

Abstract
In this article, we present a statistical analysis of the electrostatic properties of 298 protein-protein complexes and 356 domain-domain structures extracted from the previously developed database of protein complexes (ProtCom, http://www.ces.clemson.edu/compbio/protcom). For each structure in the dataset we calculated the total electrostatic energy of the binding and its two components, Coulombic and reaction field energy. It was found that in a vast majority of the cases (>90%), the total electrostatic component of the binding energy was unfavorable. At the same time, the Coulombic component of the binding energy was found to favor the complex formation while the reaction field component of the binding energy opposed the binding. It was also demonstrated that the components in a wild-type (WT) structure are optimized/anti-optimized with respect to the corresponding distributions, arising from random shuffling of the charged side chains. The degree of this optimization was assessed through the Z-score of WT energy in respect to the random distribution. It was found that the Z-scores of Coulombic interactions peak at a considerably negative value for all 654 cases considered while the Z-score of the reaction field energy varied among different types of complexes. All these findings indicate that the Coulombic interactions within WT protein-protein complexes are optimized to favor the complex formation while the total electrostatic energy predominantly opposes the binding. This observation was used to discriminate WT structures among sets of structural decoys and showed that the electrostatic component of the binding energy is not a good discriminator of the WT; while, Coulombic or reaction field energies perform better depending upon the decoy set used.
INTRODUCTION
Protein-protein interactions constitute the key mechanism maintaining the function of the cell (1). Understanding the physical principles governing these interactions (2–7) and the ability to predict both interacting partners (8–12) and three-dimensional structures of the corresponding complexes (13–16) are therefore very important tasks. Electrostatic interactions, being long-range interactions, are of particular interest for protein-protein association. Because of this, the protein-protein complexes with experimentally available three-dimensional structures were intensively studied both statistically and energetically to reveal the contribution of the electrostatic energy to the binding (17–21). It was found experimentally (22,23) and computationally (24) that most of the ionizable residues at the protein-protein interfaces contribute to the binding energy, i.e., their replacement with the alanine residue critically affects protein binding affinity. It was pointed out that electrostatic interactions play a more important role in the protein binding than they do in folding (see, e.g., (25) and references therein). In many cases, a formation of a complex could result in favorable pairwise interactions across the interface as it was demonstrated by Tidor and co-workers in case of the barnase-barstar complex (26,27) and for other complexes (28). One of the largest series of works devoted to computation of electrostatic properties for different groups of complexes is that by McCammon and co-workers (29–34) including the role of the salt bridges (30). The role of electrostatic interactions in the formation of protein-protein interfaces was thoroughly studied by Honig and co-workers (6,35,36). It was pointed out that electrostatic interactions play a dominant role in the case of complexes with small interfaces. The contribution of the electrostatic energy to the binding affinity of Rap/Raf complex was also the subject of a series of investigations (37,38). Despite the fact that all of the above studies agreed that there are many specific pairwise interactions across the interface, the conclusions about the role of electrostatics on the binding affinity remain controversial. It was found that, in some cases, the electrostatics favor the binding, but in other cases, oppose it. Since the electrostatic component of the binding energy is the difference between two large terms, namely pairwise interactions and the desolvation penalty, the outcome strongly depends on the force-field parameters, including the choice of the internal dielectric constant of proteins (39). These observations are similar to those made for the contribution of the salt bridges to the stability of proteins (40,41). In addition, as pointed out by Zhou and co-workers (42–44), the electrostatic component of the binding energy is very sensitive to the method of building the molecular surface.
The salt dependence of the binding energy is an important characteristic of the protein-protein interactions. It was extensively studied by Zhou and co-workers and it was shown that the increase of the ionic strength makes the binding weaker in the case of barnase-barstar (45) and weakens the on- and off-rates in the case of five protein-protein complexes (46). Recently, it has been pointed out that the increase of the salt concentration weakens the binding affinity of five hetero and one homo protein complexes (39). The set of protein-protein complexes used in Bertonati et al. (39) included five cases of monomers carrying opposite charges as well as two complexes made of monomers having the same polarity net charge. In the last case, if the charges of the monomers were uniformly distributed, then these interactions could be treated as interactions between entities with opposite charges and, from a macroscopic point of view, the increase of the salt concentration should screen the unfavorable interactions and thus should make the binding stronger. However, both experimental data and the numerical calculations show that the increase of the ionic strength weakens the binding for all complexes. This indirectly suggests that the charges are not distributed randomly but rather form specific interactions across the interface of protein-protein complexes.
The pKa values of ionizable groups are important indicators of the environment that proteins and protein-protein complexes provide for the ionizable groups. The formation of a complex may change the pKa values of titratable residues in respect to the pKa values in the unbound monomers, especially if these residues are located within the interface of the complex. The resulting pKa shifts can be used as an indicator of electrostatic energy contribution of a particular residue to the stability of the complex. For instance, an acidic residue, pKa of which shifts upon the complex formation toward acidic pH values (negative pKa shift), stabilizes the complex as compared to the complex with this residue replaced by a noncharged (e.g., Ala) group. Since complex formation buries interfacial residues, this will result in a desolvation penalty which can be compensated only by favorable pairwise interactions across the interface, which will require appropriate arrangement of the titratable groups at the interface. If the titratable groups are distributed randomly within the interface of protein-protein complexes, then the statistical expectation will be that the formation of the complex should increase the average pKa values of the acidic groups due to the desolvation penalty. However, a recent study shows that the pKa shifts of acidic groups induced by the complex formation are predominantly negative (7). This indicates that the complex provides a more favorable environment for these groups as compared to the monomers. This indirectly indicates that the ionizable groups are not distributed randomly, but rather their location is optimized within the protein-protein interfaces.
The pioneering work on the optimization of Coulombic interactions within monomeric proteins was done by Spassov and Karshikoff (47–49). They had shown that the Coulombic interactions are optimized in respect to the random distribution of a point charges. Recently, we applied explicit side-chain replacement in the randomization procedure to address the electrostatic energy optimization in two isoforms of plastocyanin (50). It was shown that pairwise interactions are optimized while both the reaction field energy and the interactions with mobile ions are anti-optimized (here we use the terms “optimization” and “anti-optimization” with respect to the tendency on the binding affinity, favoring or disfavoring the binding, respectively). However, the role of the electrostatic component of the binding energy on the complex formation, and how optimized these interactions are, has never been statistically addressed. The newly created large databases of three-dimensional structures of protein-protein complexes (51–56) provide the necessary pool for large-scale studies and modeling. Hence, the above-mentioned observations inspired us to study the possibility that the electrostatic energy and its components are optimized within protein-protein complexes as well. We took advantage of our previously developed large database of protein-protein complexes (ProtCom) (52) to address these questions on a set of 298 protein-protein complexes and 356 domain-domain structures, with emphasis on the optimization of the electrostatic component of the binding energy and its components.
