed to the electrostatic repulsive interaction between positively charged doubly protonated His-37 residues. Therefore, at least two residues of the His-37 tetrad presumably have to be protonated to temporarily form a continuous water wire inside the M2 channel to facilitate Grotthuss proton shuttling (42,43). Thus, the pKa values of the His-37 tetrad and its exact protonation state for both the open and closed M2 channel are vital for fully understanding the proton transport mechanism.
Recently, the protonation behavior of the four His-37 residues as a function of pH has been investigated by examining the variance of the chemical shift pattern of the 15N-NMR spectra corresponding to the different relative abundances of the His-37 protonation forms (44). These results suggest that the triply protonated His-37 tetrad is the mostly likely open state of the M2 channel. The pore opening is a consequence of the breakage of the hydrogen bond formed by two adjacent His-37 residues that otherwise occlude the proton pathway by “laying down” their imidazole rings in closed state M2. Furthermore, the reported pKa value of 8.2 for the first and second titration is high enough that two His-37 residues are expected to be protonated under most physiological conditions.
Although the experimental NMR study described above provides a direct determination of the protonation states of the His-37 tetrad, the proton transport mechanism through M2 is not well understood, partially because of the hypothesized conformational change of His-37 residues that is proposed to be a key step for pore opening. Indeed, before the experimental NMR measurements, a large number of computational studies were carried out to relate the structure of the M2 channel to its proton conductivity (20–22,45,46). However, the absence of an explicit excess proton in these studies, which is capable of hopping through water wires by the Grotthuss mechanism, has made these studies speculative in character. More recently, however, the multistate empirical valence bond (MS-EVB) methodology (42,47–49) has been employed to reliably describe the dynamics of proton translocation in TM channels (23,50–53).
The objective of this study is to utilize MS-EVB simulations to study the effects of the protonation of the gating His-37 tetrad on the proton transport behavior through the M2-TMP, to investigate the phenomenon of ion selectivity and AMT proton blockage, and to provide quantified proton transport properties, all of which can in turn be examined by further experiments.
METHODS
Systems
The starting structure of M2-TMP, obtained from the Protein Data Bank with the entry code 1MP6 (34) and as determined by solid-state NMR to have a tilt angle of 38°, has been demonstrated to destabilize the homotetramer bundle in a previous molecular dynamics (MD) simulation (25). Thus, a modified structure of M2-TMP with a smaller tilt angle of 30°, which is still within the range of the experimental value (30,41), was employed in this study. This structure manifested a substantial improvement in the stability of the homotetramer bundle (Fig. 1). Moreover, one of the four Asp-24 residues was treated as being ionized according to a pKa calculation (21) based on a generalized Born solvation model calculation (54). The simulation system was then constructed by embedding the M2-TMP tetramer bundle into a fully hydrated DMPC bilayer with 64 lipid molecules in each leaflet. The M2/DMPC/H2O system consisted of ∼11,000 atoms after 15 DMPC molecules in each leaflet were removed to avoid bad contacts. The thickness of the water layer in the direction of the channel axis was ∼13 Å on each side of the bilayer.
FIGURE 1.

Reduced system in the MS-EVB2 simulation. The M2 channel is highlighted and colored blue. The proton-gating His-37 and Trp-41 residues are colored orange and mauve, respectively. The C-terminal end is at the top. The figure was created with visual molecular dynamics software (87).
In terms of the fourfold symmetry of M2-TMP with respect to the channel axis, there are a total of six possible protonation states for the His-37 tetrads as schematically illustrated in Fig. 1 of Kass and Arkin (22) (denoted here +0, +1, +2_S, +2_A, +3, and +4). State +0 has been previously studied and revealed to be an essentially closed form (14,25). Based on the relevant pKa estimates, only four protonation states (namely, +1, +2, +3, and +4) have been investigated in this study with the same initial structure as described above. The state +2_S, which has two opposite protonated His-37 residues, is more likely to exist than state +2_A, which has two adjacent protonated His-37 residues. The +2 state is thus assumed here to be the +2_S configuration. Neutral His-37 residues were assigned to be ɛ-protonated in accord with the experimental finding that the ɛ-nitrogen of His-37 tends to have higher pKa than the δ-nitrogen under the condition of high pH (44). Chloride ions were also added to the simulation cell to neutralize the overall charge of the systems.
Classical MD simulations of M2-TMP with different protonation states
The AMBER99 parameters (55) and an AMBER-like force field (56) were used in the simulations to model the protein, ions (i.e., Na+ and Cl−), and DMPC lipids; and the water molecules were represented by the flexible TIP3P model (57). The force field for AMT was constructed by using the GAFF (58) package with AMBER atom types. The Nose-Hoover thermostat and barostat (59), both with a relaxation time of 0.2 ps, were applied to maintain the temperature and pressure of the system to 298 K and 1.0 bar, respectively. Periodic boundary conditions were employed, and the long-range electrostatics were treated by the smooth particle mesh Ewald (SPME) (60) method with a tolerance of 10−6. A 6.0 ns classical simulation was performed by using the DL_POLY MD package (61) with a time step of 1.0 fs for each of the four protonation states. With the exception of the channel pore radius profile, which was calculated by using the HOLE2 package (62), all analysis programs were coded in house.
MS-EVB simulations of proton transport in M2-TMP
In the MS-EVB model, the extent of proton charge delocalization, characterized by the EVB state amplitude, is determined by diagonalizing a Hamiltonian matrix at each time step in which the diagonal elements represent the diabatic energy of different classical hydrogen-bonding topologies. These diagonal matrix elements are called EVB states, whereas the off-diagonal elements reflect the coupling between EVB states, allowing transformations to occur in the bonding topology and hence allowing proton delocalization and Grotthuss shuttling to occur. Because the system dynamics is propagated according to the Hellmann-Feynman theorem, the proton charge delocalization can be viewed as a linear combination of different hydrogen-bond topologies weighted by their EVB amplitudes. A more detailed description of the MS-EVB model, and its second and third generation derivatives (MS-EVB2, MS-EVB3), is given in Schmitt and Voth (47), Day et al. (48), and Wu et al. (49).
