

Abstract
Amelogenin, the major extracellular enamel matrix protein, plays critical roles in controlling enamel mineralization. This generally hydrophobic protein self-assembles to form nanosphere structures under certain solution conditions. To gain clearer insight into the mechanisms of amelogenin self-assembly, we first investigated the occurrences of secondary structures within its sequence. By applying isothermal titration calorimetry (ITC), we determined the thermodynamic parameters associated with protein-protein interactions and with conformational changes during self-assembly. The recombinant porcine full length (rP172) and a truncated amelogenin lacking the hydrophilic C-terminal (rP148) were used. Circular dichroism (CD) measurements performed at low concentrations (<5 μM) revealed the presence of the polyproline-type II (PPII) conformation in both amelogenins in addition to α-helix and unordered conformations. Structural transition from PPII/unordered to β-sheet was observed for both proteins at higher concentrations (>62.5 μM) and upon self-assembly. ITC measurements indicated that the self-assembly of rP172 and rP148 is entropically driven (+ΔSA) and energetically favorable (−ΔGA). The magnitude of enthalpy (ΔHA) and entropy changes of assembly (ΔSA) were smaller for rP148 than rP172, whereas the Gibbs free energy change of assembly (ΔGA) was not significantly different. It was found that rP172 had higher PPII content than rP148, and the monomer-multimer equilibrium for rP172 was observed in a narrower protein concentration range when compared to rP148. The large positive enthalpy and entropy changes in both cases are attributed to the release of ordered water molecules and the associated entropy gain (due to the hydrophobic effect). These findings suggest that PPII conformation plays an important role in amelogenin self-assembly and that rP172 assembly is more favorable than rP148. The data are direct evidence for the notion that hydrophobic interactions are the main driving force for amelogenin self-assembly.
INTRODUCTION
During the secretory stage of enamel, the amelogenin-rich organic matrix self-assembles to form nanosphere structures that are aligned along the developing enamel crystallites (1). Amelogenin is the most abundant dental enamel protein, accounting for >90% of the total extracellular organic matrix. Other components of the organic matrix include enamelins, ameloblastins, proteinases, and amelotin (2). The primary structure of amelogenin, which has been characterized as having a bipolar nature (hydrophobic-hydrophilic), is highly conserved (>80% identity) among different species (3). In rodents, removal of amelogenin resulted in hypomineralized enamel that lacked the regular prism structure (4). It has also been observed that removal of the conserved domains in amelogenin sequence caused formation of defective enamel (5). Abnormal human enamel formation has been associated with mutations identified on amelogenin gene (AMEL, 6). These results collectively highlight the significance of amelogenin self-assembly in orchestrating the controlled and organized growth of crystals during enamel biomineralization.
Recombinant amelogenin self-assembles to form nanosphere structures with an apparent molecular mass of 2–3 million daltons in vitro (7). It has been proposed that the assembly occurs through intermolecular hydrophobic interactions with the charged C-terminus exposed on the surface and the assembly properties depend on pH, temperature, and protein concentration (8). Using small angle x-ray scattering, Aichmayer et al. have provided additional support to show that the charged C-terminal of the full-length amelogenin prevents the excessive agglomeration of the nanospheres (9). Although numerous investigators have reported structural data, a clear model for amelogenin secondary structure is still lacking. Renugopalakrishnan et al. have reported the secondary structural preferences of a native bovine amelogenin lacking the hydrophilic C-terminal residues (10). Based on the Chou-Fasman algorithm for theoretical calculations, a high probability for appearance of β-turns, their coiling into a “β-spiral” structure, and the occurrence of polyproline-type structures at the Pro-Pro segments were proposed (10). Later, Goto et al. showed that the native full-length porcine amelogenin had similar structural features and proposed that the protein consisted of three independent folding units with distinctive secondary structures (11). Despite recent studies, ambiguity regarding understanding the secondary structure of amelogenin still exists (12,13).
To better understand the molecular mechanisms of amelogenin self-assembly and study the driving forces for the formation of nanospheres, this study addresses the thermodynamics of amelogenin self-assembly and the role of secondary structures in this process. We first investigated the presence of different secondary structures in amelogenin using circular dichroism (CD). We compared the CD spectra of recombinant amelogenins rP172 and rP148 with poly(L)proline, which is known to adopt a polyproline left-handed II helix (PPII) structure. Comparison with poly(L)proline was justified because the amelogenin primary structure consists of ∼23% proline. In a number of globular proteins, the PPII structure is stabilized by the presence of proline at every third residue, i.e., (PXXP)n repeats, and many such repeats are found in the primary structure of rP172 and rP148. In addition, the sequence also contains amino acid residues that have a high propensity to form PPII helix structure (14).
Isothermal titration calorimetry (ITC) is a sensitive technique that directly determines the thermodynamic parameters of protein-protein or protein-ligand interactions (15). ITC dilution experiments have been used to investigate the monomer-dimer and monomer-heptamer equilibrium and micellar assembly of antibiotic protein, lipids, and surfactants (16–20). The large aggregation number of amelogenin molecules within nanospheres suggests that they resemble micellar assemblies of surfactants, making them appropriate for analysis by ITC. The enthalpy changes associated with amelogenin-amelogenin dissociation after dilution experiments were measured, and the entropy changes were estimated. Comparison between secondary structural changes and thermodynamic parameters of assembly between recombinant full-length porcine amelogenin rP172 and the truncated one lacking the hydrophilic C-terminal 25 amino acids (rP148) provided insight into the function of the hydrophilic domain. Determining these thermodynamic parameters along with the analysis of secondary structural changes has provided valuable insight into the driving force for protein-protein interactions and contribution of conformational changes during amelogenin self-assembly.
MATERIALS AND METHODS
Protein expression and purification
Recombinant porcine amelogenin (rP172) was expressed in Escherichia coli strain BL21-codon plus (DE3-RP, Strategene, La Jolla, CA) and precipitated by 20% ammonium sulfate (21). For the synthesis of rP148, a previously cloned pig amelogenin cDNA construct encoding P173 was used as the template for polymerase chain reaction-mediated site-directed mutagenesis. The protein was expressed, purified, and characterized as described by Sun et al. (22). Purification of the ammonium sulfate precipitate was performed on a Varian (Palo Alto, CA) Prostar high performance liquid chromatography system. The precipitate was dissolved in 0.1% trifluoro acid and loaded onto a C4 column (10 mm × 250 mm, 5 μ) and fractionated using a linear gradient of 60% acetonitrile at a flow rate of 2 mL/min. The homogeneity of the protein was confirmed by analytical chromatography (C4, 2 mm × 250 mm, 5 μ). Unlike full-length porcine amelogenin (P173), rP172 lacks a methionine residue at first position and a phosphate at Ser16. rP148 differs from C-terminal truncated amelogenin proteolytic product (“20k”, P148), as it lacks the first methionine residue, a phosphate on Ser16, but has an extra methionine at position 149.