The results obtained in this study could be used in evaluation of the quality of the structures of protein-protein complexes. Predicting three-dimensional structures of protein-protein complexes is one of the most important tasks in the post-genomic era and many efforts are currently devoted to advance the modeling techniques (7,14,57–61). However, in many cases, the same pair of sequences with unknown structures (query sequences) produces several models and hence, tools are needed to evaluate and rank these models. The same is valid for docking methods (62–64), which generate large numbers of alternative conformation of a complex, given the three-dimensional structures of the monomers. These alternative models need to be evaluated to select the nativelike three-dimensional structure of the complex. It is desirable for the scoring algorithm to be fast and not require extensive energy minimization. Here, we address such a possibility by ranking decoy protein-protein complexes with electrostatic binding energy and its components, and with an in-house-derived kernel function based on a combination of Z-scores of Coulombic and reaction field energy components of the electrostatic energy.
METHODS
The set of protein structures used in the study
Protein-protein complexes subjected to the study were extracted from the ProtCom (52) database (as of June 2006) (www.ces.clemson.edu/compbio/protcom), which contains more than 3000 entries. To avoid the bias toward overrepresented complexes, the entries were purged with CD-hit (65) at 40% sequence identity level (note that this requirement automatically removes all homo complexes). This resulted in 298 protein-protein complexes and 356 domain-domain structures. The protein-protein complexes were manually classified into five major classes: antibody-antigen complexes, enzyme-inhibitors structures, G-proteins, transport proteins, and other ensembles. All structures were subjected to the Jackal program (http://wiki.c2b2.columbia.edu/honiglab_public/index.php/Software:Jackal, which was developed in Honig's lab to fix missing atoms and side chains; note that the domain-domain structures in the PotCom database are not real complexes but are artificially made from monomeric proteins with two distinctive domains; for details, see (52)).
The vast majority of the results reported in this study were done using nonminimized structures since minimization of all 131,454 (native structures and 200 mutants of each of the 654 complexes) structures is far beyond the available computational resources. However, to test the sensitivity of the results, short minimization runs were performed in the case of the α-chymotrypsin-eglin C complex and the corresponding 200 mutants. The details of the minimization protocol are as follows: each structure was minimized with the Tinker package (66) using its “minimize.x” module by means of the quasi-Newton optimization procedure. The implicit solvent Still GB model (67) and the CHARMM27 (68) force field were used. To make the problem computationally tractable, we applied a weak convergence criteria (RMS gradient per atom = 0.5).
Shuffling of the charged side chains
The randomization of the charged side chains was done in the following manner: For each of the monomers within a particular complex, a list of charged groups (Asp, Glu, Lys, Arg, and His) was created from the corresponding Protein Data Bank (PDB) file (69). A residue from this list was randomly picked up and swapped with a residue randomly picked within the entire structure of the same monomer. The second residue can be of any type thus not restricted to charged groups only. This results in better randomization of the corresponding sequence. Hereafter a structure with shuffled residues will be referred to as a sequence decoy. In addition, two protocols were tested for creating a sequence decoy: A protocol that allows any residues to participate in the randomization procedure and a protocol that restricts the sites of possible randomization to surface residues only (surface residues are defined as residues retaining in the structure >20% of their side-chain solvent-accessible surface area). On a test set of protein-protein complexes, the side chains of the titratable groups were swapped 500 times in each of the monomers and corresponding electrostatic energies and their components (see below) were calculated. Then the calculations were repeated with 200 randomizations per monomer and the resulting energy distributions were compared to the distributions from the previous run. No significant difference was found and the rest of the calculations were performed with 200 randomizations per monomer. The side-chain replacement was done with the SCAP (70) program developed in the Honig lab with the default set of parameters.
Electrostatic calculations
The wild-type (WT) PDB files and the corresponding structures with randomized side chains of ionizable groups (sequence decoys) were protonated with the Multi-Conformational Continuum Electrostatics (MCCE) (71–73) program. It was recently demonstrated that MCCE-generated proton positions are highly accurate (74). Then the structures of the complexes and separated monomers were outputted to Delphi (75,76) to calculate the Coulombic and reaction field energies components of total electrostatic energy as described in the details in Rocchia et al. (75). Coulombic energy was calculated in the absence of salt in homogeneous media with the dielectric constant of the solute. The reaction field energy was calculated as the interaction energy between permanent and induced surface charges in the absence of salt (75). Parse charges and radii (77) were used. The dielectric constant of the solute was 2 and water phase was modeled with a dielectric constant of 80 in most of the calculations. However, to test the sensitivity of the results, the electrostatic component of the binding energy was calculated with internal dielectric constants of 4 and 20. The salt concentration was set to zero. The grid size of the finite-difference algorithm was kept at 65 to speed up the calculations. Such a grid size resulted in a resolution of 1 grid/Å or better. As it was demonstrated in the past, Delphi calculations are accurate enough at resolution higher than 1 grid/Å (76,78).
The electrostatic components of the binding energy were calculated using the rigid body approach, keeping the structure of the monomers in the same conformation as they have in the complex structure. Single point calculations were applied and the corresponding component of the binding energy was calculated as
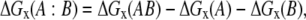 |
(1) |
where x stands for either the Coulombic (ΔGcoul), reaction field (ΔGrxn), or the total electrostatic energies (ΔGel), respectively. Hereafter, the corresponding quantities for the entire complex are marked with AB, those for the monomers with either A or B and those for the binding energy with A:B.
Z-score
The electrostatic energies of the randomized structures were used to obtain the distribution of the energy. The mean (〈ΔGx〉) of the distribution and the corresponding standard deviation (σx) were calculated with standard formulas:
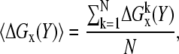 |
(2) |
and
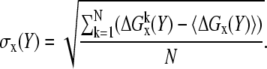 |
(3) |
In the above formulas, Y stands for either AB (entire complex), A or B (monomer), or the A:B (corresponding component of the binding energy). The number of samples was 200 in this study. The distributions of energies for randomized structures (sequence decoys) have a Gaussian, bell-like shape and therefore, it is convenient to compare energy optimization for the wild-type structure (WT) for different complexes using the Z-score calculated as
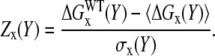 |
(4) |
RESULTS
Distributions of the electrostatic binding energy and its components
The binding energies ΔGx(A:B) were calculated using Eq. 1 for each of the 654 entries in our dataset for the total electrostatic energy (x = el), the Coulombic interactions (x = coul), and for the reaction field energy (x = rxn) using three different values of dielectric constants for the protein interior (ɛp). The distributions of these quantities are shown in Fig. 1 (for better presentation, outliers, representing <5% of the cases, were omitted from the graph). No significant difference was observed for the calculated energies of protein-protein complexes and domain-domain structures. In this section, we show them together. As it is seen, in a majority of the cases, ΔGel(A:B) is positive (Fig. 1 A), indicating that the total electrostatic interactions oppose the binding. The obtained distributions vary significantly with the internal dielectric constant, ɛp, but in all cases the energies are predominantly positive. The mean of the distributions are +90 kcal/mol, +30 kcal/mol, and +10 kcal/mol for ɛp = 2, 4, and 20, respectively. The distribution of ΔGel(A:B) calculated with ɛp = 20 is much narrower than those calculated with ɛp = 2 and 4, simply because the large dielectric constant reduces the magnitude of calculated energies. However, in all cases the distributions have a long tail stretching toward large positive energies (the right side of the graph).