To reduce the relatively demanding computational costs of the MS-EVB model, a reduced system was constructed by removing all DMPC molecules from the final configuration of the classical simulation described above. The resulting reduced system was reequilibrated for 3.0 ns under the same simulation condition as the classical simulation except with harmonic position restraints applied on backbone atoms of the M2-TMP with a force constant of 1.2 kcal/mol/Å2 to maintain the tetramer bundle stability without the presence of explicit phospholipids. The reduced system is shown in Fig. 1. A comparison of the root mean-square fluctuations (RMSFs) in Fig. 2 indicates that the reduced system yields positional fluctuation patterns with ∼50% smaller magnitudes compared to the full system. (The effect of this approximation was tested and is described later.) An excess proton and a neutralizing chloride ion were then added to the reduced system, and the MS-EVB2 model (48) was employed to allow the proton charge delocalization and Grotthuss shuttling (43). The MS-EVB simulations were then performed using the DL_EVB code (63).
FIGURE 2.

Comparison of the RMSF of the M2 protein backbone atoms between the full composite system and the reduced system in Fig. 1 for protonation states +1, +2, +3, and +4.
Potential of mean force, position-dependent diffusion coefficient, and maximum ion conductance calculations
The potential of mean force (PMF) provides the proton permeation free energy profile through the channel. The PMF was obtained by employing the weighted histogram analysis method (WHAM) (64) to connect free energy segments of the sampling windows in which the excess proton center of excess charge (CEC) (48,65) was restrained by a harmonic umbrella sampling potential. In the bulk water region, an additional harmonic radial restraint (66) was applied to the CEC with appropriate force constants to ensure that the proton can explore an area in the plane perpendicular to the channel axis with a similar radius as that inside the channel. The statistical error of the PMF was estimated by block averaging. In this study, all PMF profiles were calculated with the reduced M2-TMP systems that were constructed as described in the previous section.
Similar simulations and analyses were also conducted for the Na+ cation. Furthermore, to investigate whether the PMF results could be seriously affected by some simulation artifacts, three additional PMFs for Na+ were also calculated for the following different simulation conditions: 1), the force field for the Na+ cation was replaced by the CHARMM force field; 2), the flexibility of the protein backbone in the reduced model was increased by lowering the force constant of the restraints to 0.6 kcal/mol/Å2; and 3), the simulation box size was increased by adding more water in the channel axis direction, giving a 25 Å water layer on each side. These three changes were done separately, and overall PMFs were obtained for each particular system (see Discussion).
The position-dependent diffusion coefficient for the excess proton was calculated according to the simplified Woolf-Roux equation (67,68):
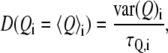 |
(1) |
where 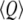 is the average of the reaction coordinate Q,
is the average of the reaction coordinate Q, 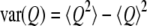 is the variance of the reaction coordinate, and
is the variance of the reaction coordinate, and 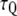 is a characteristic time obtained by exponentially fitting the autocorrelation function of Q. In the case here, the reaction coordinate Q was taken to be the CEC distance along the channel axis. The use of Eq.1 for the position-dependent diffusion coefficient calculation was validated by the observation that in the PMF simulations the restrained particle always behaves like an overdamped harmonic oscillator.
is a characteristic time obtained by exponentially fitting the autocorrelation function of Q. In the case here, the reaction coordinate Q was taken to be the CEC distance along the channel axis. The use of Eq.1 for the position-dependent diffusion coefficient calculation was validated by the observation that in the PMF simulations the restrained particle always behaves like an overdamped harmonic oscillator.
The maximum ion conductance 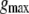 for protons can be estimated from the quasi-one-dimensional Poisson-Nernst-Planck (PNP) electrodiffusion theory (69) such that
for protons can be estimated from the quasi-one-dimensional Poisson-Nernst-Planck (PNP) electrodiffusion theory (69) such that
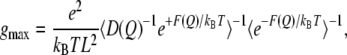 |
(2) |
where e is the elementary charge, 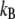 is the Boltzmann's constant, T is the temperature, L is the length of the pore region (for M2-TMP, L = 30 Å), D(Q) and F(Q) are, respectively, the diffusion coefficient and the PMF as a function of the reaction coordinate Q, and the brackets denote spatial averaging over the length of the channel in the direction of the channel axis. This equation is based upon the assumption that the channel is singly occupied and the solvent baths at the two ends of the channel have the same pH, which are consistent with our simulation conditions. Note that the relatively high pKa values of the His-37 tetrad (along with our PMFs indicating that there is no strong proton binding site other than the His-37 residues inside the M2 channel) diminishes the possibility of multiproton occupancy under physiological conditions.
is the Boltzmann's constant, T is the temperature, L is the length of the pore region (for M2-TMP, L = 30 Å), D(Q) and F(Q) are, respectively, the diffusion coefficient and the PMF as a function of the reaction coordinate Q, and the brackets denote spatial averaging over the length of the channel in the direction of the channel axis. This equation is based upon the assumption that the channel is singly occupied and the solvent baths at the two ends of the channel have the same pH, which are consistent with our simulation conditions. Note that the relatively high pKa values of the His-37 tetrad (along with our PMFs indicating that there is no strong proton binding site other than the His-37 residues inside the M2 channel) diminishes the possibility of multiproton occupancy under physiological conditions.
