

Abstract
The interaction of Saposin C (Sap C) with negatively charged phospholipids such as phosphatidylserine (PS) is essential for its biological function. In this study, Sap C (initially protonated in a weak acid) was inserted into multilamellar vesicles (MLVs) consisting of either 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-L-serine] (negatively charged, POPS) or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (neutrally charged, POPC). The MLVs were then investigated using solid-state NMR spectroscopy under neutral pH (7.0) conditions. The 2H and 31P solid-state NMR spectroscopic data of Sap C-POPS and Sap C-POPC MLVs (prepared under the same conditions) were compared using the 2H order parameter profiles of the POPC-d31 or POPS-d31 acyl chains as well as the 31P chemical shift anisotropy width and 31P T1 relaxation times of the phospholipids headgroups. All those solid-state NMR spectroscopic approaches indicate that protonated Sap C disturbs the POPS bilayers and not the POPC lipid bilayers. These observations suggest for the first time that protonated Sap C inserts into PS bilayers and forms a stable complex with the lipids even after resuspension under neutral buffer conditions. Additionally, 31P solid-state NMR spectroscopic studies of mechanically oriented phospholipids on glass plates were conducted and perturbation effect of Sap C on both POPS and POPC bilayers was compared. Unlike POPC bilayers, the data indicates that protonated Sap C (initially protonated in a weak acid) was unable to produce well-oriented POPS bilayers on glass plates at neutral pH. Conversely, unprotonated Sap C (initially dissolved in a neutral buffer) did not interact significantly with POPS phospholipids allowing them to produce well-oriented bilayers at neutral pH.
INTRODUCTION
Sapocins (a four-member family, Saposin A, B, C, and D) are derived from the single precursor protein, prosaposin (1,2). These small (∼80 amino acids) heat stable glycoproteins are necessary for the hydrolytic activity of several lysosomal enzymes involved in the catalytic pathway of glycosphingolipids (1). The focus of this study is Saposin C (Sap C); a protein that can stimulate the catalytic activity of glucosylceramidase ((GCase) EC 3.1.2.45, a lysosomal enzyme) (3,4). GCase is essential for the hydrolysis of glucosylceramide to ceramide and glucose (3,4). Additionally, Sap C participates in the fusion and destabilization of negatively charged phospholipids (5,6). Membrane fusion is a key step in the processes of secretion, endocytosis, exocytosis, fertilization, and intracellular transport (7). Also, the destabilization ability of Sap C to the membrane surface is thought to promote the insertion of the lysosomal enzymes into the bilayer (5,8). Moreover, Sap C plays a significant role in lipids trafficking and transfer (2,9). Deficiency in either Sap C or GCase leads to different variant forms of Gaucher's disease (10,11). In Gaucher's disease as well as in other saposins related pathologies including Tay-Sachs, Sandhoff, and type I Niemann-Pick diseases, saposins were reported to accumulate in the tissues of lysosomal storage disease patients (1). In a previous review article, it was hypothesized that accumulation of saposins could be related to a stimulation in their synthesis by a compensatory mechanism that results from accumulation of either lipids or defective enzyme (1). Alternatively, it was suggested that codeposition of saposins with the accumulated lipids may explain the saposin accumulation (1).
The crystal structures of Sap C and D have recently been reported (12). Also, two recent reports have determined the three-dimensional structure of Sap C in solution and in a detergent environment via solution NMR spectroscopy (13,14). The amino acid sequence of Sap C is shown in Fig. 1 A. As shown in Fig. 1 B, Sap C was found to have five α-helices (13,14). Additionally, a recent solid-state NMR article has indicated that Sap C interacts with negatively charged phospholipid membranes under acidic conditions (15). The solid-state NMR data agrees with the structural model proposed by Qi and co-workers utilizing fluorescence quenching experiments (16). This model suggests that only the amphipathic amino terminus of helix 1 and the carboxyl terminus of helix 5 of Sap C are inserted into the membrane leaving the middle region of Sap C exposed to the aqueous phase (16).
FIGURE 1.

The amino acid sequence (A) and the previously reported three-dimensional solution NMR structure of Sap C (B).
Acidic conditions help to initialize the electrostatic binding between the Sap C protein and negatively charged phospholipids such as phosphatidylserine (PS) (5,13). The protonation of the negative charges of Glu residues of Sap C at acidic conditions eliminates the repulsive forces between Sap C and the negatively charged headgroups of POPS phospholipids (13). Additionally, Sap C has many positively charged Lys residues that are predominantly located at the amino terminal portion of the protein (16). The positively charged Lys residues are important in the recognition of the negative surface charges of phosphatidylserine-containing membranes for initial binding of Sap C (16). Finally, after Sap C recognizes the negatively charged membrane surface, only the terminus regions of helix 1 and helix 5 of Sap C insert into the negatively charged membrane (16).