FIGURE 1.

Distribution of the total electrostatic binding energy and its components over 658 protein-protein complexes and domain-domain structures calculated for three different dielectric constants of proteins: (A) total electrostatic binding energy; (B) Coulombic component of the binding energy; and (C) reaction field component of the binding energy.
The distribution of ΔGcoul(A:B) is shown in Fig. 1 B and it can be seen that the mean of all distributions is shifted to negative values. This indicates that Coulombic energy favors the binding for a majority of the structures studied in this work. At the same time, ΔGrxn(A:B) shows an opposite trend. In a vast majority of the cases, it was calculated to be positive, thus opposing the binding. The tendency is even stronger as compared to the trend of the Coulombic component. The variation of the internal dielectric constant affects the magnitude of these energies, and at a high dielectric constant ɛp = 20, both distributions are narrower. In several cases, ΔGrxn(A:B) was calculated to be a negative number (the left side of Fig. 1 B). Since ΔGrxn(A:B) is the electrostatic component of the change of the solvation energy upon the binding (usually called desolvation energy), one may wonder how it could be a negative number. The analysis showed that these outliers exhibit strong repulsive Coulombic interactions due to monomers bearing a large net charge of the same polarity. Electrostatic calculations of a complex consisting of two monomers carrying large net charge of the same polarity could result to a reaction field energy more negative than the sum of the reaction field energies calculated for the separated monomers, and thus ΔGrxn(A:B) < 0. In part, that results from the assignment of default ionization states of all titratable groups and thus, in some cases, may overestimate the net charge of the monomers. However, computationally, it is almost impossible to perform thorough atomic scale electrostatic calculations (with accurate assignment of ionization states) within such a large-scale study (654 entries).
The above results were obtained using a particular set of radii and partial charges (Parse parameters (77)). To test the sensitivity of the results obtained in respect to these parameters, we performed calculations using parameters from a different force field (CHARMM (79)) on a subset of our dataset. This resulted in different magnitudes of the binding energies and their components (results not shown, but the trends were the same as described above: the electrostatic energy opposes the binding. However, the negativity of the Coulombic component of the binding energy in the vast majority of the cases suggests that the electrostatic interactions assist the monomers in their initial approach to each other (so-called steering effect). At distances of the magnitude of one water layer, the desolvation penalty rapidly increases and the role of the electrostatics depends upon the precise balance between favorable Coulombic interactions and unfavorable desolvation energy. It should be mentioned that, at such short distances, van der Waals energy, specific interactions, and the change of entropy may be the driving forces of the binding. Finally, the observation that electrostatics oppose the binding should be taken with certain precautions since the absolute value of the calculated electrostatic component of the binding energy, as pointed out by Zhou and co-workers (42), depends on how the dielectric boundary between solute and the water phase is determined. Using the van der Waals surface of the atoms as the surface of a molecule dramatically changes the results (42). All these findings indicate that the calculations of the absolute value of the electrostatic components of the binding energy are sensitive to parameters, the force field, and the method used. It should also be noted that structures in our dataset were not minimized before the energy calculations and any minimization will further affect the results.
Our results indicate that in most of the cases the electrostatics opposes binding. However, the electrostatic energy is only part of the total binding energy, which includes nonelectrostatic and entropy contributions. The total binding energy must be negative for binding to occur, but individual energy contributions do not have to. As it was pointed out in the Introduction, the discussion about the electrostatic contribution to the binding is a sensitive issue and in this article we would like to tackle the problem from a different angle. Namely, we want to see if the electrostatic energy and its components are optimized, given the amino-acid sequences composition and three-dimensional structures of the complexes. In this way, the issue of the absolute value of the energy will be avoided since we will be interested in the energy difference between WT and the set of sequence-randomized complexes. Thus the question that will be addressed is how different are the components of the electrostatic energy of the WT complexes compared to the energies calculated on a set of sequence decoys.
In further analysis below, the value of the dielectric constant will be kept as 2 and the boundary between solute and the water will be determined with water probe with a radius of 1.4 Å. From prospective of the optimization studies, the choice of these parameters is not crucial, since we will be interested in the difference between energies of WT complexes and of complexes with randomized charge groups; thus, the absolute value of the energy is not important.
Z-scores of energies
The concept of this study will be illustrated by analyzing a particular complex (α-chymotrypsin complex with eglin C, PDB code 1ACB) in detail. Following the algorithm described in Methods, we generated a set of 500 sequence decoys (the rest of the results are done with 200 randomizations) by shuffling the side chains of the charged amino acids but keeping the backbone unchanged. For each of decoys, we calculated binding energy components ΔGel(A:B), ΔGcoul(A:B), and ΔGrxn(A:B), and then these energies were used to build the corresponding distributions. Fig. 2 shows resulting distributions of these three electrostatic components of the binding energy (the data points are grouped into 11 equally valued intervals and are fitted with a smooth curve). It is clearly seen from the figure that all distributions are of the symmetric Gaussian type (some small deviations from the Gaussian curve observed in the figure are caused by the limited sampling). On the same figures, we show the corresponding energy component calculated using the WT complex (marked with vertical arrows in Fig. 2). If the electrostatic energy of the WT complex happens to be far away from the mean of the distribution, then this will illustrate that the WT energy is not the result of random distribution of charges, but rather it requires specific organization of the charged groups within the complex. In this particular example, the total electrostatic binding energy ΔGel(A:B) is +25.4 kcal/mol, and that is not much different from the mean of the distribution (+32.4 kcal/mol, Fig. 2 A). In contrast, the Columbic and reaction field components are away from the corresponding means (Fig. 2, B and C). The ΔGcoul(A:B) is −31.8 kcal/mol for the WT structure while the mean of the distribution is ∼+19.5 kcal/mol (Fig. 2 B). This clearly indicates that WT Coulombic interactions in this complex are not random but rather they are highly optimized to favor the stability of the complex. This effect will be referred to throughout the article as optimization of the interaction energy. The reaction field component of the WT structure is also far away from the mean of the distribution (ΔGrxn(A:B)) = +57.2 kcal/mol for the WT structure (and mean is ∼+38.2 kcal/mol, Fig. 2 C), but is located to the right from the mean, i.e., the WT reaction field energy is more positive than expected by chance. Since such tendency opposes the binding further, we will refer to that as anti-optimization.
FIGURE 2.

Distribution of the electrostatic binding energy and its component within a set of 500 decoys. The energy of the WT complex is shown with an arrow. The energies were calculated for Protein Data Bank entry 1acb, bovine α-chymotrypsin-eglin C complex.
To test the sensitivity of the results with respect to the exact side-chain positions and possible structural imperfections, short minimization runs were performed on the native α-chymotrypsin-eglin C complex and each of its mutants (in this case only 200 randomizations instead of 500 were generated to reduce the computational demands). The structures of the monomers were kept as they were in the complex. The minimization of the native complex resulted in Cα RMSD 0.35 Å with respect to the nonminimized structure. The corresponding energy components reported above slightly changed their magnitudes to ΔGel(A:B) = +23.4 kcal/mol, ΔGcoul(A:B) = −39.4 kcal/mol, and ΔGrxn(A:B) = +62.8 kcal/mol. The total electrostatic binding energy becomes slightly less unfavorable, the Coulombic energy becomes more favorable, and reaction field energy becomes less favorable due to the minimization of the structures. However, the changes are small. The resulting energy distributions are smoother and the minimization removes the long tails (very favorable and unfavorable energies). Thus, despite the small changes in the magnitude of the energy components and in the mean/standard deviation of the corresponding distributions, the resulting Z-scores are practically the same as for the nonminimized structures.