Estimation of pKa values of the His-37 residue
The pKa values of the His-37 residues for the four protonation states were estimated by applying the H++ (70,71) program, a Poisson-Boltzmann equation solver, to the final structure of M2-TMP obtained from the classical simulation as described above. The dielectric constants inside and outside the channel lumen were assigned to be 4 and 80, respectively. Because further protonation, if possible, only occurs when the pH is lower than the highest pKa value of the neutral His-37 residues, that pKa value is denoted as the transition pH and summarized in row a of Table 1.
TABLE 1.
Properties for different protonation states of the His-37 tetrad
| +1 | +2 | +3 | +4 | |
|---|---|---|---|---|
| pH(a) | 6.6 | 6.0 | 5.5 | 2.1 |
| dHIS-37(Å)(b) | 9.09 ± 0.75 | 9.47 ± 0.90 | 9.65 ± 0.90 | 10.04 ± 0.87 |
| dTRP-41 (Å)(c) | 9.40 ± 0.54 | 10.20 ± 0.52 | 10.39 ± 0.45 | 11.08 ± 1.37 |
| Percent (%)(d) | 0% | 0.64% | 2.22% | 3.38% |
| gmax(pS)(e) | 4.7 × 10−8 | 3.6 × 10−4 | 53 | 1.3 × 10−3 |
Transition pH for His-37 tetrad.
Average distance between adjacent backbone α-carbon atoms of His-37 residues.
Average distance between adjacent backbone α-carbon atoms of Trp-41 residues.
Percentage of hydrogen-bond forming for facilitation proton transport.
Maximum ion conductance.
RESULTS
Effects of the His-37 protonation state on the structure of M2-TMP
The proton conductivity through the M2 channel is believed to be a consequence of the protonation of the pH gating His-37 tetrad and the corresponding channel conformational change. However, because of its rather low proton conductance of 6.0 pS and small open-state probability of 0.03 (38), the short-lived structure of the open-state M2 channel has not yet been accurately characterized by experiments. Thus, it was essential to explore the structures of M2 channels with different His-37 protonation states in DMPC lipid (denoted +1, +2, +3, and +4 for each successive His-37 protonated in the tetrad) before examining the behavior of the proton transport.
The deviation of the M2-TMP backbone with respect to the initial structure was studied by calculating the RMSD (data not shown). The overall RMSD reaches a plateau value of ∼2.0 Å after its rise during the first 1.0 ns, indicating a stable equilibrated backbone. The α-helix integrity of each monomer of the M2 tetramer was checked via the Ramachandran plot (data not shown), which demonstrates a stable right-handed α-helix with the exception of a few terminal residues, which are of a result of weak interresidue hydrogen bonds.
In these cases, the helix bundle stability of the M2 tetramer was investigated by plotting the distance of the center of mass (COM) between the α-helices as a function of time (Fig. 3). Although a distance between two adjacent α-helices are well maintained at a value of 1.0 nm except the H1-H2 of the +4 protonation state, the disparity of the distance between two opposing α-helices indicates two distinct bundle-packing patterns, namely a tetramer conformational for states +2 and +3 and a dimer-of-dimers conformational for states +1 and +4, which have also been observed in other MD simulations (21). The tilt angle (Fig. 4) with respect to the DMPC bilayer varies from 20° to 40° and is consistent with the experimental value of 30° ∼ 38° (30,34). The primary exception is the +3 state, which was found to have the whole bundle tilt by ∼10° and thus has a low tilt angle value of ∼15° for one helix that may result from the asymmetric triple His-37 protonation.
FIGURE 3.

Distance of COM between any two of the four α-helices as a function of time during 6.0 ns classical MD simulations for protonation states +1, +2, +3, and +4.
FIGURE 4.

Tilt angle of the four α-helices with respect to the DMPC bilayer normal as a functions of time during 6.0 ns classical MD simulations for protonation states +1, +2, +3, and +4.
The pore radius profile (Fig. 5), obtained by examining 5000 configurations over a 5.0 ns trajectory for each His-37 protonation state, indicates a substantial enlargement of the channel pore at the His-37 domain for the +3 and +4 states relative to the +1 and +2 states. This pore radius change results mainly from a side-chain conformational change of one of the four His-37 residues characterized by χ1 (defined as the dihedral angle of N-CA-CB-CG) as shown on Fig. 6. This is different than the backbone relaxation as quantified by the average distance between adjacent backbone α-carbon atoms of the His-37 residues given in Table 1 b. The latter exhibits only a slight expansion in going from state +2 to state +3, whereas the strong electrostatic repulsion of the state +4 results in a considerable swelling of the proximate Trp-41 domain as shown in Table 1 c.
FIGURE 5.

Profile of the average pore radius during 6.0 ns classical MD simulations as calculated by the HOLE2 program (62) for protonation states +1, +2, +3, and +4. The water Van der Waals radius of 1.4 Å is marked by a straight dashed line. The locations of the His-37 and Trp-41 domains are indicated by rectangular bars with overhead legends “HIS37” and “TRP41”, respectively.
FIGURE 6.

Torsion angle χ1 of the four His-37 residues as a function of time during 6.0 ns classical MD simulations for protonation states +1, +2, +3, and +4.
Because the pore radius of the gating His-37 domain of states +3 and +4 is significantly larger than 1.4 Å, i.e., the Van der Waals radius of water, a hydrogen-bond network is able to form through a water chain across this narrow region of the channel. By examining the oxygen-oxygen distance R and the largest intermolecular hydrogen-oxygen-hydrogen angle θ between two closest water molecules from opposite sides of the His-37 domain, the percentage of hydrogen-bond formation (Table 1 d) that can effectively facilitate the proton transport can be assessed by counting the number of water configurations of M2 channel with the criteria of R < 2.6 Å and θ > 120.0°. Even for the pore-opening states of +3 and +4, the probability of establishing such a continuous proton-transport ready water wire is still rare at 2% ∼ 3%, which is consistent with the experimental lower value of 3% (38). Therefore, it is not surprising that the M2 channel has the smallest conductance of any known ion channel (72).