Peptide synthesis and characterization
The C-terminal (M149-D173) 25 amino acid residue peptide (25 C-term) was synthesized at the DNA core facilities at the University of Southern California, using the Pioneer peptide synthesizer (Applied Biosystems, Foster City, CA) using the Nα-Fmoc-L-aminoacid pentafluorophyenyl ester/HOBt coupling method. The crude peptide was purified on a C4 RP-HPLC column and the homogeneity was confirmed by analytical chromatography and ESI-MS measurements. The theoretical and experimental masses of the peptide were 2883.27 and 2884.0 Da, respectively.
Circular dichroism spectroscopy
Far ultraviolet-CD spectra (260–190 nm) of amelogenins were recorded on a Jasco (Tokyo, Japan) J-810 spectropolarimeter after calibrating the instrument with (−) camphor sulfonic acid. All the measurements were carried out at room temperature using 0.1/0.01 cm path length stoppered cuvettes. The instrument optics were flushed with N2 gas at a flow rate of 20 L/min. The proteins/peptide were dissolved in cold 50 mM Tris-HCl buffer at a pH = 5.8 and refrigerated overnight. This pH was selected to avoid problems associated with solubility of amelogenin at higher concentrations. The concentration of rP172 and rP148 varied from 3 μM to 75 μM. The spectra were recorded at a scan speed of 50 nm/min, 0.2 nm resolution, and 2 nm bandwidth at 25°C. For each spectrum, a total of four scans was recorded, averaged, and baseline subtracted. Poly-L-proline from Sigma-Aldrich (St. Louis, MO) with degree of polymerization (DP) = 206 was used for comparison. The mean residual weight (MRW) ellipticity (θMRW) was estimated using the equation
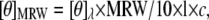 |
where [θ]λ is the observed ellipticity, MRW is defined as the M/N − 1 where M is the molecular mass (Mrp172 = 19572.5; Mrp148 = 16838.5; M25C-term = 2883.3 Da) and N is the number of amino acid residues. l is the optical path length, and c is the concentration of the protein (mg/mL). The α-helix content in the presence of various amounts of 2,2,2-trifluoro ethanol (TFE) was estimated as reported by Scholtz et al. (23). The helical content is proportional to the MRW ellipticity at 222 nm, [θ]222. The percentage helicity is calculated as
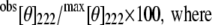 |
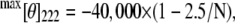 |
where N is the number of amino acid residues.
ATR-FTIR spectroscopy
Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) spectra of the proteins at various concentrations were recorded in Tris-HCl buffer (pH = 5.8) on a Jasco-4100 spectrophotometer (Jasco, Easton, MD) with a KRS-5 interferometer and DTGS detector; ∼10 μL of the protein sample was placed on an AMTIR (Pike Technologies, Madison, WI) crystal and the spectra were collected using 64 scans at 4 cm−1 resolution, averaged, and the background subtracted. The contributions of water and the background were subtracted from amelogenin solutions. The resultant spectra were baseline corrected by a two-point line between 1550 and 1800 cm−1 and normalized over the concentration. The difference spectra was obtained using the formula A − B * coefficient, where A and B represent the spectra at higher and lower concentrations, respectively, and the coefficient is the average intensity ratio of the internal standard (sodium perchlorate).
Isothermal titration calorimetry
ITC dilution experiments were performed using a CSC 4200 titration calorimeter (Calorimetry Sciences, Provo, UT). The mixing cell had a volume of 1.3 mL. The calorimetric syringe was filled with a concentrated solution of rP172 (200 μM) and rP148 (240 μM) in Tris-HCl buffer at pH = 5.8 at 25°C (for rP172) and at 20°C (for rP148). The sample and the reference cells were also filled with the same buffer. The concentration solutions were prepared in cold condition as mentioned before and degassed before the titration. A 10 μL aliquot of the solution was injected into the calorimetric cell (Vcell = 1.34 mL) containing the same buffer and results in ∼130 times dilution of amelogenins. Each injection was carried out at 300-s intervals to allow the solution to reach the equilibrium and the cell was stirred continuously at 300 rpm. The assembled amelogenins disintegrated into monomer/oligomer, a process accompanied by absorption or evolution of heat. As more and more amelogenin was injected into the cell, the injected assembled amelogenin no longer disintegrated and the heat of disassembly decreased. The midpoint of the transition was determined by first derivative curve. Since a nanosphere is assumed to be a micellar aggregate, the midpoint of the transition was taken as the critical micellar concentration (CMC). The CMC is related to the Gibbs free energy by the equation
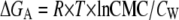 |
where R is the gas constant and T is the absolute temperature in Kelvin. The factor CW denotes the concentration of water and taken as 55.5 M. Using the equation
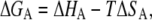 |
the entropy of aggregation has been determined. The enthalpy of disassembly was estimated using the equation provided by Heerklotz et al. (24). The validity of measurement was defined by the dimensionless parameter c = Cinj/Kd, which must lie between 10 and 10,000, where Cinj and Kd are the concentration of the protein in the syringe and dissociation constant, respectively (25). The values of 1905 and 2243 were obtained for rP172 and rP148, respectively.
Dynamic light scattering
Dynamic light scattering (DLS) measurements were carried out on a DynaPro Protein Solutions (Wyatt Technologies, Santa Barbara, CA) instrument equipped with a solid-state laser operating at 655 nm with a temperature-controlled microsampler at 20°C. Proteins were dissolved (5 and 200 μM for rP172, and 5 and 240 μM for rP148) in 50 mM Tris-HCl buffer at pH 5.8. The autocorrelation function of the signal from the scattered intensity was measured by a multifunctional digital correlator. The normalized intensity correlation function was analyzed by a regularization method included in the data analysis software (Dynamics V6.3.01) to give the information on the distribution of the exponential decay function with decay rate Γ. The translational diffusion coefficient can be determined through Γ = Dq2. The hydrodynamic radius RH = kT/(6πηD), where k is the Boltzmann constant, η the solvent viscosity, and T the temperature. The mass distribution of the radius is calculated from the intensity distribution via a simplified form of the Rayleigh equation. A data-collecting strategy of multiple runs at short acquisition time was employed. The acquisition time was set at 10 s. The experiment continued for 40–50 runs that corresponded to 6–8 min. The data analysis software was able to analyze the correlation function of each individual run as well as the whole experiment. The latter gave the statistically averaged size distribution analysis.