Z-score distributions
To access the statistical significance of the effects described above we carried out similar calculations on a large set of proteins (for all 654 entries in our dataset). This requires random shuffling of the side chains of all of these complexes and obtaining the corresponding Z-scores for ΔGel(A:B), ΔGcoul(A:B), and ΔGrxn(A:B). During these calculations, we also computed the Z-scores of the Coulombic and reaction field energies of the WT complexes (ΔGcoul(AB), (ΔGrxn(AB)) and separated monomers “A” (ΔGcoul(A) and ΔGrxn(A)) and “B” (ΔGcoul(B), ΔGrxn(B)). It was found that the Z-scores of the total electrostatic binding energy do not have clear tendency and because of that they will not be discussed further. However, the optimization/anti-optimization effects were found to be statistically considerable for both Coulombic interactions and reaction field energy. Below we present details and discuss the results separately for each of these components.
Z-scores distribution of the Coulombic energy
The Z-score distributions of the Coulombic components of the electrostatic energy for all 654 entries in this study are shown in Fig. 3. The Z-scores of the monomers and the complexes are quite similar with and for vast majority of the cases (>90% of proteins in the studied dataset) Z-score of the WT Coulombic energy is a negative number. The mean for all three cases is ∼−2.6, which indicates strong optimization of the Coulombic interactions in monomers and in the complexes. The Coulombic component of the binding energy is also optimized as seen in Fig. 3 D, but the optimization is not as strong as in other three cases (the mean of the Z-score is now ∼−1). However, there is still significant degree of optimization since >90% of the complexes and domains studied in this work have a negative Z-score.
FIGURE 3.

Distribution of the Z-score of the Coulombic component of the electrostatic energy of (A) monomers A (ΔGcoul(A)); (B) monomers B (ΔGcoul(B)); (C) complexes (ΔGcoul(AB)); and (D) binding (ΔGcoul(A:B)).
Reaction field energy
The Z-scores of the reaction field energy components are shown in Fig. 4. Strong anti-optimization can be seen for reaction field energy of the monomers and the complexes (Fig. 4, A–C). In all three cases the mean is ∼+2.0 and very few complexes have a negative Z-score. However, the Z-score of binding energy, (ΔGrxn(A:B)) is almost symmetrical around the zero (Fig. 4 D). There is a slight preference toward small negative Z-scores (the bars on the left side of the zero are much taller than on the right side), which indicates that the anti-optimization of the reaction field energy observed for complexes and monomers is suppressed and even slightly reversed for the binding component of the reaction field energy.
FIGURE 4.

Distribution of the Z-score of the reaction field component of the electrostatic energy of (A) monomers A (ΔGrxn(A)); (B) monomers B (ΔGrxn(A)); (C) complexes (ΔGrxn(A)); and (D) binding (ΔGrxn(A)).
Electrostatic optimization within four classes of protein-protein complexes
Fig. 5 shows Z-scores of Coulombic (ΔGcoul(A:B)) and reaction field (ΔGrxn(A:B)) components of the binding energy separately for the four types of protein-protein complexes in our dataset. The annotation was done manually using the description provided in the header of the corresponding PDB files and thus is not exclusive. Many entries were not annotated and are not shown in Fig. 5. The number of annotated antibody-antigen and G-protein complexes is very small. We will show the results, but their Z-scores cannot be analyzed from a statistical standpoint. The Z-scores of the Coulombic component of the binding energy shows the same trend among all types of protein-protein complexes. It is shifted to negative Z-scores with a maximum of ∼−1. The tail of the distribution for some of the complexes runs to very negative values of <−11. This indicates very prominent optimization of the Coulombic component of the binding energy. The distribution of the Z-scores of the reaction field component of the binding energy is not homogeneous among the different types of complexes. ΔGrxn(A:B) is optimized for the enzyme-inhibitor complexes (negative Z-score), while it is anti-optimized for the transport proteins (slightly positive Z-score). Comparison of Figs. 4 D and 5 B brings forward the conclusion that in terms of ΔGrxn(A:B) most of the complexes in our dataset perform as enzyme-inhibitor complexes, since there is slight tendency of optimization in Fig. 4 D.
FIGURE 5.

Distribution of the Z-score of the electrostatic component of the binding energy for four types of protein complexes: Anti, antibody-antigen; enzyme, enzyme-inhibitor; G, G-protein complexes; and transp, transport proteins.
Using Z-score of the electrostatic energy to rank decoys
Finding the WT structure among structural decoys is usually considered to be an exercise that evaluates the quality of either force fields or scoring functions. In the case of decoy complexes delivered with rigid body approach, the structures of the monomers are the same among the decoys and WT and the only difference is the binding interface. Thus, from an electrostatic point of view, the main difference between WT and decoy complexes is the electrostatic interactions across the interface. Since the decoys are usually generated to have a similar interfacial area, the variability of the charge-charge interactions among structural decoys should have similar trends as the above studied sequence variability (sequence decoys). Here we study the performance of the components of the electrostatic energy and the corresponding Z-scores to rank structural decoys. Since our statistical study found that Coulombic interactions within WT complexes are predominantly negative, it is plausible to rank decoys in respect to their Coulombic energy, assuming that the complexes with lowest Coulombic energy are nativelike (in this case, the Coulombic energy of the complex or the Coulombic component of the binding energy will give the same result because the monomeric structures are the same for all decoys). Similarly, it was demonstrated that reaction field energy is predominantly positive for all complexes in this study. Then, it is plausible to rank the decoys with respect to the most positive reaction field energy of the complex (since the reaction field component of the binding energy was found not to have a clear trend (Fig. 4 D), we will not discuss it here). The performance of these two ranking criteria will be compared with the performance of the corresponding Z-scores. For that purpose we will calculate the Z-score of Coulombic and reaction field energy of WT and decoy complexes. Decoys with most negative Z-score of the Coulombic energy will be considered nativelike, while decoys with most positive Z-score of the reaction field energy will be ranked the best. In addition, since the effects are opposite for Coulombic and reaction field energies, we will test the performance of two kernel functions
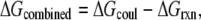 |
(5) |
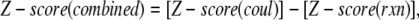 |
(6) |
where ΔGcombined is the energy difference of the Coulombic and reaction field energy of the complex, a quantity that does not have physical meaning, and Z-score(combined) is the difference of the Z-scores of the Coulombic and reaction field energy of the same complex. The minus sign comes from the observation that these two energy terms (Coulombic and reaction field energy) show opposite trends. In Discussion, we talk about that issue because of the strong statistical correlation between Coulombic and reaction field energies.