Proton transport behavior as a function of the His-37 protonation state
The PMFs (free energy profiles) of the channel proton permeation for the four His-37 protonation states, obtained from the all-atom MS-EVB2 MD simulation trajectories for the reduced systems, are depicted in Fig. 7. The primary PMF peaks are all located in the His-37/Trp-41 region, which is believed to be the gating domain (41). The +1 state has an overall free energy barrier of ∼18 kcal/mol with a large elevation of ∼14 kcal/mol, caused by strong dehydration penalty, occurring at the N-terminus and Trp-41 domains. These regions are substantially narrower than corresponding domains of the +2 state, the latter exhibiting a somewhat smoother free energy rise. The +3 state has the lowest free energy barrier, ∼7 kcal/mol, and shows a free energy profile very similar to the +4 state, except at the His-37 domain. The additional positively charged His-37 residue region of the +4 state likely imposes a stronger electrostatic repulsion without a further dehydration penalty for the excess proton (Fig. 7).
FIGURE 7.

Free energy profile of the excess proton CEC along the channel permeation pathway for protonation states +1 (solid), +2 (dotted), +3 (dot-dashed), and +4 (dashed). The locations of the His-37 and Trp-41 domains are indicated by rectangular bars with overhead legends “HIS37” and “TRP41”, respectively.
The position-dependent diffusion coefficient and the maximum ion conductance gmax were evaluated and are shown in Fig. 8 and Table 1 e, respectively. The calculated gmax of the +3 state is 53 pS, which is considerably greater than the conductance of any other protonation state and in good agreement with the experimental value of 6.0 pS (38), considering the approximations inherent in the simulation model and in PNP theory (73–75). Thus, our results indicate that the +3 state is most likely the open state of the M2 channel at the experimental pH conditions, in agreement with a recent experimental suggestion (44) that the triple protonation of the His-37 tetrad results in pore-opening and pH-gated proton conductivity.
FIGURE 8.

Profile of the position-dependent diffusion coefficient of the excess proton for protonation states +1, +2, +3, and +4.
Estimation of transition pH for the His-37 tetrad
The transition pHs for the four protonation states, estimated by continuum electrostatic theory (70,71), are 6.6, 6.0, 5.5, and 2.1, respectively. These values are only weakly affected by the parameters; for instance, changes of only up to 0.5 were observed when the dielectric constant inside the channel lumen was varied from 2.0 to 20.0, a range generally applied to the protein interior in simulations with implicit solvent models. The decremental change of these values as associated with the protonation of the channel is qualitatively similar to the experimental results, where they were determined to be 8.2, 8.2, 6.3, and <5.0 by solid-state NMR (43). However, our results are consistently smaller than the corresponding experimental ones. One possible reason accounting for this underestimation is that the effects of the lipid bilayer are not included in our continuum electrostatics calculations. Explicit inclusion of the bilayer should otherwise increase the pKas by creating a more hydrophobic environment for the protein. Note that if the triply protonated state is the open state as suggested by our calculations of the PMF profile and gmax as described above, the gating pH for proton conductivity is expected to be ∼5.5, which is also in good agreement with the value of 5.77 from electrophysiological experiments (13).
Ion selectivity
The M2 channel is a proton-selective channel that is nearly impermeable to any other ions (6,76). Despite the difficulties in directly measuring the extremely low single-channel ion current, several methods, such as comparison of equilibrium proton potential (11) and application of the proteoliposome assay system (72), have been employed to measure the proton selectivity over sodium cation. A comparison of the permeation free energy profiles between proton and sodium for the proposed +3 open state is shown in Fig. 9. The primary free energy barrier of ∼10 kcal/mol for Na+, located at the His-37 domain, which is the narrowest part of the channel, is substantially higher than that of the excess proton. The latter is capable of shuttling through the water wire without significant atomistic displacements according to the Grotthuss mechanism (43). The lack of Grotthuss shuttling for Na+ can also explain the considerably slower self-diffusion of that cation, as seen in Fig. 10. These results are consistent with experimental mutagenesis studies (13,77), which have shown that the M2 channel loses its proton selectivity with the His-37 substituted by Gly, Ala, Glu, Ser, and Thr. By comparing the estimated conductance gmax of proton and Na+ (Table 2), the proton selectivity is seen to be a factor of 6.0 × 103, which is qualitatively comparable with experimental values of 2 × 106 and 3 × 106 determined by electrophysiological (11) and proteoliposome assay methods (72), respectively. Because the quasi-one-dimensional PNP theorem assumes all ions enter into the conductive channel with no barrier, the relatively low calculated ion selectivity could be attributed to the proton antenna effect (78–82) present in real systems, which may facilitate proton transport across the phospholipid bilayer surface relative to bulk diffusion.
FIGURE 9.

Free energy profile along the channel permeation pathway of excess proton (dashed) and proton with the presence of AMT (dotted) and sodium cation (solid) for the proposed open state +3. The locations of the Ala-29, His-37, and Trp-41 domains are indicated by rectangular bars with overhead legends “ALA29”, “HIS37”, and “TRP41”.
FIGURE 10.

Profile of the position-dependent diffusion coefficient of excess proton (dashed) and proton with the presence of AMT (dotted) and sodium cation (solid) for the proposed open state +3.
TABLE 2.