The proton pumps in the lysosomes are key to making the interior of these intracellular digestive organelles acidic (17). Many lysosomal enzymes have acidic pH optimums for activity (18,19). Previously, we demonstrated with solid-state NMR spectroscopy that Sap C interacts with a mixture of negatively charged PS/PG phospholipids under acidic conditions (15). However, some lysosomal enzymes have neutral pH optimums (20,21). To make such enzymes active, the pH of the lysosome fluctuates from 4.5 to 7.0 or more (22,23). This variation helps the hydrolytic enzymes to efficiently degrade macromolecules (22). Additionally, some physiological disorders related to lipid accumulation have been connected with pH elevation in the lysosomes (24,25). To extend and complement our previous experiments, we designed new solid-state NMR experiments to probe the interaction of Sap C (initially protonated in a weak acid) with both the neutral 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and negatively charged phospholipids such as 1-palmitoyl-2-oleoyl-sn-glycero-3- [phospho-L-serine] (POPS). Both randomly dispersed multilamellar vesicles (MLVs), as well as mechanically oriented bilayers were resuspended in a neutral buffer to mimic the less common neutral lysosomal pH conditions. For the first time, solid-state NMR data of both Sap C-POPS and Sap C-POPC MLVs, prepared under similar neutral pH conditions, were compared from the perspective of the lipids using 2H order parameter profiles, 31P chemical shift anisotropy (CSA) width and 31P T1 relaxation times.
MATERIAL AND METHODS
Materials
All synthetic phospholipids such as 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC); 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-L-serine] (POPS) were purchased from Avati Polar Lipids (Alabaster, AL). The phospholipids were dissolved in chloroform and stored at −20°C; 2,2,2 triflouroethanol (TFE), N-[2-hydroxyethyl]piperazine-N′-2-ethane sulfonic acid (HEPES) and EDTA were obtained from Sigma-Aldrich (St. Louis, MO).
Sap C preparation and purification
The biologically active nonglycosylated recombinant form of Sap C (26,27) has been chosen in this study, since it has a high protein production yield. Preparation and purification of Sap C was performed according to procedures described previously in the literature (28). In brief, recombinant Sap C was overexpressed in Escherichia coli cells using an isopropyl-1-thio-β-D-galactopyranoside-induction pET system. Next, after Sap C was dialyzed and lyophilized, the dried protein was dissolved in 0.1% trifluoroacetic acid and further purified via HPLC C4 reverse-phase column. Then Sap C (with >98% purity) was lyophilized, the correct molecular weight was determined using MALDI-TOF, Western blot (data not shown), and Sap C concentrations were determined as previously reported (29). No lipids and glucosylceramidase were found in the Sap C preparation (GCase EC 3.1.2.45).
NMR sample preparation
Two different MLV samples of POPC and POPS containing 0 mol % and 2 mol % of Sap C were prepared using a slightly modified protocol (30). The phospholipids (dissolved in chloroform) were mixed with Sap C, dissolved in a minimal amount of triflouroethanol ((TFE) ∼50 μl), in a small test tube (total of 40 mg lipids) and dried using a steady steam of N2 gas. After placing the test tube in a vacuum desiccator overnight, the peptide/lipid sample was resuspended in 85 μL of HEPES buffer (pH = 7.0). The buffer was prepared using deuterium-depleted water from Isotec/Sigma-Aldrich (St. Louis, MO). Then, the test tube was shortly heated in a water bath at 50°C along with slight frequent sample agitation to avoid frothing of the suspension. After the phospholipid mixture was completely dissolved, the sample was transferred to a NMR sample tube. POPC-d31 or POPS-d31 (5 mg) was added to the samples when performing the 2H NMR experiments.
The mechanically oriented bilayers were prepared using three different procedures, in which TFE, HEPES neutral buffer or methanol were used to dissolve Sap C. Weak acids such as TFE can protonate proteins (31). In this report, the term protonated Sap C will be used to describe Sap C that was dissolved in the TFE (weak acid). Conversely, the term unprotonated Sap C will be used to describe Sap C that was dissolved in methanol or directly in a neutral buffer.
In the first procedure, to insure that Sap C is incorporated within the bilayers, Sap C was dissolved in 50 μl of TFE, mixed with POPS or POPC phospholipids (15 mg), dissolved in chloroform, and the final volume was reduced to approximately one-half. Then the lipid sample was added via a syringe onto 15 glass plates (1 mg/glass plate) and allowed to dry. The final Sap C concentration on each glass plate was 2 mol % with respect to the lipids. Next, the glass plates were left in a desiccator overnight. The following day, 2 μl of a neutral HEPES buffer (pH 7.0) was added to each glass plate and after 4–5 min the glass plates were stacked and placed in a humidity chamber of saturated ammonium monophosphate at a relative humidity of 93% at 45°C for 2 days. Finally, the glass plates were wrapped in parafilm and placed inside polyethylene bags to minimize dehydration.