The test was performed using all available decoy sets from the Vakser lab (http://vakser.bioinformatics.ku.edu/files/decoys/database.html). These include trypsin-BPTI, subtilisin-chymotrypsin inhibitor, chymotrypsin-ovomucoid 3rd domain, and barnase-barstar. Each set includes the native structure and 100 decoys. Both the WT and decoys are not minimized, which is suitable for our approach.
Commonly used criterion for ranking rigid-body-generated decoys is to rank them according to the nonbonded interaction energy. Since the internal structure of the monomers is the same for all of the decoys, the internal mechanical energy is a constant and does not affect the ranking. The nonbonded energies include electrostatic (Coulombic and reaction field) energy, van der Waals (vdW) energy, and so-called surface tension energy proportional to the interface of the complexes. In our case, the complexes are not minimized and thus the vdW cannot be reliably calculated and is not taken into account below. The performances of the total electrostatic binding energy (ΔGel(A:B)) and of the total binding energy excluding vdW (ΔGtot) are shown in the Supplementary Material for all four complexes. In most of cases, the WT complex is calculated to have energy less favorable than most of the decoys, indicating that this ranking criterion does not work for these four complexes. For comparison, applying the Z-score (combined) as a criterion drastically improves the ranking of the WT complexes (see Supplementary Material).
The performance of all above-defined ranking criteria is summarized in Table 1, where we show the ranks of four WT complexes among hundreds of decoys. It can be seen that the ranking of the WT complexes with the total electrostatic binding energy (ΔGel(A:B)) is not good. The most pronounced is the effect for the barnase-barstar complex, where the ranking of the WT with ΔGel(A:B) is 86 while WT ranks first or second with the Z-score of reaction field energy and combined Z-score, respectively. Among the direct energy criteria, the reaction field energy of the complex (ΔGrxn(AB)) performs the best and even outperforms the combined Z-score in two cases. Despite that, these four sets of decoys are not enough to draw definite conclusions. It seems that there is no significant difference for the performance of the direct energy and the Z-score methods. In two cases, the direct energy method generates the best ranking while, in the other two, the Z-score does. It is not surprising that they perform similarly, since in this case of rigid-body-generated decoys, the structural and sequence variation should give very similar effects.
TABLE 1.
Rank of wild-type (WT) structure among 100 structural decoys with respect to four different ranking schemes for four protein-protein complexes
| Rank
|
|||||||
|---|---|---|---|---|---|---|---|
| WT protein complex | ΔGel(AB) | ΔGcoul(AB) | ΔGrxn(AB) | ΔGcoul(AB) - ΔGrxn(AB) | Z-score of ΔGcoul(AB) | Z-score of ΔGrxn(AB) | Z-score (combined) |
| Trypsin-BPTI | 93 | 16 | 1 | 11 | 17 | 27 | 15 |
| Subtilisin-chymotrypsin inhibitor | 64 | 34 | 20 | 21 | 7 | 47 | 16 |
| Chymotrypsin-ovomucoid 3rd domain | 88 | 45 | 14 | 16 | 59 | 42 | 50 |
| Barnase-barstar | 86 | 10 | 5 | 9 | 9 | 1 | 2 |
The best ranking for each of the complexes is shown as bold number.
The calculations were repeated using different dielectric constants and it was found that the ranking does not change significantly for all of the scoring methods. The results with ep = 4 and 20 are very similar to those shown in the Table 1 (results not shown).
To further address the possibility of using electrostatic energy components to rank decoys, we performed a test using the Boston University benchmark set (80) (Benchmark 2.0; http://zlab.bu.edu/zdock/benchmark.shtml) and selecting only binary complexes (see Table 1S in the Supplementary Material). This resulted in a set of 41 protein complexes, and for each complex we generated 1000 decoys using ZDOCK 2.3 (81) and the bound structures of the monomers. Then the WT complexes were ranked with respect to the decoys using the aforementioned electrostatic energy components (see Table 1S in the Supplementary Material section). In contrast to the benchmarks done on Vakser's unbound decoys, the strongest signal was obtained with the Coulombic component of the binding energy. On average, the WT complex was ranked at the top 16–17% of the decoys with the Coulombic energy, while using the reaction field energy ranked the WT within top 38–49%, which is unsatisfactory. The electrostatic component of the binding energy as well as the difference between Coulombic and reaction field energies did not perform well, resulting in ranks from 20 to 26% and 25 to 33%, respectively. This confirms our previous finding that the electrostatic component of the binding energy is not a good criterion for finding the WT complex. However, in contrast to the results on the Vakser's decoys, the reaction field energy does not perform well while the Coulombic energy results are the best. This difference could be due to the fact that Vakser's decoys set is based on unbound structures, while ZDOCK constructed decoys were generated using bound structures. Alternatively, this may simply reflect the difference of the GRAMM (63) and ZDOCK algorithms. However, consistently in both cases, we found that the electrostatic component of the binding energy is not a good discriminator of the WT complexes.
The results of this paragraph suggest that total electrostatic binding energy is not a good criterion for discriminating decoys of protein-protein complexes. Ranking based on the Coulombic interactions performs much better, but not as good as the ranking based on the most positive reaction field energy of the complexes. The kernel function that is the difference between Coulombic and reaction field energies shows a medium performance. Among the Z-score ranking methods, the kernel function of the combined Z-score performs the best. It reaches the performance of the reaction field energy function. These results indicate that reaction field energy is an important factor in ranking decoys and should not be omitted from the ranking algorithms. In addition, the Z-scores provide an alternative method for ranking decoys of protein-protein complexes that, in some cases, outperforms the energy ranking.
DISCUSSION
This large-scale study of the role of the electrostatics on the protein-protein interactions indicates that the electrostatic energy do not necessarily favor the binding. For the vast majority (>90%) of the complexes in our dataset, the calculated total electrostatic binding energy is positive. The results were found to be independent of the internal dielectric constant value. The choice of which is the subject of many debates in the literature (see review (82)). While the results presented here were obtained with the Parse force field, we also tested the outcome of our calculations with the parameters from the CHARMM force field and found no qualitative difference, although the magnitudes of the binding energies were quite different. It should be noted, however, that we did not test the sensitivity of our results with respect to other parameters of the computational protocol such as the dielectric boundary presentation, which could make the results different (42,43). In addition, the structures were not minimized and one can argue that eventual minimization may further optimize the electrostatic interactions and may make the electrostatic contribution into the binding more favorable. Nevertheless, the calculations on a large (654 entries) set of nonrefined x-ray structures resulted in electrostatic energy opposing the binding, and hence, it is plausible to suggest that perhaps electrostatics in WT complexes plays a role mostly in steering the monomers into the complex structure rather than having significant contribution to the affinity.