Maximum ion conductance for proton (with and without the presence of AMT) and sodium
| H+ | H+_AMT | Na+ | |
|---|---|---|---|
| gmax(pS) | 53 | 0.12 | 8.8 × 10−3 |
AMT binding and blocking
The first step to investigate the effects of the presence of AMT on the proton conductance of the M2 channel is to locate the AMT binding site. This site has been determined by neutron diffraction (8) and solid-state NMR (83) to come from the extracellular side of the channel and fall in the peptide sequence between Val-27 and Ser-31. By examining the 3.0 ns trajectory of the COM of the AMT, which is initially placed 8 Å away from the His-37 tetrad to the extracellular side, the AMT binding site was revealed to be near the Ala-29, presumably because this domain is relatively hydrophobic and spacious. The strong affinity for AMT binding in the M2 channel during the simulation is in agreement with the irreversible proton inhibition by AMT in whole-cell systems (33) and in planar lipid bilayers (38). The presence of AMT relocates the primary proton permeation free energy (Fig. 9) barrier in the +3 state from the His-37/Trp-41 domain to Ala-29, i.e., the AMT binding site. The height of the barrier also increases to ∼10 kcal/mol. Moreover, the secondary free energy peak of ∼8.0 kcal/mol residing at the His-37/Trp-41 domain is significantly higher than that without the presence of AMT, suggesting a possible allosteric blocking behavior (6) in addition to the intrachannel inhibition which disrupts the continuous water wire. The proton conductance, gmax, is predicted to be reduced by 99.8% in the presence of AMT (Table 2). It should be noted, however, that this molecular model for AMT is relatively primitive, so these results can be viewed as preliminary.
DISCUSSION
The primary goal of this study was to investigate the explicit proton transport behavior through the M2 channel, which in turn depends on the protein structure as a function of the protonation state of the pH-gating His-37 tetrad. The permeation free energy profile for an explicit excess proton and the maximum proton conductance have been calculated from the MS-EVB simulation data combined with PNP theory. These results indicate that the +3 state and +4 state undergo a pore opening mainly by a significant side-chain conformational change of a His-37 residue due to the strong electrostatics repulsion. Although the opened pore is wide enough to accommodate a water molecule, the probability of forming a continuous water wire that can efficiently facilitate the proton transport via Grotthuss shuttling is still rare. By examining the permeation free energy profile and maximum ion conductance, gmax, the state +3 is found to be the open state. The predicted protonation states from continuum electrostatics calculations are also found to be in reasonable agreement with experiment.
As described earlier, to test the approximations inherent in this simulation model, several variants on the system were simulated for Na+ permeation (largely because these simulations are considerably faster than full MS-EVB simulations of the excess proton permeation). As seen in Fig. 11, some differences in the PMF profiles for sodium cation arise from varying the force field, the flexibility of protein backbone in the reduced model, and the thickness of solvating water layers. However, the overall peaks are all located at the His-37 domain with a similar magnitude of ∼10 kcal/mol. This relative consistency provides additional confidence in these simulations (see also the work of Allen et al. (84)).
FIGURE 11.

Permeation free energy profiles for sodium cation along the channel axis for the proposed open state +3 by applying all the AMBER force field (solid), the AMBER protein force field combined with the CHARMM ion force field (dotted), the AMBER force field with enhanced greater protein backbone flexibility (dashed), and the AMBER force field with a thicker water layer (dot-dashed). The reduced model was used in all simulations here.
Another important role found in the calculations by the His-37 tetrad is the proton selectivity that effectively blocks Na+ permeation while allowing excess proton to pass through in the open state. The primary binding site of AMT was found to be at the hydrophobic Ala-29 domain to which the primary free energy peak barrier for proton permeation relocates upon AMT binding. The proton conductance is then significantly reduced by the presence of AMT. These results are in agreement with the experimental observation that the BM2 channel of the Influenza B virus, which has a similar gating domain motif of HXXXW but with less hydrophobic pore-lining residues compared to the M2 channel here, is less sensitive to the AMT inhibition of proton transport (85).
In this study, our PMFs suggest that the additional positive charges in the triply protonated M2 channel seems not to cause a prohibitively high barrier for protons; rather they open up the His-37/Trp-41 domain, assisting the proton transport by reducing the desolvation penalty. To elucidate the possible connection between the proton transport and the histidine tautomerization (41) as well as the histidine-histidine resonance (44), a protonatable histidine MS-EVB model will be explored in the future with multiple reaction sites that allow the proton association/dissociation on the imidazole ring of the His-37 residues. Moreover, further investigation of the proton transport behavior of M2 mutants, which either lack proton selectivity (77) or AMT sensitivity (86), will be a target for future research.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) (R01-GM53148). We also gratefully acknowledge the computational resources supported in part by the following National Science Foundation programs: Partnerships for Advanced Computational Infrastructure, Distributed Terascale Facility, and Terascale Extensions: Enhancements to the Extensible Terascale Facility. These computational resources were provided by Texas Advanced Computing Center at the University of Texas, the National Center for Supercomputing Applications at the University of Illinois Urbana-Champaign, and the Arches Metacluster administered by the Center for High Performance Computing at the University of Utah (funded by NIH grant NCRR 1 S10 RR17214-01).
Editor: Peter C. Jordan.