For the second procedure, the phospholipids dissolved in chloroform (15 mg) were spotted onto 15 glass plates, air-dried, and left in the desiccator overnight. The following day, 2 μl of the HEPES buffer (pH 7.0) was added. In this procedure, Sap C was completely dissolved in the buffer; therefore, Sap C would interact with the oriented bilayers only after the addition of buffer. Finally, for the third procedure, Sap C was dissolved in a minimal amount of methanol (∼50 μl) and mixed with POPS phospholipids (15 mg), dissolved in chloroform, and then the glass plates samples were prepared similar to the first procedure described above.
NMR spectroscopy
A Bruker Avance 500 MHz WB solid-state NMR spectrometer was used to run all of the experiments. For the mechanically oriented bilayers samples, a static double-resonance flat coil probe from Doty Scientific (Columbia, SC) was used. The flat coil in the probe can be varied to adjust the angle at which the bilayer normal makes with respect to the magnetic field. The 31P NMR spectra were recorded with 1H decoupling using a 5-μs π/2 for 31P and a 4-s recycle delay. For the 31P NMR spectra 512 scans were taken and the free induction delay was processed using 100 Hz of line broadening. The spectral width was set to 250 ppm. In addition, a Bruker 4 mm triple resonance CP-MAS probe (Bruker, Billerica, MA) was used to record the 31P and 2H NMR spectra of MLVs. The 31P NMR spectra were recorded with 1H decoupling using a 4-μs π/2 for 31P and a 4-s recycle delay. A 55-kHz proton decoupling field was uniformly applied for all static 31P NMR samples. For the 31P NMR spectra 1-k scans were taken and the free induction delay was processed using 300 Hz of line broadening. The spectral width was set to 300 ppm. All 31P NMR spectra were referenced by assigning the 85% H3PO4 31P peak to 0 ppm.
2H NMR spectra were collected at a frequency of 76.77 MHz. A quadrupolar echo pulse sequence was employed using quadrature detection with complete phase cycling of the pulse pairs (32). The 90° pulse length was 3.8 μs, the interpulse delay was 40 μs, the recycle delay was 0.5 s, and the spectral width was set to 100 kHz. A total of 20,480 transients were averaged for each spectrum and processed using 300-Hz line broadening. 31P T1Z longitudinal relaxation experiments were conducted using an inversion-recovery pulse sequence 180°-T-90°-acq with the sample spinning (4 kHz) at the magic angle. After the 180° pulse, the delay time (T) was varied from 10.0 ms to 6.0 s. The recycle delay was set to 10 s. To measure T1, the change in the area under a particular peak was fitted to a single exponential function: I(t) = I(0) − Aexp(−t/T1), where I(0) is close to 1 and A is ∼2 (33).
Solid-state NMR data analysis
POPC-d31 and POPS-d31 were used to probe the changes in the order of the acyl chains of POPC and POPS MLVs, respectively, upon Sap C insertion when compared to the control (without Sap C). Powder-pattern 2H NMR spectra of the multilamellar dispersions were numerically deconvoluted (dePaked) using the algorithm of McCable and Wassall (15,34). The spectra were deconvoluted such that the bilayer normal was perpendicular with respect to the direction of the static magnetic field. SCD order parameters were calculated from the expression previously described in the literature (15,30). The 2H peaks in the NMR spectra were assigned based upon dynamic properties of the individual CD3 and CD2 groups. The quadrupolar splitting of the CD3 methyl groups at the end of the acyl chains are the smallest and closest to 0 kHz and the 2H attached to the C-14 have the next smallest splitting followed by the 2H attached to the C-13, and so forth along the acyl chain. According to the literature, the quadrupolar splittings of the plateau region can be estimated by integration of the last broad peak in the 2H NMR spectra and the order parameters calculated for the CD3 quadrupolar splitting were multiplied by three (15,34). Finally, the simulations of the static 31P NMR spectra were carried out using the DMFIT software program (35). The principle elements of the CSA tensors are represented according to the convention σ33 ≥ σ22 ≥ σ11.