It is very well known that the amino-acid sequence determines the fold of the proteins. Nevertheless, proteins can, to a certain degree, tolerate amino-acid substitutions and still retain the same fold. Especially, the solvent-exposed charged groups may not be very important in determining three-dimensional structures of monomers and their complexes, but they could, at the same time, be just as important for the solubility of molecules and their complexes. Then their exact locations at the protein surface would be not so important and should not affect the energy of protein and their complexes. However, this study shows that the arrangement of the charged groups is not random in proteins and protein complexes. In particular, it was shown that the Z-score of the Coulombic component of electrostatic interactions in the wild-type structures exhibits strong optimization for both the energies of the entire structures (monomers and/or complexes) and for the energies related to the complex interface (binding energy). The reaction field component was found to be anti-optimized for energies of entire structures only. The anti-optimization tendency is suppressed for the binding energy, and for some proteins the reaction field component of the binding energy is also optimized. However, it should be emphasized that optimization/anti-optimization are measured in respect to the mean of the energy of sequence randomized decoys (sequence decoys), and thus do not reflect the absolute contribution of the electrostatic to the binding. Thus, the given energy component may oppose the binding, but still be optimized with respect to the mean of the energy of a randomized sequence.
The finding that the Coulombic component of binding energy is optimized confirms our previous studies of electrostatic properties of protein-protein complexes (7,39). We have shown, using a set of six protein-protein complexes, that increase of the salt concentration makes the binding weaker (39)—an effect that is experimentally measured. Since, from the point of view of nonspecific interaction, the electrostatics is the only energy component sensitive to the ion concentration, the above finding indicates that electrostatic Coulombic interactions favor the binding for the complexes studied in Bertonati et al. (39). Screening of these favorable Coulombic interactions as the ionic strength increases makes the binding weaker (note that, in our approach, the reaction field energy is salt-independent; for the energy decomposition of the electrostatic energy, see (75)). The observation that the formation of complexes lowers the pKa values of acidic groups (7) also indicates electrostatic optimization. Since the pKa shifts are caused by the new (presumably favorable) interactions across the interface and the loss of solvation energy (desolvation) upon complex formation, a negative pKa shift for acidic groups indicates that the gain of favorable Coulombic interactions overcompensates the desolvation penalty. Such an effect requires specific organization of the charged groups at the interface of the complexes and reflects the optimization of the charge-charge interactions.
The optimization of the Coulombic interactions and anti-optimization of the reaction field energy for entire structures (monomers and their complexes) deserves a special discussion. The reaction field energy depends on the Coulombic interactions, and in principle, the stronger the electrostatic field, the larger the magnitude of the reaction field energy. Statistical studies have shown that the correlation between Coulombic and reaction field energies results in a coefficient of ∼0.8 and many studies aimed at high performance speed had used that correlation to avoid the time-consuming calculations of the reaction field energy (83–85). Our large-scale statistical study indirectly confirms that observation. A plot of the Z-score for the Coulombic energy versus Z-scores for the reaction field energy shows a very strong correlation (correlation coefficient of 0.92, data not shown) for the energies of either the monomers or the complexes. However, the components of the binding energy behave differently. There is no strong correlation between the Z-scores for the Coulombic and reaction field components of the binding energies, and in this case the correlation coefficient is only 0.24 (data not shown).
In the test of detecting structural decoys we have used several ranking functions ranging from the components of the electrostatic energy to the corresponding Z-scores of these energies in respect to sequence randomization. It was shown that the total electrostatic binding energy does not perform well, while the individual components (especially the reaction field component) do. The ranking with the corresponding Z-scores performs better in two of the cases indicating the potential of this approach. Additional benchmarking was done on a set of 41 protein complexes extracted from Boston University benchmark and for each complex we generated 1000 decoys using ZDOCK 2.3 (81) and the bound structures of the monomers. The results confirmed that the electrostatic component of the binding energy is not as good a criterion for discriminating decoys, while the Coulombic component performs the best.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Barry Honig for the continuous support.
This research was supported by an award to Clemson University from the Howard Hughes Medical Institute Undergraduate Science Education Program.
K. Brock, K. Talley, and K. Coley contributed equally to this work.
Editor: Ruth Nussinov.
References
- 1.Alberts, B., D. Bray, J. Lewis, M. Raff, K. Roberts, and J. Watson. 1994. Molecular Biology of the Cell. Garland Publishing, New York.
- 2.Sham, Y., Z. Chu, H. Tao, and A. Warshel. 2000. Examining methods for calculations of binding free energies: LRA, LIE, PDLD-LRA, and PDLD/S-LRA calculations of ligands binding to an HIV protease. Proteins. 39:393–407. [PubMed] [Google Scholar]
- 3.Keskin, O., B. Y. Ma, K. Rogale, K. Gunasekaran, and R. Nussinov. 2005. Protein-protein interactions: organization, cooperativity and mapping in a bottom-up Systems Biology approach. Phys. Biol. 2:S24–S35. [DOI] [PubMed] [Google Scholar]
- 4.McDonald, I. K., and J. M. Thornton. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238:777–793. [DOI] [PubMed] [Google Scholar]
- 5.Jones, S., and J. Thornton. 1996. Principles of protein-protein interactions derived from structural studies. Proc. Natl. Acad. Sci. USA. 93:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheinerman, F., R. Norel, and B. Honig. 2000. Electrostatics aspects of protein-protein interactions. Curr. Opin. Struct. Biol. 10:153–159. [DOI] [PubMed] [Google Scholar]
- 7.Kundrotas, P. J., and E. Alexov. 2006. Electrostatic properties of protein-protein complexes. Biophys. J. 91:1724–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shoemaker, B. A., and A. R. Panchenko. 2007. Deciphering protein-protein interactions. Part I. Experimental techniques and databases. PLoS Comput. Biol. 3:e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou, H. X., and Y. Shan. 2001. Prediction of protein interaction sites from sequence profiles and residue neighbor list. Proteins. 44:336–343. [DOI] [PubMed] [Google Scholar]
- 10.Chen, H., and H. X. Zhou. 2005. Prediction of interface residues in protein-protein complexes by a consensus neural network method: test against NMR data. Proteins. 61:21–35. [DOI] [PubMed] [Google Scholar]
- 11.Ofran, Y., and B. Rost. 2003. Predicted protein-protein interaction sites from local sequence information. FEBS Lett. 544:236–239. [DOI] [PubMed] [Google Scholar]
- 12.Kundrotas, P., and E. Alexov. 2007. Predicting protein-protein interactions using continuous interacting residue segments Biophys. J. Abstracts of the Biophysical Meeting, Supplementary issue: 367A–368A.