RESULTS
Secondary structural preferences of amelogenins analyzed by CD and ATR-FTIR spectroscopy
Poly(L)proline, which was used as a positive control, exhibited a strong negative band at 206 nm (π − π*) and a weak positive band at 229 nm (n − π*), which is considered to be the hallmark of the left-handed PPII helix structure (Fig. 1 A). For both rP172 and rP148, the spectra were characterized by the presence of a negative dichroic minima at 202 nm and a weak shoulder in the n − π* region (215–230) nm, as was observed in the native porcine amelogenins (11). The position of the Cotton band at 206 nm for polyproline was, however, blue shifted to 202 nm for rP172 and rP148, due to the smaller amounts of tertiary amide groups. The absence of a positive band in the n − π* region and the presence of a weak shoulder suggests the coexistence of an ordered structure (α-helix/β-sheet), along with the unordered/PPII structure or due to the contribution from aromatic amino acids (Fig. 1 A) (26). Two additional experiments were performed to confirm the presence of PPII structure in rP172 and rP148. Unlike other secondary structures, the PPII conformation does not depend on intramolecular hydrogen bonding; therefore, addition of chaotropic agents such as guanidine hydrochloride or urea will enhance the appearance of PPII structure (27).
FIGURE 1.

CD spectra of recombinant porcine amelogenins in (A) Tris-HCl buffer at pH = 5.8 at 25°C. The concentrations of the protein used are [rP172] = 5 μM; [rP148] = 3 μM; [25 C-term] = 34.5 μM; and [poly(proline)] = 6 μM. (B) Tris-HCl buffer containing 8 M guanidine hydrochloride; (C) in Tris-HCl buffer containing 2 M CaCl2.
As shown in Fig. 1 B, the negative band at 226 nm for rP172 and rP148 became positive in the presence of 8 M guanidine hydrochloride. The increase intensity is higher for rP172 compared to rP148, indicating a higher amount of PPII structure in rP172. Also consistent with the proteins that contain a significant amount of PPII conformation, addition of 2 M CaCl2 decreased the ellipticity at 226 nm (Fig. 1 C). The detail of molecular conformation of the C-terminal 25 amino acid was obtained by subtracting the CD spectra of rP172 from rP148, which showed a random-coil structure (data not shown). Consistent with the predicted random coil structure, the synthetic C-terminal peptide (25 C-term) showed a negative peak at 198 nm and no shoulder was observed in the n − π* region (Fig. 1 A). Addition of 8 M guanidine hydrochloride resulted in a negligible increase in the intensity at 226 nm confirming that the C-terminus as an isolated entity exists as a truly unordered conformation (Fig. 1 B).
The N-terminus and the central region of amelogenin contain a significant amount of hydrophobic amino acid residues (28). To probe the environment around the hydrophobic residues, we analyzed the CD spectra of rP172 and rP148 in TFE. TFE is known to induce helical structures in peptides and proteins that have an intrinsic propensity to form native-like helical structures. The dielectric constant of TFE resembles that of the interior of the protein and has been shown to promote the structure formation by minimizing the exposure of the peptide backbone to water (29–31). Fig. 2 shows the CD spectra of rP172 and rP148 at various concentrations of TFE. Addition of small amounts of TFE (10%) had a dramatic effect on the conformation of rP172 (Fig. 2 A). The spectrum was characterized by a negative minimum at 207 nm, a shoulder around 222 nm, and a weak maximum at 192 nm, indicating the presence of a helical structure. Increasing the concentration of TFE induced double minima at 207 nm and 222 nm and a large increase in the positive maximum at 192 nm, suggesting the increased population of helical structures. Maximum helical content was reached at 30% TFE, as shown by the increase in the intensity at [θ]222 (Table 1). Further addition of TFE decreased the helical population, and a plateau was reached at concentrations above 50%. In the presence of 10% TFE, the conformation of rP148 was not altered significantly (Fig. 2 B).
FIGURE 2.

CD spectra of rP172 (A) and rP148 (B) at various amounts of TFE at 25°C. The concentration of [rP172] = 5 μM and [rP148] = 6 μM.
TABLE 1.
Estimated α-helix content and R-values for rP172 and rP148 in various amount of trifluoroethanol
|
R-value (=θ222/θ207)
|
−θ222, deg cm2 dmol−1
|
% α-helix
|
||||
|---|---|---|---|---|---|---|
| % TFE | rP172 | rP148 | rP172 | rP148 | rP172 | rP148 |
| 10 | 0.5 | 0.35 | 3140 | 3231 | 8.0 | 8.2 |
| 20 | 0.55 | 0.55 | 4461 | 4854 | 11.3 | 12.3 |
| 30 | 0.74 | 0.54 | 7601 | 5793 | 19.3 | 14.7 |
| 40 | 0.61 | 0.56 | 5416 | 5780 | 13.7 | 14.7 |
| 50 | 0.63 | 0.56 | 4561 | 6190 | 11.6 | 15.7 |
At increasing concentrations of TFE, however, rP148 exhibited behavior similar to rP172. The maximum helical content was observed at 50% TFE for rP148. The CD spectral characteristics of rP172 and rP148 were different from a typical spectrum for a α-helix, suggesting a significant population of 310-helix (32,33). The criterion for distinguishing conformationally related 310 and α-helix by CD is the intensity ratio at 222 nm over 208 nm, R-value (= [θ]222/[θ]208). The right-handed 310-helix displays a negative band at 207 nm and a weak shoulder around 222 nm with the R-value being ∼0.4; for an α-helix this ratio is close to unity, and a value between 0.4 and 1.0 suggests the presence of interconverting populations of α-helix and 310-helix. The R-value varied from 0.5 to 0.75 for rP172 and rP148, indicating the presence of lower populations of α-helical structures. The estimated α-helical content was low for both rP172 and rP148 even at high amounts of TFE (Table 1). Both rP172 and rP148 exhibited TFE-dependent isoelliptic points at 204 nm and 207 nm, respectively, indicating a two-state equilibrium between unordered/PPII and helical conformations. The transition from a flexible to α-helical structure suggests the intrinsic tendency of amelogenin to self-orient to more ordered structures. The presence of lower amounts of helical structures at the initial addition of TFE for rP148 indicates a reduced tendency to form helical structures after removal of the C-terminal 24 amino acid residues.
Analysis of amelogenin particle size distribution by DLS of diluted (5 μM) solutions of rP172 at 20°C indicated the existence of monomers or dimers of amelogenin of size 4.1 nm, whereas in the concentrated solution (200 μM) two scattering populations were observed: a higher amount of 25.2 nm and smaller amount of larger 103.1 nm particles (Fig. 3, A and C). rP148, on the other hand, contained 3.6 nm particles at low concentrations (6 μM) and higher amounts of 33.3 nm particles with smaller amounts of larger sized particles in concentrated solutions (240 μM) (Fig. 3, B and D). These results demonstrate that both rP172 and rP148 existed as higher order protomers (nanospheres) in concentrated solutions at pH 5.8. To investigate possible conformational changes upon self-assembly, we have recorded the CD spectrum of rP148 and rP172 as a function of protein concentration. The results show that both rP172 and rP148 undergo significant conformational changes upon self-assembly.