RESULTS AND DISCUSSION
2H NMR study of Sap C interacting with both POPC and POPS MLVs
Previously, Sap C was studied using different biophysical techniques that includes Trp fluorescence and quenching experiments (16), fluorescent dye leakage (8), direct atomic force microscopy studies (36), solution NMR in a detergent environment (13), and fluorescence resonance energy transfer (6). In many of these studies, lipid vesicles or large unilamellar vesicles LUVs were used to study the effect of Sap C on the fusion, leakage, or distribution of these vesicles. Conversely, in this study, only the interaction of Sap C with POPC and POPS lipids as well as its perturbation effect on the lipid acyl chains and headgroups were investigated; therefore, MLVs (commonly used in solid-state NMR spectroscopic experiments) and not LUVs were chosen as a model membrane system.
The effect of Sap C on the order and dynamics of the acyl chains of both POPC-d31 and POPS-d31 bilayers was studied using 2H solid-state NMR spectroscopy in the absence (Figs. 2, A and C) and in the presence (Fig. 2, B and D) of 2 mol % Sap C at 25°C. The addition of 2 mol % Sap C to POPC does not significantly alter the 2H NMR spectra (Fig. 2 B) when compared to the control (Fig. 2 A). Inspection of (Fig. 2 D) clearly indicates that the addition of 2 mol % Sap C to the POPS-d31 bilayers alters the lineshape and the spectral resolution of the 2H NMR spectra. The loss in spectral resolution is manifested by the disappearance of sharp edges of the peaks of 2 mol % Sap C in POPS bilayers. The changes in spectral resolution of the 2H NMR spectra confirm that Sap C interacts significantly with the POPS-d31-containing MLVs and not with the POPC-d31-containing MLVs. The central resonance doublet corresponds to the terminal CD3 groups and the remaining overlapped doublets result from the different CD2 segments of the acyl chain of the POPC-d31 and POPS-d31.
FIGURE 2.

2H NMR powder-pattern spectra of the both POPC and POPS bilayers in the absence (A and C) and in the presence (B and D) of 2 mol % Sap C with respect to the lipids at 25°C.
In this study, POPC-d31 and POPS-d31 segmental SCD order parameters describe the local orientation or dynamic perturbations of the C-D bond vector from its standard state due to perturbations of the POPC-d31 and POPS-d31 phospholipid conformations or dynamics as a result of the addition of Sap C to the lipid bilayers. The smoothed segmental C-D bond order parameters (SCD) can be calculated by dePakeing the powder 2H NMR spectra represented in Fig. 2 for both POPC-d31 and POPS-d31. Fig. 3 reveals a characteristic profile of decreasing order (SCD) with increasing distance from the glycerol backbone for the pure bilayer and 2 mol % Sap C-bound bilayer at 25°C. In (Fig. 3 A), there is no significant difference in the order parameter profile of neutral phospholipids (POPC-d31) in the presence of 2 mol % of Sap C when compared to the control sample (without Sap C). Conversely, for the POPS sample (Fig. 3 B), there is a significant decrease in the order parameter profile values (mainly in the first half and not the second half of the acyl chain) upon interaction of negatively charged phospholipids (POPS) with 2 mol % of Sap C when compared to the control sample (without Sap C). Therefore, the current 2H solid-state NMR data indicate that Sap C disrupts the negatively charged phospholipids (POPS) and not the neutrally charged (POPC) lipids.
FIGURE 3.

The smoothed acyl chains orientational order SCD profiles calculated from the dePaked 2H NMR spectra (Fig. 2) of both POPC (A) and POPS (B) phospholipid bilayers with and without 2 mol % Sap C with respect to the lipids at 25°C.
Additionally, the effect of temperature (25–45°C) on the order and dynamics of the acyl chains of POPC and POPS MLVs was investigated. The 2H solid-state NMR spectra indicate that as the temperature increases, the quadrupolar splittings decrease for all samples with and without the Sap C protein, implying that the mobility of the acyl chains increases as the temperature increases (Supplementary Material, Fig. 1S).
To confirm the current 2H solid-state NMR spectroscopic results, additional 31P NMR spectroscopic studies were conducted as well.
31P NMR studies of Sap C interacting with both POPC and POPS MLVs
In several cases, the lineshapes of the static 31P NMR spectra and its corresponding CSA values have been successfully used to study the perturbation effect induced by proteins on phospholipids (15,33,34). In this study, the static 31P NMR spectra of both POPC and POPS lipid bilayers prepared in the absence and presence of 2 mol % of Sap C at 25°C are shown in Fig. 4. At 25°C, the motionally averaged powder pattern spectra are characteristic of MLVs in the liquid crystalline phase (Lα) and are expected for POPC and POPS bilayers at a temperature well above the different chain melting point transition temperatures (Tm). The Tm values are −2 and 14°C for POPC and POPS, respectively (37). For both POPS and POPC control samples (Fig. 4, A and C), the phospholipid molecules in the MLVs can be characterized by an axially symmetric motion with fast reorientation about the long molecular axis (gives the sharp peak at the σ-perpendicular (σ⊥) edge) and a slower reorientation rate perpendicular to that same axis (38). The POPC bilayers containing 2 mol % Sap C (Fig. 4 B) have the same 31P lineshape as the pure POPC bilayers (Fig. 4 A). Moreover, the CSA width of pure POPC MLVs is 48 ± 1 ppm (Fig. 4 A) and the CSA width of POPC MLVs interacting with Sap C is 47 ± 1 ppm (Fig. 4 B), indicating that Sap C is not significantly interacting with the POPC bilayers. The error associated with measuring the CSA width was +/− 1 ppm.