- 13.Shoemaker, B. A., and A. R. Panchenko. 2007. Deciphering protein-protein interactions. Part II. Computational methods to predict protein and domain interaction partners. PLoS Comput. Biol. 3:e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu, L., H. Lu, and J. Skolnick. 2005. MULTIPROSPECTOR: an algorithm for the prediction of protein-protein interactions by multimeric threading. Proteins. 15:350–364. [DOI] [PubMed] [Google Scholar]
- 15.Aloy, P., M. Pichaud, and R. B. Russell. 2005. Protein complexes: structure prediction challenges for the 21st century. Curr. Opin. Struct. Biol. 15:15–22. [DOI] [PubMed] [Google Scholar]
- 16.Kundrotas, P. J., and E. Alexov. 2006. Predicting 3D structures of transient protein-protein complexes by homology. Biochim. Biophys. Acta. 1764:1498–1511. [DOI] [PubMed] [Google Scholar]
- 17.Bordner, A. J., and R. Abagyan. 2005. Statistical analysis and prediction of protein-protein interfaces. Proteins. 60:353–366. [DOI] [PubMed] [Google Scholar]
- 18.Jones, S., and J. Thornton. 1997. Prediction of protein-protein interaction sites using patch analysis. J. Mol. Biol. 272:113–143. [DOI] [PubMed] [Google Scholar]
- 19.Jones, S., and J. Thornton. 1996. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA. 93:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nooren, I. M. A., and J. M. Thornton. 2003. Structural characterization and functional significance of transient protein-protein interactions. J. Mol. Biol. 325:991–1018. [DOI] [PubMed] [Google Scholar]
- 21.Ofran, Y., and B. Rost. 2003. Analyzing six types of protein-protein interfaces. J. Mol. Biol. 325:377–387. [DOI] [PubMed] [Google Scholar]
- 22.Clackson, T., and J. A. Wells. 1995. A hot-spot of binding-energy in a hormone-receptor interface. Science. 267:383–386. [DOI] [PubMed] [Google Scholar]
- 23.Wells, J. A. 1991. Systematic mutational analyses of protein-protein interfaces. Methods Enzymol. 202:390–411. [DOI] [PubMed] [Google Scholar]
- 24.Massova, I., and P. A. Kollman. 1999. Computational alanine scanning to probe protein-protein interactions: a novel approach to evaluate binding free energies. J. Am. Chem. Soc. 121:8133–8143. [Google Scholar]
- 25.Hu, Z., B. Ma, H. Wolfson, and R. Nussinov. 2000. Conservation of polar residues as hot spots at protein interfaces. Proteins. 39:331–342. [PubMed] [Google Scholar]
- 26.Lee, L. P., and B. Tidor. 2001. Optimization of binding electrostatics: charge complementarity in the barnase-barstar protein complex. Protein Sci. 10:362–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee, L. P., and B. Tidor. 2001. Barstar is electrostatically optimized for tight binding to barnase. Nat. Struct. Biol. 8:73–76. [DOI] [PubMed] [Google Scholar]
- 28.Lee, L., and B. Tidor. 2001. Optimization of binding electrostatics: charge complementarity in the barnase-barstar protein complex. Protein Sci. 10:362–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerutti, D. S., L. F. Ten Eyck, and J. A. McCammon. 2005. Rapid estimation of solvation energy for simulations of protein-protein association. J. Chem. Theory Comput. 1:143–152. [DOI] [PubMed] [Google Scholar]
- 30.Elcock, A. H., D. Sept, and J. A. McCammon. 2001. Computer simulation of protein-protein interactions. J. Phys. Chem. B. 105:1504–1518. [Google Scholar]
- 31.Ma, C. S., N. A. Baker, S. Joseph, and J. A. McCammon. 2002. Binding of aminoglycoside antibiotics to the small ribosomal subunit: a continuum electrostatics investigation. J. Am. Chem. Soc. 124:1438–1442. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen, J. E., and J. A. McCammon. 2003. Calculating pKa values in enzyme active sites. Protein Sci. 12:1894–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sims, P. A., C. F. Wong, and J. A. McCammon. 2004. Charge optimization of the interface between protein kinases and their ligands. J. Comput. Chem. 25:1416–1429. [DOI] [PubMed] [Google Scholar]
- 34.Sims, P. A., C. F. Wong, D. Vuga, J. A. McCammon, and B. M. Sefton. 2005. Relative contributions of desolvation, inter- and intramolecular interactions to binding affinity in protein kinase systems. J. Comput. Chem. 26:668–681. [DOI] [PubMed] [Google Scholar]
- 35.Norel, R., F. Sheinerman, D. Petrey, and B. Honig. 2001. Electrostatic contribution to protein-protein interactions: fast energetic filters for docking and their physical basis. Protein Science. 10:2147–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheinerman, F. B., and B. Honig. 2002. On the role of electrostatic interactions in the design of protein-protein interfaces. J. Mol. Biol. 318:161–177. [DOI] [PubMed] [Google Scholar]
- 37.Muegge, I., T. Schweins, and A. Warshel. 1998. Electrostatic contribution to protein-protein binding affinities: application to Rap/Raf interaction. Proteins. 30:407–423. [DOI] [PubMed] [Google Scholar]
- 38.Gohlke, H., C. Kiel, and D. Case. 2003. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J. Mol. Biol. 330:891–913. [DOI] [PubMed] [Google Scholar]
- 39.Bertonati, C., B. Honig, and E. Alexov. 2007. Poisson-Boltzmann calculations of nonspecific salt effects on protein-protein binding free energies. Biophys. J. 92:1891–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hendsch, Z., and B. Tidor. 1994. Do salt bridges stabilize proteins? A continuum electrostatics analysis. Protein Science. 3:211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luisi, D. L., C. D. Snow, J. J. Lin, Z. S. Hendsch, B. Tidor, and D. P. Raleigh. 2003. Surface salt bridges, double-mutant cycles, and protein stability: an experimental and computational analysis of the interaction of the Asp 23 side chain with the N-terminus of the N-terminal domain of the ribosomal protein l9. Biochemistry. 42:7050–7060. [DOI] [PubMed] [Google Scholar]
- 42.Dong, F., and H.-X. Zhou. 2006. Electrostatic contribution to the binding stability of protein-protein complexes. Proteins. 65:87–102. [DOI] [PubMed] [Google Scholar]
- 43.Dong, F., M. Vijayakumar, and H. X. Zhou. 2003. Comparison of calculation and experiment implicates significant electrostatic contributions to the binding stability of barnase and barstar. Biophys. J. 85:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qin, S., and H. X. Zhou. 2007. Do electrostatic interactions destabilize protein-nucleic acid binding? Biopolymers. 86:112–118. [DOI] [PubMed] [Google Scholar]
- 45.Boschitsch, A. H., M. O. Fenley, and H.-X. Zhou. 2002. Fast boundary element method for the linear Poisson-Boltzmann equation. J. Phys. Chem. B. 202:2741–2754. [Google Scholar]
- 46.Zhou, H.-X. 2001. Disparate ionic-strength dependence of on and off rates in protein- protein association. Biopolymers. 59:427–433. [DOI] [PubMed] [Google Scholar]
- 47.Spassov, V., R. Ladenstein, and A. Karshikoff. 1997. Optimization of the electrostatic interactions between ionized groups and peptide dipoles in proteins. Protein Science. 6:1190–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spassov, V. Z., and B. P. Atanasov. 1994. Spatial optimization of electrostatic interactions between the ionized groups in globular proteins. Proteins. 19:222–229. [DOI] [PubMed] [Google Scholar]