FIGURE 3.

Particle size distribution analysis of rP172 and rP148 in diluted and concentrated solutions. Regularization distribution graphs of rP172 at 5 μM (A) and 200 μM (C); rP148 at 6 μM (B) and 240 μM (D) in Tris-HCl buffer (pH = 5.8) at 20°C.
For rP172, at the highest concentration the negative ellipticity observed at 202 nm became slightly positive, and a new negative minimum was observed around 212 nm (Fig. 4 A). For rP148 a negative minimum around 212 nm was observed (Fig. 4 B). The difference spectra, obtained by subtracting the CD spectra at high concentrations from the one at low concentrations, give details of the conformational changes (Fig. 4 C). Both rP172 and rP148 exhibited a negative minimum at 212 nm and a maximum at 198/200 nm. The presence of a positive band around 198 nm and a negative minimum around 212 nm indicated the presence of β-sheet structure. We have also recorded the CD spectra by diluting a concentrated stock solution of amelogenin (data not shown). A structural transition from β-sheet to PPII/unordered structure was observed upon dilution, indicating that the β-structured aggregates can be dissociated by dilution in a reversible process.
FIGURE 4.

CD spectra of rP172 (A) and rP148 (B) at two different concentrations in Tris-HCl buffer (pH = 5.8). The difference spectra obtained by subtracting the spectrum at higher concentration from the one at lower concentration is shown in C.
To further confirm the CD observations regarding the conformational changes of amelogenin upon self-assembly, we analyzed the ATR-FTIR spectra of rP172 and rP148. Fig. 5, A and B, shows the amide I band of rP172 and rP148 at two different concentrations. The band maximum around 1642 cm−1 indicates the presence of an unordered structure. Assembly resulted in an apparent broadening of the amide I band. The difference spectrum for rP172 indicates a maximum around 1630 cm−1, suggesting a β-sheet structure (Fig. 5 A, inset). On the other hand, the difference spectrum of rP148 shows a number of peaks in the amide I region (Fig. 5 B, inset). A peak around 1612 cm−1 and 1680 cm−1 indicates the presence of β-sheet and antiparallel β-sheet structures, respectively, whereas an additional peak at 1651 cm−1 indicates the presence of α-helical structures. These results indicate that although various conformations are observed in rP148 assemblies, rP172 assemblies exhibit a more uniform conformation.
FIGURE 5.

ATR-FTIR spectra of rP172 (A) and rP148 (B) in Tris-HCl buffer (pH = 5.8) at various concentrations. The difference spectra obtained by subtracting the spectrum at higher concentration from the one at lower concentration is shown in the insets.
Thermodynamic parameters for self-assembly as analyzed by ITC
Concentrated solutions of rP172 and rP148 were taken in the syringe and 10 μL aliquots were injected in a stepwise manner into an adiabatic cell that contained the buffer. Each injection produced exothermic heat change caused by the disintegration of amelogenin nanospheres into monomer/dimers. After the addition of amelogenin and after a particular concentration was reached in the cell, the nanospheres were no longer disassembled and the heat change remained constant due to the dilution of the nanospheres (Fig. 6 A). Integration of the power peaks and normalization of the heat by injected mole number gave the molar heat of disassembly (Fig. 6 B). The changes in enthalpy and free energy of assembly (that is equal in magnitude to but has the opposite sign of disassembly) were calculated using the equation reported by Heerklotz et al. (19,25). The results show that self-assembly of amelogenin was driven by favorable entropy (positive) and unfavorable enthalpy (endothermic) changes (Table 2). The enthalpy and entropy changes involved in rP172 were much higher than rP148. Removal of the C-terminal 24 amino acids resulted in a slight decrease of the thermodynamic stability (∼0.7 kJ/mole). The midpoint of the titration was obtained by taking the first derivative of the plot in Fig. 6 B against the total concentration in the cell (Fig. 6 C). The derivative curve shows that at pH 5.8 and temperature 20°C/25°C amelogenin started to assemble at 6.1 μM (for rP172) and 5.8 μM (for rP148) and that the monomer-multimer equilibrium for rP148 took place over a broader concentration range than that of rP172.
FIGURE 6.

ITC dilution studies on rP172 and rP148. (A) ITC raw data for the dilution of rP172 in Tris-HCl buffer (pH = 5.8) at 25°C. (B) The integrated normalized heat per mole of the injectant is plotted against the total protein concentration in the cell. (C) The progress of the transition is obtained as the first derivative of the plot in B. The maximum in the curve is taken as the critical aggregation concentration.
TABLE 2.
Thermodynamic parameters for the monomer-multimer equilibria for rP172 and rP148 obtained by the ITC dilution experiments
| Protein | ΔGA, kJ/mole | ΔHA, kJ/mole | ΔSA, kJ/mole |
|---|---|---|---|
| rP172 | −39.8 | +200.7 | 807.2 |
| rP148 | −39.1 | +32.4 | 243.9 |
DISCUSSION
Organized assembly of proteins at the mineral interface is key to the formation of hierarchical structures observed in a number of mineralized tissues (34–37). In the case of enamel, and as documented by numerous in vivo and in vitro studies, amelogenin self-assembly is critical to the oriented and elongated growth of crystallites within enamel prisms and therefore for normal enamel formation (3,7,8,38). To better understand the molecular mechanisms of amelogenin self-assembly, we used CD and FTIR spectroscopy to analyze secondary structural preferences within amelogenin sequences. We then applied ITC to analyze thermodynamic parameters for protein-protein interactions as well as conformational changes during self-assembly. We report that besides α-helix and unordered conformations, PPII conformations are present in amelogenin, a secondary structure we suggest to be critical for amelogenin self-assembly. Amelogenin undergoes secondary structural transition from PPII/unordered structure to β-sheet upon self-assembly, and the main driving force for amelogenin self-assembly is the entropy gain due to hydrophobic effect.