FIGURE 4.

31P NMR powder-pattern spectra of both POPC and POPS MLVs in the absence (A and C) and in the presence (B and D) of 2 mol % Sap C with respect to the lipids at 25°C.
Conversely, the POPS MLVs containing 2 mol % Sap C (Fig. 4 D) has a broader 31P lineshape than the pure POPS MLVs (Fig. 4 C). The decrease in the 31P spectral resolution (lineshape) of the Sap C-POPS MLVs (Fig. 4 D) indicates that the axially symmetric motion of the phospholipids has been altered upon protein incorporation (38). Additionally, the CSA width of the pure POPS MLVs is 55 ± 1 ppm (Fig. 4 C) and the CSA width of POPS MLVs interacting with Sap C is 51 ± 1 ppm (Fig. 4 D). The decrease in 31P CSA width of the POPS MLVs upon Sap C insertion could be due to several factors, including changes in orientation, conformation, or dynamics of the phospholipid headgroups. An increase in the axial rotational motion of the lipids as well as other wobbling motions of the phospholipid headgroups may also contribute to this decrease (15,33,34,39,40). Taking into consideration the observed decrease in the 2H order parameter profile values (see Fig. 3), the overall increase in motion and disorder of the upper half of the acyl chain of Sap C-POPS MLVs may be another reason for the decrease in the 31P CSA width of the POPS MLVs. The 31P lineshape and CSA width data indicate that the phospholipid headgroups are only perturbed by the insertion of Sap C into the negatively charged POPS bilayers and not into the neutral POPC lipids.
Additionally, the 31P powder pattern NMR spectra of both POPC and POPS MLVs, with and without 2 mol % Sap C, were recorded at temperatures ranging from 25°C to 45°C (Supplementary Material, Fig. 2S) and the chemical shift anisotropy width were measured by simulation using the DIMFIT software program (Supplementary Material, Fig. 3S) (35). The data indicate that as the temperature increases, the CSA width of both POPC and POPS bilayers (with and without the Sap C) decreases, indicating that the axial rotational motion of the phospholipid headgroups increases with temperature.
To further probe the interaction of Sap C with the headgroups of POPC and POPS phospholipids, 31P MAS T1 relaxation experiments were conducted (Fig. 5). 31P T1 spin-lattice relaxation rates are sensitive to rapid conformational changes of lipid acyl chains and polar headgroups as well as to changes in the long axis rotation and diffusion of lipids upon peptide interaction (41). The 31P T1 is directly related to the correlation time (τC) of fast molecular motion and the larmor frequency (ωo) (42,43). This frequency (ωo) is fixed when the same magnetic field is used in the whole study. The relation between T1, τC, and ωo can be summarized according to 1/T1 ∝ τC/(1 + 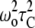 ) (42,43). The relaxation mechanism of certain phospholipid membrane systems is efficient when the molecular correlation time (τC) fits the equation (ωoτC ≈ 1), which results in T1 to be at its minimum (42,43). When τC moves away from 1/ωo, the T1 value increases (33,42,43). It has been previously reported that the temperature can also modulate τC (42,43). The 31P MAS isotropic peak positions of POPC and POPS MLVs with and without 2 mol % Sap C were recorded. POPC MLVs showed sharp isotropic peaks at −1.17 and −1.11 ± 0.05 ppm, with and without Sap C at 25°C, respectively. Conversely, POPS MLVs showed a sharp isotropic peak at −0.57 ± 0.05 ppm for the pure lipid sample and a broader isotropic peak at −0.63 ± 0.05 ppm for the sample with 2 mol % Sap C (Supplementary Material, Fig. 4S). At higher temperatures (35°C and 45°C), the peak positions slightly decreased (within 0.15 ppm range; data not shown).