References
- 1.Martin, K., and A. Helenius. 1991. Nuclear transport of influenza virus ribonucleoproteins: the viral matrix protein (M1) promotes export and inhibits import. Cell. 67:117–130. [DOI] [PubMed] [Google Scholar]
- 2.Helenius, A. 1992. Unpacking the incoming influenza virus. Cell. 69:577–578. [DOI] [PubMed] [Google Scholar]
- 3.Sugrue, R. J., and A. J. Hay. 1991. Structural characteristics of the M2 protein of influenza A viruses: evidence that it forms a tetrameric channel. Virology. 180:617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeuchi, K., and R. A. Lamb. 1994. Influenza virus M2 protein ion channel activity stabilizes the native form of fowl plague virus hemagglutinin during intracellular transport. J. Virol. 68:911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grambas, S., and A. J. Hay. 1992. Maturation of influenza A virus hemagglutinin—estimates of the pH encountered during transport and its regulation by the M2 protein. Virology. 190:11–18. [DOI] [PubMed] [Google Scholar]
- 6.Chizhmakov, I. V., F. M. Geraghty, D. C. Ogden, A. Hayhurst, M. Antoniou, and A. J. Hay. 1996. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J. Physiol. 494:329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimbo, K., D. L. Brassard, R. A. Lamb, and L. H. Pinto. 1996. Ion selectivity and activation of the M2 ion channel of influenza virus. Biophys. J. 70:1335–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duff, K. C., P. J. Gilchrist, A. M. Saxena, and J. P. Bradshaw. 1994. Neutron diffraction reveals the site of amantadine blockade in the influenza A M2 ion channel. Virology. 202:287–293. [DOI] [PubMed] [Google Scholar]
- 9.Pinto, L. H., and R. A. Lamb. 1995. Understanding the mechanism of action of the anti-influenza virus drug amantadine. Trends Microbiol. 3:271. [DOI] [PubMed] [Google Scholar]
- 10.Hu, J., T. Asbury, S. Achuthan, C. Li, R. Bertram, J. R. Quine, R. Fu, and T. A. Cross. 2007. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from influenza A virus. Biophys. J. 92:4335–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mould, J. A., J. E. Drury, S. M. Frings, U. B. Kaupp, A. Pekosz, R. A. Lamb, and L. H. Pinto. 2000. Permeation and activation of the M2 ion channel of influenza A virus. J. Biol. Chem. 275:31038–31050. [DOI] [PubMed] [Google Scholar]
- 12.Pinto, L. H., L. J. Holsinger, and R. A. Lamb. 1992. Influenza virus M2 protein has ion channel activity. Cell. 69:517–528. [DOI] [PubMed] [Google Scholar]
- 13.Wang, C., R. A. Lamb, and L. H. Pinto. 1995. Activation of the M2 ion channel of influenza virus: a role for the transmembrane domain histidine residue. Biophys. J. 69:1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishimura, K., S. Kim, L. Zhang, and T. A. Cross. 2002. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR. Biochemistry. 41:13170–13177. [DOI] [PubMed] [Google Scholar]
- 15.Tian, C., P. F. Gao, L. H. Pinto, R. A. Lamb, and T. A. Cross. 2003. Initial structural and dynamic characterization of the M2 protein transmembrane and amphipathic helices in lipid bilayers. Prot. Sci. 12:2597–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torres, J., and I. T. Arkin. 2002. C-Deuterated alanine: a new label to study membrane protein structure using site-specific infrared dichroism. Biophys. J. 82:1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torres, J., A. Kukol, and I. T. Arkin. 2000. Use of a single glycine residue to determine the tilt and orientation of a transmembrane helix. A new structural label for infrared spectroscopy. Biophys. J. 79:3139–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang, Y., F. Zaitseva, R. A. Lamb, and L. H. Pinto. 2002. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J. Biol. Chem. 277:39880–39886. [DOI] [PubMed] [Google Scholar]
- 19.Okada, A., T. Miura, and H. Takeuchi. 2001. Protonation of histidine and histidine-tryptophan interaction in the activation of the M2 ion channel from influenza A virus. Biochemistry. 40:6053–6060. [DOI] [PubMed] [Google Scholar]
- 20.Sansom, M. S. P., I. D. Kerr, G. R. Smith, and H. S. Son. 1997. The influenza A virus M2 channel: a molecular modeling and simulation study. Virology. 233:163–173. [DOI] [PubMed] [Google Scholar]
- 21.Forrest, L. R., A. Kukol, I. T. Arkin, D. P. Tieleman, and M. S. P. Sansom. 2000. Exploring models of the influenza A M2 channel: MD simulations in a phospholipid bilayer. Biophys. J. 78:55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kass, I., and I. T. Arkin. 2005. How pH opens a H+ channel: the gating mechanism of influenza A M2. Structure. 13:1789–1798. [DOI] [PubMed] [Google Scholar]
- 23.Smondyrev, M., and G. A. Voth. 2002. Molecular dynamics simulation of proton transport through the influenza A virus M2 channel. Biophys. J. 83:1987–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu, Y., and G. A. Voth. 2003. Computational studies of proton transport through the M2 channel. FEBS Lett. 552:23–27. [DOI] [PubMed] [Google Scholar]
- 25.Wu, Y., and G. A. Voth. 2005. A computational study of the closed and open states of the influenza A M2 proton channel. Biophys. J. 89:2402–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamb, R. A., S. L. Zebedee, and C. D. Richardson. 1985. Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface. Cell. 40:627–633. [DOI] [PubMed] [Google Scholar]
- 27.Duff, K. C., and R. H. Ashley. 1992. The transmembrane domain of influenza A M2 protein forms amantadine-sensitive protein channels in planar lipid bilayers. Virology. 190:485–489. [DOI] [PubMed] [Google Scholar]
- 28.Schroeder, C., C. M. Ford, S. A. Wharton, and A. J. Hay. 1994. Functional reconstitution in lipid vesicles of influenza virus M2 protein expressed by baculovirus: evidence for proton transfer activity. J. Gen. Virol. 75:3477–3484. [DOI] [PubMed] [Google Scholar]