Secondary structures in amelogenin
Evidence for the presence of PPII conformation
Proline is an alicyclic amino acid, i.e., the side chain is cyclized back to the amide backbone. The presence of an alicyclic ring has three important consequences: i), the dihedral angle φ (= −65°) becomes more restricted, ii), the bulkiness of the N-CH2 group restricts the residue preceding proline, and iii), the nitrogen of the proline lacks the hydrogen bonding because of the presence of N-CH2, which together with the bulkiness produces the well-known “helix/β-sheet breaker” effect (39). These place restrictions on the Xaa-Pro bond, where Xaa is any amino acid other than proline. The Pro-Pro bond is even more constrained with φ = −78° and ψ = +146° and adopts a PPII conformation (39). Increasingly compelling evidence suggests that PPII is the major conformation in a number of proteins that have unordered structures (40). PPII is a left-handed helix structure with three residues per turn and does not contain intramolecular hydrogen bonds.
By performing CD experiments in the presence of neutral salts we showed that amelogenin has a PPII structure. However, the absence of a positive band near 220 nm without the denaturant at room temperature suggests that PPII is not a dominant conformation in amelogenin. It has been shown that decreasing the temperature increases the population of this conformation with a well-defined isoelliptic point at 213 nm (41,42). This has been previously observed for native full-length porcine (25 kDa) and recombinant murine (rM179) amelogenins, further supporting the presence of PPII structure in rP172 and rP148 (11,12). The well-defined isoelliptic point indicates two-state equilibrium between PPII and truly unordered conformation (41). Our CD data here show that both rP172 and rP148 contain a PPII-extended helix and unordered conformations and that rP172 has a higher PPII content than rP148.
Since the amide group in PPII conformation is hydrogen bonded to solvent water rather than with the other amide groups, addition of poorly hydrogen-bonded solvent removes this conformation. Therefore, addition of various amounts of TFE shifted the conformation of rP172 and rP148 to right-handed helix conformations with an isoelliptic point at 204 nm for rP172 and 207 nm for rP148. R-values between 0.4 and 1.0 suggest the coexistence of 310- and α-helix conformations in rP172 and rP148. The presence of 310-helix and smaller amounts of α-helix suggest the intrinsic propensity of amelogenin to form an ordered structure in a membrane-like environment where other macromolecules are available for binding. It is likely that, under in vivo conditions, portions of rP172 can adopt 310-/α-helix conformations that may depend on phosphorylation and intermolecular interactions with other proteins/cellular components.
PPII/unordered to β-sheet conformational transition upon self-assembly
We have used DLS to confirm that at pH 5.8 and at higher protein concentration (200 μM) the self-assembly of amelogenin is favored, compared to diluted solutions (5 μM) where amelogenins mostly exist as monomers or dimers. At higher concentrations, rP172 and rP148 undergo considerable structural transformation from a relatively flexible PPII/unordered conformation to an ordered conformation. Such a structural transition is strong support for the notion that amelogenin nanospheres are the result of organized assembly of amelogenin monomers (43). Since the CD spectra of a protein is a linear combination of secondary structural elements present, difference spectra recorded at different conditions would highlight the conformational changes involved. The difference spectra recorded at high concentrations of amelogenin indicate the transformation to a β-sheet structure. Owing to the flexibility and proximity of the dihedral angle values, PPII conformation is always in equilibrium with β-sheet, β-turn, and unordered conformations (44). This geometrical flexibility of PPII conformation may allow the polypeptide chain to progress from this conformation to an ordered β-sheet conformation through intermolecular interaction (45). Such behavior has been exhibited by elastin and a number of proteins/polypeptide under a wide variety of conditions (46–49).
Thermodynamics of amelogenin self-assembly and the role of PPII conformation
ITC dilution studies have clearly demonstrated that assembly of amelogenin involves favorable entropy (positive) and unfavorable (positive) enthalpy changes. The overall negative free energy changes indicated that the assembly of amelogenin is a thermodynamically favorable process. Analysis of the ΔSA and ΔHA values derived from the studies allowed us to better understand the molecular interactions involved in amelogenin self-assembly. In general, for a protein-protein interaction the entropy changes upon association involve three major factors: i), the hydrophobic effect (ΔSHE) (i.e., partitioning of nonpolar molecules from water to nonpolar phase); ii), the conformational entropy (ΔSother) (i.e., intramolecular conversion of a random polypeptide chain into an organized structure or vice versa); and iii), the reduction in available rotational and translational degrees of freedom (ΔSrt) upon association (i.e., ΔSA ≈ ΔSHE + ΔSrt + ΔSother, (50)). Based on our CD findings, amelogenin self-assembly is accompanied by a transition from an unordered to an ordered structure; the ΔSother is negative. ΔSrt involves unfavorable entropy (negative) changes, and the overall positive entropy of amelogenin self-assembly is, therefore, contributed to by a large positive ΔSHE. The large positive entropy observed for both rP172 and rP148 are similar to the classical entropically driven processes, which is partitioning the nonpolar molecule from water to a nonpolar phase (51–53).
Similarly, the enthalpy of association (ΔHA) is contributed to by three major terms: i), conformational enthalpy that arises due to formation of an ordered secondary structure; ii), interaction enthalpy that arises due to the noncovalent interactions such as hydrogen bonding, van der Waals forces, and electrostatic forces; and iii), solvation (hydration/dehydration) enthalpy that arises due to the removal or intake of water molecules at the interface (i.e., ΔHA ≈ ΔHsolv + ΔHconf + ΔHinteraction). In the case of amelogenin the first two terms are exothermic (since protein-protein interaction and protein folding involve favorable (negative) enthalpy changes); therefore, the overall endothermic enthalpy of association is contributed to by large positive solvation enthalpy that arises due to the release of ordered water molecules from the monomer/oligomer interface (i.e., dehydration). Thermodynamic data from various sources, such as a), the nonpolar phase to water, b), protein folding, and c), ligand binding to protein through hydrophobic effect are accompanied by burial of the nonpolar surface from water (54–56). It has been suggested that the hydrophobic surfaces induce orientation in the water-water hydrogen bonds in the first hydration shell and that this ordered water is released on burial of the surface (55,56). Recent studies on extracellular matrices highlight the importance of PPII conformation in the self-assembly, elastic properties, and protein-protein interactions (57).
Although the overall ΔGA of rP148 was found to be comparable to that of rP172 (Table 2), the magnitude of enthalpy and entropy contribution to the self-assembly was ∼6 and 4 times lower than rP172, respectively. This could be due to the considerable folding of the central domain upon self-assembly, which is enthalpically favorable (negative) and entropically unfavorable (negative). ATR-FTIR difference spectra of rP148 indicated the presence of various conformations in the assembled state. The presence of these ordered conformations contributes to this significantly and has resulted in an overall decrease of ΔSA and ΔHA. Another explanation for thermodynamic differences observed between rP172 and rP148 could be related to the higher amount of PPII content observed in rP172 than in rP148. The water molecules around the PPII conformation may be removed as the monomers with considerable amounts of PPII structure are converted to nanospheres with β-sheet structure as the dominant conformation. Molecular dynamic simulations and experimental evidence suggests that PPII conformation is considerably hydrated in water (58–60).