) (42,43). The relaxation mechanism of certain phospholipid membrane systems is efficient when the molecular correlation time (τC) fits the equation (ωoτC ≈ 1), which results in T1 to be at its minimum (42,43). When τC moves away from 1/ωo, the T1 value increases (33,42,43). It has been previously reported that the temperature can also modulate τC (42,43). The 31P MAS isotropic peak positions of POPC and POPS MLVs with and without 2 mol % Sap C were recorded. POPC MLVs showed sharp isotropic peaks at −1.17 and −1.11 ± 0.05 ppm, with and without Sap C at 25°C, respectively. Conversely, POPS MLVs showed a sharp isotropic peak at −0.57 ± 0.05 ppm for the pure lipid sample and a broader isotropic peak at −0.63 ± 0.05 ppm for the sample with 2 mol % Sap C (Supplementary Material, Fig. 4S). At higher temperatures (35°C and 45°C), the peak positions slightly decreased (within 0.15 ppm range; data not shown).
FIGURE 5.

31P longitudinal relaxation time (T1) as a function of temperature (25–45°C) for both POPC (A) and POPS (B) phospholipid bilayers with and without 2 mol % Sap C with respect to the lipids. The open squares (A) and triangles (B) represent POPC and POPS bilayer controls (without protein), respectively. The solid squares (A) and triangles (B) represent POPC and POPS bilayers with 2 mol % Sap C, respectively. The error bars were obtained by averaging 31P T1 values from two different samples.
In Fig. 5 A, the 31P T1 value increases as the temperature increases from 25 to 45°C in the presence and absence of Sap C. This indicates that the POPC lipid's minimum T1 value would be lower than 25°C with or without Sap C. The 31P T1 value of pure POPC MLVs has been reported to increase with temperature from 25 to 45°C, which is typical for such hydrated phospholipid bilayers (33,42,43). This result confirms our static 31P solid-state NMR data, which indicate that Sap C is not significantly interacting with the POPC bilayers. The 31P T1 value, 31P CSA width, and SCD order parameters of pure POPC MLVs are very close (within 6% deviation) to what was reported previously (34).
Conversely, Fig. 5 B reveals that the 31P T1 relaxation time of the 2 mol % Sap C-POPS sample is considerably less than the POPS control at temperatures ranging from 25 to 45°C. At 25°C, the 31P T1 relaxation value difference between the 2 mol % Sap C-POPS sample and the POPS control is ∼250 ms. In addition, Fig. 5 B indicates that this 31P T1 gap increases as the temperature increases and that a minimum T1 would occur at higher temperatures (when τC ≈ 1/ωo). In a recent article, the antimicrobial peptide G15 was studied using similar approaches (39). Opposite to Sap C-POPS MLVs, G15 induces an increase in the CSA width, as well as an increase in the T1 relaxation times of the G15 containing MLVs (39). The increase in the T1 relaxation times was explained as a result of a decrease in the axial rotational motion of the lipids leading to a less efficient relaxation (39). Similar to G15, the incorporation of another antimicrobial peptide (KIGAKI) to phospholipid MLVs has been reported to increase the 31P T1 relaxation values (33). This less efficient relaxation was also attributed to a reduction in the fast axial rotation motion of the lipids caused by the interaction between the peptide and the lipid (33). Conversely, in the case of Sap C, the dramatic decrease in the 31P T1 values of Sap C/POPS bilayers when compared to the Sap C/POPS control sample is obviously caused by the Sap C protein perturbing the headgroup region and increasing the axial rotational motion of the POPS negatively charged bilayers (more efficient relaxation). The decrease in the 31P T1 values was not observed with the neutral POPC lipids/Sap C system. This agrees with the results of the static 2H and 31P solid-state NMR spectroscopic experiments discussed above.
To further probe the effect of Sap C on POPC and POPS phospholipids, mechanically oriented bilayers were prepared and investigated as well.
31P solid-state NMR study of mechanically aligned POPC and POPS bilayers interacting with Sap C
Solid-state NMR spectroscopic studies of mechanically oriented phospholipid bilayers/protein systems on glass plates can provide pertinent structural information on the protein embedded inside the membrane (44). In this study, this method was used to probe the specific interaction of Sap C with aligned phospholipid bilayers. Fig. 6 shows the 31P solid-state NMR spectra of oriented POPC and POPS phospholipid bilayers in the absence (A) and presence (B) of Sap C, when the bilayer normal is aligned parallel to the static magnetic field at 25°C. All the samples in Fig. 6 were prepared exactly under the same experimental conditions and repeated twice for consistency. Additionally, similar spectra were collected for the same samples when the bilayer normal was aligned parallel to the static magnetic field (data not shown).
FIGURE 6.