- 29.Holsinger, L. J., and R. A. Lamb. 1991. Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds. Virology. 183:32–43. [DOI] [PubMed] [Google Scholar]
- 30.Kukol, A., P. D. Adams, L. M. Rice, A. T. Brunger, and I. T. Arkin. 1999. Experimentally based orientational refinement of membrane protein models: a structure for the influenza A M2 H+ channel. J. Mol. Biol. 286:951–962. [DOI] [PubMed] [Google Scholar]
- 31.Bauer, C. M., L. H. Pinto, T. A. Cross, and R. A. Lamb. 1999. The influenza virus M2 ion channel protein. Probing the structure of the transmembrane domain in intact cells by using engineered disulfide cross-linking. Virology. 254:196–209. [DOI] [PubMed] [Google Scholar]
- 32.Lamb, R. A., and C.-J. Lai. 1981. Conservation of the influenza virus membrane protein (M1) amino acid sequence and an open reading frame of RNA segment 7 encoding a second protein (M2) in H1N1 and H3N2 strains. Virology. 112:746–751. [DOI] [PubMed] [Google Scholar]
- 33.Wang, C., K. Takeuchi, L. H. Pinto, and R. A. Lamb. 1993. Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J. Virol. 67:5585–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang, J., S. Kim, F. Kovacs, and T. A. Cross. 2001. Structure of the transmembrane region of the M2 protein H+ channel. Prot. Sci. 10:2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cristian, L., J. D. Lear, and W. F. DeGrado. 2003. Use of thio-disulfide equilibria to measure the energetics of assembly of transmembrane helices in phospholipid bilayers. Proc. Natl. Acad. Sci. USA. 100:14772–14777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duong-Ly, K. C., V. Nanda, W. F. DeGrado, and K. P. Howard. 2005. The conformation of the pore region of the M2 proton channel depends on lipid bilayer environment. Prot. Sci. 14:856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Decoursey, T. E. 2003. Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 83:475–579. [DOI] [PubMed] [Google Scholar]
- 38.Vijayvergiya, V., R. Wilson, A. Chorak, P. F. Gao, T. A. Cross, and D. D. Busath. 2004. Proton conductance of influenza virus M2 protein in planar lipid bilayers. Biophys. J. 87:1697–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chizhmakov, I. V., D. C. Ogden, F. M. Geraghty, A. Hayhurst, A. Skinner, T. Betakova, and A. J. Hay. 2003. Differences in conductance of M2 proton channels of two influenza viruses at low and high pH. J. Physiol. 546:427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lear, J. D. 2003. Proton conduction through the M2 protein of the influenza A virus; a quantitative, mechanistic analysis of experimental data. FEBS Lett. 552:17–22. [DOI] [PubMed] [Google Scholar]
- 41.Pinto, L. H., G. R. Dieckmann, C. S. Gandhi, C. G. Papworth, J. Braman, M. A. Shaughnessy, J. D. Lear, R. A. Lamb, and W. F. Degrado. 1997. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc. Natl. Acad. Sci. USA. 94:11301–11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swanson, J. M. J., C. M. Maupin, H. Chen, M. K. Petersen, J. Xu, Y. Wu, and G. A. Voth. 2007. Proton solvation and transport in aqueous and biomolecular systems: Insights from computer simulations. J. Phys. Chem. B. 111:4300–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Agmon, N. 1995. The Grotthuss mechanism. Chem. Phys. Lett. 244:456–462. [Google Scholar]
- 44.Hu, J., R. Fu, K. Nishimura, L. Zhang, H.-X. Zhou, D. D. Busath, V. Vijayvergiya, and T. A. Cross. 2006. Histidines, heart of the hydrogen ion channel from influenza A virus: toward an understanding of conductance and proton selectivity. Proc. Natl. Acad. Sci. USA. 103:6865–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong, Q., D. M. Newns, P. Pattnaik, J. D. Lear, and M. L. Klein. 2000. Two possible conducting states of the influenza A virus M2 ion channel. FEBS Lett. 473:195–198. [DOI] [PubMed] [Google Scholar]
- 46.Stouffer, A. L., V. Nanda, J. D. Lear, and W. F. DeGrado. 2005. Sequence determinants of a transmembrane proton channel: an inverse relationship between stability and function. J. Mol. Biol. 347:169–179. [DOI] [PubMed] [Google Scholar]
- 47.Schmitt, U. W., and G. A. Voth. 1999. The computer simulation of proton transport in water. J. Chem. Phys. 111:9361–9381. [Google Scholar]
- 48.Day, T. J. F., A. V. Soudakov, M. Cuma, U. W. Schmitt, and G. A. Voth. 2002. A second generation multistate empirical valence bond model for proton transport in aqueous systems. J. Chem. Phys. 117:5839–5849. [Google Scholar]
- 49.Wu, Y., H. Chen, F. Wang, F. Paesani, and G. A. Voth. 2007. An improved multistate empirical valence bond model for aqueous proton solvation and transport. J. Phys. Chem. B. In press. [DOI] [PubMed]
- 50.Xu, J., and G. A. Voth. 2005. Computer simulation of explicit proton translocation in cytochrome c oxidase: the D-pathway. Proc. Natl. Acad. Sci. USA. 102:6795–6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen, H., Y. Wu, and G. A. Voth. 2006. Origins of proton transport behavior from selectivity domain mutations of the aquaporin-1 channel. Biophys. J. 90:L73–L75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen, H., B. Ilan, Y. Wu, F. Zhu, K. Schulten, and G. A. Voth. 2007. Charge delocalization in proton channels. I. The aquaporin channels and proton blockage. Biophys. J. 92:46–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu, Y., B. Ilan, and G. A. Voth. 2007. Charge delocalization in proton channels. II: The synthetic LS2 channel and proton selectivity. Biophys. J. 92:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adcock, C., G. R. Smith, and M. S. P. Sansom. 1998. Electrostatics and the selectivity of ligand-gated ion channels. Biophys. J. 75:1211–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- 56.Smondyrev, A. M., and M. L. Berkowitz. 1999. United atom force field for phospholipid membranes. Constant pressure molecular dynamics simulation of DPPC/water system. J. Comput. Chem. 20:531–545. [Google Scholar]