- 49.Spassov, V. Z., A. D. Karshikoff, and R. Ladenstein. 1994. Optimization of the electrostatic interactions in proteins of different functional and folding type. Protein Sci. 3:1556–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shosheva, A., A. Donchev, M. Dimitrov, G. Kostov, G. Toromanov, V. Getov, and E. Alexov. 2005. Comparative study of the stability of poplar plastocyanin isoforms. Biochim. Biophys. Acta. 1748:116–127. [DOI] [PubMed] [Google Scholar]
- 51.Marchler-Bauer, A., J. B. Anderson, C. DeWeese-Scott, N. D. Fedorova, L. Y. Geer, S. He, D. I. Hurwitz, J. D. Jackson, A. R. Jacobs, C. J. Lanczycki, C. A. Liebert, C. Liu, T. Madej, G. H. Marchler, R. Mazumder, A. N. Nikolskaya, A. R. Panchenko, B. S. Rao, B. A. Shoemaker, V. Simonyan, J. S. Song, P. A. Thiessen, S. Vasudevan, Y. Wang, R. A. Yamashita, J. J. Yin, and S. H. Bryant. 2003. CDD: a curated Entrez database of conserved domain alignments. Nucleic Acids Res. 31:383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kundrotas, P. J., and E. Alexov. 2007. PROTCOM: searchable database of protein complexes enhanced with domain-domain structures. Nucleic Acids Res. 35:D575–D579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stein, A., R. B. Russell, and P. Aloy. 2005. 3DID: interacting protein domains of known three-dimensional structure. Nucleic Acids Res. 33:D413–D417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gong, S., C. Park, H. Choi, J. Ko, I. Jang, J. Lee, D. M. Bolser, D. Oh, D. S. Kim, and J. Bhak. 2005. A protein domain interaction interface database: InterPare. BMC Bioinformatics. 6:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davis, F. P., and A. Sali. 2005. PIBASE: a comprehensive database of structurally defined protein interfaces. Bioinformatics. 21:1901–1907. [DOI] [PubMed] [Google Scholar]
- 56.Douguet, D., H. C. Chen, A. Tovchigrechko, and I. A. Vakser. 2006. DOCKGROUND resource for studying protein-protein interfaces. Bioinformatics. 22:2612–2618. [DOI] [PubMed] [Google Scholar]
- 57.Smith, G. R., and M. J. E. Sternberg. 2002. Prediction of protein-protein interactions by docking methods. Curr. Opin. Struct. Biol. 12:28–35. [DOI] [PubMed] [Google Scholar]
- 58.McCammon, J. A. 1998. Theory of biomolecular recognition. Curr. Opin. Struct. Biol. 8:245–249. [DOI] [PubMed] [Google Scholar]
- 59.Topf, M., and A. Sali. 2005. Combining electron microscopy and comparative modeling. Curr. Opin. Chem. Biol. 15:578–585. [DOI] [PubMed] [Google Scholar]
- 60.Russell, R. B., F. Alber, P. Aloy, F. P. Davis, D. Korkin, M. Pichaud, M. Topf, and A. Sali. 2004. A structural perspective on protein-protein interactions. Curr. Opin. Struct. Biol. 14:313–324. [DOI] [PubMed] [Google Scholar]
- 61.Aloy, P., and R. B. Russell. 2003. InterPreTS: protein interaction prediction through tertiary structure. Bioinformatics. 19:161–162. [DOI] [PubMed] [Google Scholar]
- 62.Kozakov, D., R. Brenke, S. R. Comeau, and S. Vajda. 2006. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 65:392–406. [DOI] [PubMed] [Google Scholar]
- 63.Tovchigrechko, A., and I. A. Vakser. 2006. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 34:W310–W314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vajda, S., I. Vakser, M. Steinberg, and J. Janin. 2002. Modeling of protein interactions in genomes. Proteins. 47:444–446. [DOI] [PubMed] [Google Scholar]
- 65.Li, W., L. Jaroszewski, and A. Godzik. 2001. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics. 17:282–283. [DOI] [PubMed] [Google Scholar]
- 66.Ponder, J. W. 1999. TINKER—Software Tools for Molecular Design, V. 3.7. Washington University, St. Louis, MO.
- 67.Still, W. C., A. Tempczyk, R. C. Hawley, and T. Hendrickson. 1990. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 112:6127–6129. [Google Scholar]
- 68.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 69.Kouranov, A., L. Xie, J. de la Cruz, L. Chen, J. Westbrook, P. E. Bourne, and H. M. Berman. 2006. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 34:D302–D305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xiang, Z., and B. Honig. 2001. Extending the accuracy limits of prediction for side-chain conformations. J. Mol. Biol. 311:421–430. [DOI] [PubMed] [Google Scholar]
- 71.Alexov, E., and M. Gunner. 1997. Incorporating protein conformation flexibility into the calculation of pH-dependent protein properties. Biophys. J. 74:2075–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Georgescu, R., E. Alexov, and M. Gunner. 2002. Combining conformational flexibility and continuum electrostatics for calculating residue pKas in proteins. Biophys. J. 83:1731–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alexov, E., and M. Gunner. 1999. Calculated protein and proton motions coupled to electron transfer: electron transfer from QA− to QB in bacterial photosynthetic reaction centers. Biochemistry. 38:8253–8270. [DOI] [PubMed] [Google Scholar]
- 74.Forrest, L., and B. Honig. 2005. An assessment of the accuracy of methods for predicting hydrogen positions in protein structures. Proteins. 61:296–309. [DOI] [PubMed] [Google Scholar]
- 75.Rocchia, W., E. Alexov, and B. Honig. 2001. Extending the applicability of the nonlinear Poisson-Boltzmann equation: multiple dielectric constants and multivalent ions. J. Phys. Chem. 105:6507–6514. [Google Scholar]
- 76.Rocchia, W., S. Sridharan, A. Nicholls, E. Alexov, A. Chiabrera, and B. Honig. 2002. Rapid grid-based construction of the molecular surface and the use of induced surface charges to calculate reaction field energies: applications to the molecular systems and geometrical objects. J. Comput. Chem. 23:128–137. [DOI] [PubMed] [Google Scholar]
- 77.Sitkoff, D., K. A. Sharp, and B. Honig. 1994. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 98:1978–1988. [Google Scholar]
- 78.Feig, M., A. Onufriev, M. S. Lee, W. Im, D. A. Case, and C. L. Brooks 3rd. 2004. Performance comparison of generalized Born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J. Comput. Chem. 25:265–284. [DOI] [PubMed] [Google Scholar]
- 79.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization and dynamic calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 80.Chen, R., J. Mintseris, J. Janin, and Z. Weng. 2003. A protein-protein docking benchmark. Proteins. 52:88–91. [DOI] [PubMed] [Google Scholar]
- 81.Chen, R., L. Li, and Z. Weng. 2003. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 52:80–87. [DOI] [PubMed] [Google Scholar]
- 82.Schulz, C., and A. Warshel. 2001. What are the dielectric “constants” of proteins and how to validate electrostatic models. Proteins. 44:400–417. [DOI] [PubMed] [Google Scholar]
- 83.Marshall, S., C. Morgan, and S. Mayo. 2002. Electrostatics significantly affect the stability of designed homeodomain variants. J. Mol. Biol. 316:189–199. [DOI] [PubMed] [Google Scholar]
- 84.Zollars, E. S., S. A. Marshall, and S. L. Mayo. 2006. Simple electrostatic model improves designed protein sequences. Protein Sci. 15:2014–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vizcarra, C. L., and S. L. Mayo. 2005. Electrostatics in computational protein design. Curr. Opin. Chem. Biol. 9:622–626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.