The 31P NMR spectra of oriented bilayer samples of both POPC and POPS bilayers in the absence (A and C) and in the presence (B–F) of 2 mol % Sap C with respect to the lipids at 25°C. (B) Sap C was dissolved in a minimal amount of TFE and mixed with the lipids dissolved in chloroform. After application onto the glass plates and drying of the protein-lipid complex overnight, a neutral buffer (pH 7.0) was added at the top of the dried bilayers before stacking of the glass plates. Sample D was prepared exactly the same as sample B except that POPS was used instead of POPC phospholipids. (E) POPS alone was spotted (on the glass plates) and dried. Then the protein dissolved in a neutral buffer (pH 7.0) was added at the top of the dried phospholipids before stacking. Sample F was prepared exactly the same as samples B and D except that methanol was used to initially dissolve Sap C instead of TFE. All spectra were conducted with the bilayer normal of the sample aligned parallel to the static magnetic field.
In Fig. 6 A, the spectrum exhibits one predominant peak at ∼30 ± 1 ppm for the parallel orientation. Also, one predominant peak at approximately at −15 ± 1 ppm for the perpendicular orientation was observed (data not shown). The sharp peak and the lack of any powder components, indicate that the pure POPC phospholipids were well aligned. In Fig. 6 B, the Sap C (dissolved and protonated in TFE) and POPC (dissolved in chloroform) lipids were mixed together before spotting and drying on top of the glass plates, and the buffer used to stack the glass plates was added last. For this sample, the spectra reveal one predominant peak at ∼29 ± 1 ppm (Fig. 6 B) for the parallel orientation and one peak at about −15 ± 1 ppm for the perpendicular orientation (data not shown), indicating that all Sap C/POPC phospholipids bilayers are still well aligned. In addition, the 31P lineshapes of Sap C/POPC bilayers (Fig. 6 B) did not change when compared to the control sample (Fig. 6 A). Fig. 6 B indicates that Sap C did not induce any serious perturbations in the structure and alignment of the POPC bilayer at pH 7.0.
Conversely, Fig. 6, C–F, shows the 31P solid-state NMR spectra of oriented POPS phospholipid bilayers with (D–F) and without (C) Sap C when the bilayer normal is aligned parallel to the magnetic field. In Fig. 6 C, the spectrum shows one predominant peak at ∼39 ± 1 ppm, implying that all POPS pure phospholipids are well aligned. Fig. 6, D–F, represents three methods of Sap C addition to the POPS phospholipid bilayers. For comparison, it is important here to emphasize the presence of the protonated form of Sap C (to initialize its electrostatic binding with negatively charged phospholipids) in one sample (Fig. 6 D) as well as the presence of the unprotonated Sap C form in the other two samples (Fig. 6, E and F). It is also important to distinguish between two types of mixing of the Sap C with the POPS bilayers, specifically the insertion (Fig. 6, D and F) versus membrane association (Fig. 6 E). In the first method (Fig. 6 D), Sap C (dissolved and protonated in TFE) was added to the POPS phospholipids before addition to the glass plates. Then, after the protein-bilayer mixture was dried, the neutral buffer was added and the sample was placed into the humidity chamber. This first method of sample preparation is common in the literature and has been used before for the β-hairpin antimicrobial peptides studied using oriented samples (45). Also, the helix tilt angle of the M2 transmembrane peptide from the influenza A virus has been investigated using the protein and the lipid samples initially cosolubilized in TFE (46). Also, gramicidin D, a transmembrane peptide and ovispirin, a surface peptide were initially dissolved in TFE and used as model systems to determine the membrane peptide orientation using solid-state NMR spectroscopy (47).
Using a similar method, the spectrum in Fig. 6 D indicates that Sap C was able to significantly disrupt the alignment of the POPS bilayers (demonstrated by the splitting of the single peak of the control sample (Fig. 6 C) into several peaks (Fig. 6 D)). This splitting may indicate that the POPS bilayers were fragmented into different components as a result of Sap C insertion. Each component has a local bilayer normal that has a distinguished angle with respect to the magnetic field. These data indicate that after the neutral buffer addition, the Sap C-POPS suspension (and unlike Sap C-POPC suspension) was unable to form intact well-oriented bilayers. Thus, the POPS bilayer is significantly perturbed. Conversely, a different type of perturbation effect was observed in the 31P spectrum of the 2 mol % Sap C-POPS MLVs sample (see Fig. 4 D). Only the 31P line shape and CSA widths of the POPS MLVs changed significantly when compared to the control (pure POPS MLVs). Unlike oriented samples (Fig. 6 D), no additional components or isotropic peaks were observed in Fig. 4 D. This inconsistency between oriented samples and MLVs has been observed in the literature (40). In a recent report, the inconsistency was attributed to differences in the method used to investigate the protein perturbation effect on the membranes (40). Additional factors such as the ability of the lipid to align in the presence of the protein as well as differences in hydration levels (70% and 30–50%) between MLVs and oriented bilayers may lead to this inconsistency (40). Taking together with the inconsistency in the 31P CSA values between MLVs and oriented bilayers, it can be proposed that the POPS oriented bilayers are exclusively more sensitive to perturbation effects induced by Sap C when compared to the corresponding POPS MLVs. Also, this suggests that the headgroups of the negatively charged POPS oriented bilayers have different conformation, order, architecture, and/or dynamics when compared to the headgroups of the corresponding POPS MLVs (48).