Function of the hydrophilic C-terminal on self-assembly
During the secretory stage of enamel formation, amelogenin is instantaneously cleaved by enamel proteases into various proteolytic products (61). The “20 kDa” amelogenin that lacks the C-terminal 25 amino acid residues has been identified as the major proteolytic product in developing pig enamel. The timely cleavage and the removal of proteolytic products are essential to enamel development. The charged C-terminal amelogenin has significant apatite binding affinity and the potential to control morphology and organization of the crystals (43,62–64). As documented by a series of in vitro studies, removal of the C-terminal hydrophilic residue of amelogenin does not affect its ability to control the morphology of calcium phosphate crystals (64,65). These findings were significant, since during enamel maturation the thickening of crystals occurs in a matrix that is enriched in the “20 kDa” and not the full-length amelogenin.
Here we report that the rP148 which is used as an analog to the “20 kDa” amelogenin has the same driving force (hydrophobic interactions) for self-assembly, but with different characteristics. These characteristics include the differences in secondary structure, mainly the PPII content. Although the pH conditions used in our studies are different from physiological conditions, we believe that the information gained is valuable and provides direct support for the notion that amelogenin self-assembly occurs via hydrophobic interactions. Based on the differences observed between rP148 and rP172 (Tables 1 and 2), we suggest that the large dehydration required for the self-assembly of the full-length amelogenin is compensated for by the removal of the C-terminal amino acid residues without much loss in thermodynamic stability, which is expressed as the free energy for assembly. This finding may have biological significance when considering that the assembly of the “20 kDa” amelogenin may be as essential as that of the full-length amelogenin in controlling the morphology of the crystals during the postsecretory stages on enamel formation (38).
Acknowledgments
We thank Prof. Ralf Langen of Zilka Neurogenetic Institute, University of Southern California, for use of the CD spectroscopy. We thank Sujith Chacko, Loker Hydrocarbon Research Institute and Department of Chemistry, University of Southern California, for his help in the ATR-FTIR spectroscopy. We thank Dr. Martin Phillips, lab manager, DOE-Biochemistry Instrumentation Facility at the University of California, Los Angeles, for the use of ITC.
This work was supported by the National Institutes of Health-NIDCR (DE-13414, DE-15644) to J.M.O.
Editor: Jane Clarke.
References
- 1.Fincham, A. G., J. Moradian-Oldak, and J. P. Simmer. 1999. The structural biology of the developing dental enamel matrix. J. Struct. Biol. 126:270–299. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett, J. D., B. Ganss, M. Goldberg, J. Moradian-Oldak, M. L. Paine, M. L. Snead, X. Wen, S. N. Whinte, and Y. L. Zhou. 2006. Protein-protein interactions of the developing enamel matrix. Curr. Top. Dev. Biol. 74:57–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paine, M. L., S. N. White, W. Luo, H. Fong, M. Sarikaya, and M. L. Snead. 2001. Regulated gene expression dictates enamel structure and tooth formation. Matrix Biol. 20:273–292. [DOI] [PubMed] [Google Scholar]
- 4.Gibson, C. W., Z. A. Yuan, B. Hall, G. Longenecker, E. Chen, T. Thyagarajan, T. Sreenath, J. T. Wright, S. Decker, R. Piddington, G. Harrison, and A. B. Kulkarni. 2001. Amelogenin-deficient mice display an amelogenesis imperfecta phenotype. J. Biol. Chem. 276:31871–31875. [DOI] [PubMed] [Google Scholar]
- 5.Zhu, D. H., M. L. Paine, W. Luo, P. Bringas, and M. L. Snead. 2006. Altering biomineralization by protein design. J. Biol. Chem. 281:21173–21182. [DOI] [PubMed] [Google Scholar]
- 6.Collier, P. M., J. J. Sauk, J. Rosenbloom, Z. A. Yuan, and C. W. Gibson. 1997. An amelogenin gene defect associated with human X-linked amelogenesis imperfecta. Arch. Oral Biol. 42:235–242. [DOI] [PubMed] [Google Scholar]
- 7.Fincham, A. G., J. Moradian-Oldak, J. P. Simmer, P. Sarte, E. C. Lau, T. Diekwisch, and H. C. Slavkin. 1994. Self-assembly of recombinant amelogenin protein generated supramolecular structures. J. Struct. Biol. 112:103–109. [DOI] [PubMed] [Google Scholar]
- 8.Moradian-Oldak, J., and M. Goldberg. 2005. Amelogenin supra-molecular assembly in vitro compared with the architecture of the forming enamel matrix. Cells Tissues Organs. 181:202–218. [DOI] [PubMed] [Google Scholar]
- 9.Aichmayer, B., H. C. Margolis, R. Sigel, Y. Yamakoshi, J. P. Simmer, and P. Fratzl. 2005. The onset of amelogenin nanosphere aggregation studied by small-angle x-ray scattering and dynamic light scattering. J. Struct. Biol. 151:239–249. [DOI] [PubMed] [Google Scholar]
- 10.Renugoplakrishnan, V., E. S. Strawich, P. M. Horowitz, and M. J. Glimcher. 1986. Studies of the secondary structures of amelogenin from bovine tooth enamel. Biochemistry. 25:4879–4887. [DOI] [PubMed] [Google Scholar]
- 11.Goto, Y., E. Kogure, T. Takagi, S. Aimoto, and T. Aoba. 1993. Molecular conformation of porcine amelogenin in solution: three folding units at the N-terminal, central, and C-terminal regions. J. Biol. Chem. 113:55–60. [DOI] [PubMed] [Google Scholar]
- 12.Oobatake, M., T. Yamasaki, J. P. Simmer, and V. Renugopalakrishnan. 2006. Thermal denaturation of a recombinant mouse amelogenin: circular dichroism and differential scanning calorimetric studies. Proteins. 62:461–469. [DOI] [PubMed] [Google Scholar]
- 13.Margolis, H. C., E. Beniash, and C. E. Fowler. 2006. Role of macromolecular assembly of enamel matrix proteins in enamel formation. J. Dent. Res. 85:775–793. [DOI] [PubMed] [Google Scholar]