- 57.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 58.Wang, J., W. Wang, P. A. Kollman, and D. A. Case. 2006. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Mod. 25:247–260. [DOI] [PubMed] [Google Scholar]
- 59.Hoover, W. G. 1985. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 31:1695–1697. [DOI] [PubMed] [Google Scholar]
- 60.Essman, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- 61.Smith, W., and T. Forester. 1996. DL_POLY_2.0: a general-purpose parallel molecular dynamics simulation package. J. Mol. Graph. 14:136–141. [DOI] [PubMed] [Google Scholar]
- 62.Smart, O. S., J. M. Goodfellow, and B. A. Wallace. 1993. The pore dimensions of Gramicidin A. Biophys. J. 65:2455–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smondyrev, A. M., and G. A. Voth. 2002. Molecular dynamics simulation of proton transport near the surface of a phospholipid membrane. Biophys. J. 82:1460–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kumar, S., D. Bouzida, R. H. Swendsen, P. A. Kollman, and J. M. Rosenberg. 1992. The weighted histogram analysis method for free-energy calculations. J. Comp. Chem. 13:1011–1021. [Google Scholar]
- 65.Cuma, M., U. W. Schmitt, and G. A. Voth. 2001. A multi-state empirical valence bond model for weak acid dissociation in aqueous solution. J. Phys. Chem. A. 105:2814–2823. [Google Scholar]
- 66.Allen, T. W., O. S. Andersen, and B. Roux. 2004. Energetics of ion conduction through the gramicidin channel. Proc. Natl. Acad. Sci. USA. 101:117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Woolf, T. B., and B. Roux. 1994. Conformational flexibility of o-phosphorylcholine and o-phosphorylethanolamine: a molecular dynamics study of solvation effects. J. Am. Chem. Soc. 116:5916–5926. [Google Scholar]
- 68.Hummer, G. 2006. Position-dependent diffusion coefficients and free energies from Bayesian analysis of equilibrium and replica molecular systems. N. J. Phys. 7:34–47. [Google Scholar]
- 69.Levitt, D. 1991. General continuum theory for multiion channel. I. Theory. Biophys. J. 59:271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gordon, J. C., J. B. Myers, T. Folta, V. Shoja, L. S. Heath, and A. Onufriev. 2005. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 33:W368–W371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bashford, D., and M. Karplus. 1990. pKa of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochem. 29:10219–10225. [DOI] [PubMed] [Google Scholar]
- 72.Lin, T.-I., and C. Schroeder. 2001. Definitive assignment of proton selectivity and attoampere unitary current to the M2 ion channel protein of influenza A virus. J. Virol. 75:3647–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Furini, S., F. Zerbetto, and S. Cavalcanti. 2006. Application of the Poisson-Nernst-Plank theory with space-dependent diffusion coefficients to KcsA. Biophys. J. 91:3162–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Noskov, S. Y., W. Im, and B. Roux. 2004. Ion permeation through the α-hemolysin channel: theoretical studies based on Brownian dynamics and Poisson-Nernst-Plank electrodiffusion theory. Biophys. J. 87:2299–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kurnikova, M. G., R. D. Coalson, P. Graf, and A. Nitzan. 1999. A lattice relaxation algorithm for three-dimensional Poisson-Nernst-Plank theory with application to ion transport through the Gramicidin A channel. Biophys. J. 76:642–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ogden, D., I. V. Chizhmakov, F. M. Geraghty, and A. J. Hay. 1999. Virus ion channels. Methods Enzymol. 294:490–506. [DOI] [PubMed] [Google Scholar]
- 77.Venkataraman, P., R. A. Lamb, and L. H. Pinto. 2005. Chemical rescue of histidine selectivity filter mutants of the M2 ion channel of influenza A virus. J. Biol. Chem. 280:21463–21472. [DOI] [PubMed] [Google Scholar]
- 78.Williams, R. J. P. 1978. The multifarious couplings of energy transduction. BBA. 505:1–44. [DOI] [PubMed] [Google Scholar]
- 79.Brändén, M., T. Sandén, P. Brzezinski, and J. Widengren. 2006. Localized proton microcircuits at the biological membrane-water interface. Proc. Natl. Acad. Sci. USA. 103:19766–19770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gogievskii, Y., E. S. Medvedev, and A. A. Stuchebrukhov. 2002. Proton transport via the membrane surface. Biophys. J. 82:2833–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gutman, M., A. B. Kotlyar, N. Borovok, and E. Nachliel. 1993. Reaction of bulk protons with a mitochondrial inner membrane preparation: time-resolved measurements and their analysis. Biochemistry. 32:2942–2946. [DOI] [PubMed] [Google Scholar]
- 82.Gutman, M., E. Nachliel, and S. Moshiach. 1989. Dynamics of proton diffusion within the hydration layer of phospholipid membrane. Biochemistry. 28:2936–2940. [DOI] [PubMed] [Google Scholar]
- 83.Song, Z., F. A. Kovacs, J. Wang, J. K. Denny, S. C. Shekar, J. R. Quine, and T. A. Cross. 2000. Transmembrane domain of M2 protein from influenza A virus studied by solid-state 15N polarization inversion spin exchange at magic angle NMR. Biophys. J. 79:767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Allen, T. W., O. S. Andersen, and B. Roux. 2006. Ion permeation through a narrow channel: using gramicidin to ascertain all-atom molecular dynamics potential of mean force methodology and biomolecular force fields. Biophys. J. 90:3447–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pinto, L. H., and R. A. Lamb. 2006. The M2 proton channels of influenza A and B viruses. J. Biol. Chem. 281:8997–9000. [DOI] [PubMed] [Google Scholar]
- 86.Mould, J. A., R. G. Paterson, M. Takeda, Y. Ohigashi, P. Venkataraman, R. A. Lamb, and L. H. Pinto. 2003. Influenza B virus BM2 protein has ion channel activity that conducts protons across membranes. Dev. Cell. 5:175–184. [DOI] [PubMed] [Google Scholar]
- 87.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: Visual Molecular Dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]