- 14.Kelly, M. A., B. W. Chellgren, A. L. Rucker, J. M. Troutman, M. G. Fried, and T. P. Creamer. 2001. Host-guest study of left-handed polyproline helix formation. Biochemistry. 40:14376–14383. [DOI] [PubMed] [Google Scholar]
- 15.Russell, D. J., and L. D. Hansen. 2006. Calorimeter for biotechnology. Thermochim. Acta. 445:141–159. [Google Scholar]
- 16.Burrows, S. D., M. L. Doyle, K. P. Murphy, S. G. Franklin, J. R. White, I. Brooks, D. E. McNulty, M. O. Scott, J. R. Knutson, D. Porter, P. R. Young, and P. Hensley. 1994. Determination of monomer-dimer equilibrium of interleukin-8 reveals it is monomer at physiological concentration. Biochemistry. 33:12741–12745. [DOI] [PubMed] [Google Scholar]
- 17.Luke, K., D. Apiyo, and P. Wittung-Stafshede. 2005. Dissecting the homo-heptamer thermodynamics by isothermal titration calorimetry: entropy-driven assembly of co-chaperonin protein 10. Biophys. J. 89:3332–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heerklotz, H., and J. Seelig. 2001. Detergent-like action of the antibiotic peptide surfactin on lipid membranes. Biophys. J. 81:1547–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heerklotz, H., and J. Seelig. 2000. Titration calorimetry of surfactant-membrane partitioning and membrane solubilization. Biochim. Biophys. Acta. 1508:69–85. [DOI] [PubMed] [Google Scholar]
- 20.Paula, S., W. Sus, J. Tuchtenhagen, and A. Blume. 1995. Thermodynamics of micelle formation as a function of temperature—a high-sensitivity titration calorimetry study. J. Phys. Chem. 99:11742–11751. [Google Scholar]
- 21.Hu, C. C., J. D. Bartlet, C. H. Zhang, Q. Qian, O. H. Ryu, and J. P. Simmer. 1996. Cloning, cDNA sequence, and alternative splicing of porcine amelogenin mRNAs. J. Dent. Res. 75:1735–1741. [DOI] [PubMed] [Google Scholar]
- 22.Sun, Z., M. M. Ahsan, H. J. Wang, C. Du, C. Abbott, and J. Moradian-Oldak. 2006. Assembly and processing of an engineered amelogenin proteolytic product (rP148). Eur. J. Oral Sci. 114(Suppl. 1):59–63. [DOI] [PubMed] [Google Scholar]
- 23.Scholtz, J. M., H. Qian, E. J. York, J. M. Stewart, and R. L. Baldwin. 1991. Parameters of helix-coil transition theory for alanine-based peptides of varying chain lengths in water. Biopolymers. 31:1463–1470. [DOI] [PubMed] [Google Scholar]
- 24.Heerklotz, H., A. Tsamaloukas, K. Kita-Tokarczyk, P. Stunz, and T. Gutberlet. 2004. Structural, volumetric, and thermodynamic characterization of micellar sphere to rod transition. J. Am. Chem. Soc. 126:16544–16552. [DOI] [PubMed] [Google Scholar]
- 25.Velazquez-Campoy, A., S. A. Leavitt, and E. Freire. 2004. Characterization of protein-protein interactions by isothermal titration calorimetry. In Protein-Protein Interactions: Methods and Applications. H. Fu, editor. Humana Press, Totowa, NJ. 35–54. [DOI] [PubMed]
- 26.Andersson, D., U. Carlsson, and P. O. Freskgard. 2001. Contribution of tryptophan residues to the CD spectrum of the extracellular domain of human tissue factor. Eur. J. Biochem. 268:1118–1128. [DOI] [PubMed] [Google Scholar]
- 27.Parrot, I., P. C. Huang, and C. Khosla. 2002. Circular dichroism and nuclear magnetic resonance spectroscopy of immunogenic gluten peptides and their analogues. J. Biol. Chem. 277:45572–45578. [DOI] [PubMed] [Google Scholar]
- 28.Snead, M. L., M. Zeichner-David, T. Chandra, K. J. Robson, S. L. Woo, and H. C. Slavkin. 1983. Construction and identification of mouse amelogenin cDNA clones. Proc. Natl. Acad. Sci. USA. 80:7254–7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cammers-Goodwin, A., T. J. Allen, S. L. Oslick, K. F. McClure, J. H. Lee, and D. S. Kemp. 1996. Mechanism of stabilization of helical conformations of polypeptides by water containing trifluoroethanol. J. Am. Chem. Soc. 118:3082–3090. [Google Scholar]
- 30.Luo, P. Z., and R. L. Baldwin. 1997. Mechanism of helix induction by trifluoroethanol: a framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry. 36:8413–8421. [DOI] [PubMed] [Google Scholar]
- 31.Rajan, R., and P. Balaram. 1996. A model for the interaction of trifluoroethanol with peptides and proteins. Int. J. Pept. Protein Res. 48:328–336. [DOI] [PubMed] [Google Scholar]
- 32.Milhauser, G. L. 1995. Views of helical peptides: a proposal for the position of 310-helix along the thermodynamic folding pathway. Biochemistry. 34:3873–3877. [DOI] [PubMed] [Google Scholar]
- 33.Toniolo, C., A. Polese, F. Formaggio, M. Crisma, and J. Kamphuis. 1996. Circular dichroism spectrum of a peptide 310-helix. J. Am. Chem. Soc. 118:2744–2745. [Google Scholar]
- 34.Aizenberg, J., J. C. Weaver, M. S. Thanawala, V. C. Sundar, D. E. Morse, and P. Fratzl. 2005. Skeleton of Euplectella sp.: structural hierarchy from the nanoscale to the macroscale. Science. 309:275–278. [DOI] [PubMed] [Google Scholar]
- 35.Sarikaya, M. 1999. Biomimetics: materials fabrication through biology. Proc. Natl. Acad. Sci. USA. 96:14183–14185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nudelman, F., B. A. Gotliv, L. Addadi, and S. Weiner. 2006. Mollusk shells formation: mapping the distribution of organic matrix components underlying single aragonitic tablet in nacre. J. Struct. Biol. 153:176–187. [DOI] [PubMed] [Google Scholar]
- 37.Hassenkam, T., G. E. Fantner, J. A. Cutroni, J. C. Weaver, D. E. Morse, and P. K. Hansma. 2004. High resolution AFM imaging of intact and fractured trabecular bone. Bone. 35:4–10. [DOI] [PubMed] [Google Scholar]
- 38.Moradian-Oldak, J. 2001. Amelogenins: assembly, processing and control of crystal morphology. Matrix Biol. 20:293–305. [DOI] [PubMed] [Google Scholar]
- 39.Wilson, M. P. 1994. The structure and function of proline-rich regions in proteins. Biochem. J. 297:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi, Z. S., K. Chen, Z. G. Liu, and N. R. Kallenbach. 2006. Conformation of the backbone in unfolded proteins. Chem. Rev. 106:1877–1897. [DOI] [PubMed] [Google Scholar]
- 41.Drake, A. F., G. Siligardi, and W. A. Gibbons. 1988. Reassessment of the electronic circular dichroism criteria for random coil conformations of poly(L-lysine) and the implications for protein folding and denaturation. Biophys. Chem. 31:143–146. [DOI] [PubMed] [Google Scholar]
- 42.Woody, R. W. 1992. Circular dichroism and conformation of unordered polypeptides. Advan. Biophys. Chem. 2:37–79. [Google Scholar]
- 43.Du, C., G. Falini, S. Fermani, C. Abbott, and J. Moradian-Oldak. 2005. Supramolecular assembly of amelogenin nanospheres into birefringent microribbons. Science. 307:1450–1454. (Erratum in Science. 2005. 309:2166). [DOI] [PubMed] [Google Scholar]
- 44.Tamburro, A. M., A. Pepe, B. Bochicchio, D. Quaglino, and I. P. Ronchetti. 2005. Supramolecular amyloid-like assembly of the polypeptide sequence coded by exon 30 of human tropoelastin. J. Biol. Chem. 280:2682–2690. [DOI] [PubMed] [Google Scholar]
- 45.Bochicchio, B., A. Pepe, and A. M. Tamburro. 2001. On (GGLGY) synthetic repeating sequences of lamprin and analogous sequences. Matrix Biol. 20:243–250. [DOI] [PubMed] [Google Scholar]
- 46.Valery, C., M. Patemostre, B. Robert, T. Gulik-Krzywicki, T. Narayanan, J. C. Dedieu, G. Keller, M. L. Torres, R. Cherif-Cheikh, P. Calvo, and F. Artzner. 2002. Biomimetic organization: octapeptide self-assembly into nanotubes of viral capsid-like dimension. Proc. Natl. Acad. Sci. USA. 100:10258–10262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Terzi, E., G. Holzemann, and J. Seelig. 1995. Self-association of β-amyloid peptide (1–40) in solution and binding to lipid membranes. J. Mol. Biol. 252:633–642. [DOI] [PubMed] [Google Scholar]
- 48.von Bergen, M., P. Friedhoff, J. Biernat, J. Heberle, E. M. Mandelkow, and E. Mandelkow. 2000. Assembly of τ protein into Alzheimer paired helical filaments depends on local sequence motif (306VQIVYK311) forming β-structure. Proc. Natl. Acad. Sci. USA. 97:5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li, G., P. Zhou, Z. Shao, X. Xie, X. Chen, H. Wang, L. Chunyu, and T. Yu. 2001. The natural silk spinning process: a nucleation dependent mechanism. Eur. J. Biochem. 268:6600–6606. [DOI] [PubMed] [Google Scholar]
- 50.Spolar, R. S., and T. M. Record. 1994. Coupling of local folding to site-specific binding of proteins to DNA. Science. 263:777–784. [DOI] [PubMed] [Google Scholar]
- 51.Kadler, K. E., Y. Hojima, and D. J. Prockop. 1987. Assembly of collagen fibrils de novo by cleavage of the type I pC-collagen with procollagen C-proteinase. Assay of critical concentration demonstrates that collagen self-assembly is a classical example of an entropy-driven process. J. Biol. Chem. 262:15696–15701. [PubMed] [Google Scholar]
- 52.Kadler, K. E., Y. Hojima, and D. J. Prockop. 1988. Assembly of type I collagen fibrils de novo. Between 37 and 41°C the process is limited by micro-unfolding of monomers. J. Biol. Chem. 263:10517–10523. [PubMed] [Google Scholar]
- 53.Friedhoff, P., A. Schneider, E. M. Mandelkow, and E. Mandelkow. 1998. Rapid assembly of Alzheimer-like paired helical filaments from microtubule-associated protein tau monitored by fluorescence in solution. Biochemistry. 37:10223–10230. [DOI] [PubMed] [Google Scholar]
- 54.Blokzijl, W., and J. B. F. N. Engberts. 1993. Hydrophobic effects—opinion and facts. Angew. Chem. Int. Ed. Engl. 32:1545–1579. [Google Scholar]
- 55.Xu, H., and K. A. Dill. 2005. Water's hydrogen bond in the hydrophobic effect: a simple model. J. Phys. Chem. B. 109:23611–23617. [DOI] [PubMed] [Google Scholar]
- 56.Sharp, K. A., and B. Madan. 1997. Hydrophobic effect, water structure, and heat capacity changes. J. Phys. Chem. B. 101:4343–4348. [Google Scholar]
- 57.Bochicchio, B., and A. M. Tamburro. 2002. Polyproline II structure in proteins: identification by chiroptical spectroscopies, stability and functions. Chirality. 14:782–792. [DOI] [PubMed] [Google Scholar]
- 58.Sreerama, N., and R. W. Woody. 1999. Molecular dynamics simulation of polypeptide conformations in water: a comparison of α, β, and poly(pro)II conformations. Proteins. 36:400–406. [PubMed] [Google Scholar]
- 59.Adzhubei, A. A., and M. Sternberg. 1993. Left handed polyproline II helices commonly occur in globular proteins. J. Mol. Biol. 229:472–493. [DOI] [PubMed] [Google Scholar]
- 60.Gough, C. A., R. W. Anderson, and R. S. Bhatnagar. 1988. The role of bound water in the stability of the triple-helical conformation of (Pro-Pro-Gly)10. J. Biomol. Struct. Dyn. 15:1029–1037. [DOI] [PubMed] [Google Scholar]
- 61.Yamakoshi, Y., T. Tanabe, M. Fukae, and M. Shimizu. 1994. Porcine amelogenins. Calcif. Tissue Int. 54:69–75. [DOI] [PubMed] [Google Scholar]
- 62.Moradian-Oldak, J., N. Bouropoulos, L. Wang, and N. Gharakhanian. 2002. Analysis of self-assembly and apatite binding properties of amelogenin proteins lacking the hydrophilic C-terminal. Matrix Biol. 21:197–205. [DOI] [PubMed] [Google Scholar]
- 63.Fan, Y. W., R. Z. Wang, C. Abott, and J. Moradian-Oldak. 2007. Enamel inspired nano-composite fabrication through amelogenin supramolecular self-assembly. Biomaterials. 28:3034–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beniash, E., J. P. Simmer, and H. C. Margolis. 2005. The effect of recombinant mouse amelogenins on the formation of and organization of hydroxyl apatite crystals in vitro. J. Struct. Biol. 149:182–190. [DOI] [PubMed] [Google Scholar]
- 65.Iijima, M., Y. Moriwaki, H. B. Wen, A. G. Fincham, and J. Moradian-Oldak. 2002. Elongated growth of octacalcium phosphate crystals in recombinant gels under controlled ionic flow. J. Dent. Res. 81:69–73. [DOI] [PubMed] [Google Scholar]