In the spectrum shown in Fig. 6 E, a sample was prepared using an alternative method, where Sap C was mixed with the neutral buffer and added to the dried phospholipids bilayers that were spotted at the top of the glass plates and the spectrum was recorded. Clearly, the spectrum show one predominant peak at ∼39 ± 1 ppm for the parallel orientation, implying that all POPS/Sap C phospholipids bilayers are still well aligned. Similarly, one 31P peak at −19 ± 1 ppm was observed for the perpendicular orientation (data not shown). Fig. 6 E indicates that the addition of 2 mol % of Sap C to the negatively charged POPS bilayer did not significantly disturb the alignment of the POPS phospholipid bilayers. These data are expected because unprotonated Sap C has been reported not to interact significantly with PS lipids under neutral conditions (5,13). This alternative method is not commonly used to prepare protein-lipid oriented samples for solid-state NMR spectroscopic studies; however, it is commonly used to prepare Sap C samples studied using other techniques that include direct atomic force microscopy observation (36), solution NMR in a detergent environment (13), Trp fluorescence and quenching experiments (16), fluorescent dye leakage (8), and fluorescence resonance energy transfer (6). In all of these reports, Sap C was mixed with previously prepared lipid vesicles in the buffer at the desired pH. Then, the effect of Sap C association on the fusion, leakage, or distribution of these vesicles was measured.
Finally, a sample was prepared similar to the first method (Fig. 6 D) with a slight modification. In the modified method (Fig. 6 F), methanol was used to initially dissolve Sap C. Using methanol to dissolve proteins is common for preparing oriented samples for solid-state NMR spectroscopic studies and has been used to study the protein-lipid interaction of a magainin analog (MSI-78) and the synthetic lipopeptide MSI-843 (49,50). In addition, the orientational behavior of phospholipid membranes with the toxic peptide mastoparan (isolated from wasp venom) was studied on oriented samples prepared from the peptide and DMPC initially cosolubilized in a methanol/chloroform mixture (51). Also, the target selectivity of the antimicrobial peptide G15 was investigated using an oriented sample of protein initially dissolved in a mixture of methanol and chloroform (39).
Similarly, this third method was used in this study and the spectra show one predominant peak at ∼39 ± 1 ppm for the parallel orientation (Fig. 6 F) and one at about −19 ± 1 ppm for the perpendicular orientation (data not shown), indicating that all POPS/Sap C phospholipid bilayers are well aligned. Additionally, the spectra in Fig. 6 F indicate that unprotonated Sap C was unable to significantly disrupt the alignment of the POPS bilayers when compared to the spectra shown in Fig. 6 D. This result is similar to the data of the second method (see Fig. 6 E) and is expected as discussed above (5,13).
Finally, most of the spectra shown in Fig. 6 have additional small peaks besides the predominant component of the most intense peak (other than those discussed for Fig. 6 D). The smaller peaks represent additional minor components that were imperfectly aligned or minor powder-pattern components that were not aligned at all.
Sap C initially protonated in weak acid forms a complex with POPS and not POPC lipids
It is clear based upon the results of the 2H order parameter profiles, 31P chemical shift anisotropy width, and 31P T1 relaxation times of the MLVs, as well as the 31P NMR data of the oriented bilayers, that protonated Sap C perturbs the POPS bilayers and not the POPC when compared to the control (pure MLVs) under the conditions presented in this study. Additionally, since protonated Sap C does not interact strongly with the neutral POPC lipids as indicated by the 31P and 2H solid-state NMR data, POPC can be used as a control model membrane. The initial acidic binding conditions between Sap C and POPS lipids was provided using the weak Bronsted acids (TFE) which have been reported to be a good H-bond donor and to have a protonation effect on proteins (31). The measured pH of TFE used in this report was ∼5.3. This pH is enough to initiate the Sap C interaction with the negatively charged lipids (13). Acidic conditions have been reported to be necessary to initialize the electrostatic interaction between the Sap C protein and negatively charged phospholipids (5,13). Chloroform is more volatile than TFE; therefore, the protein-lipid suspension was given more time under the steady steam of N2 gas to completely dry the sample when compared to the controls (no protein or TFE). Interestingly, although the pH of the HEPES buffer added to the dried protein-phospholipid complex was neutral (pH 7.0), the perturbation effect induced by Sap C on the POPS MLVs was observed using all our solid-state NMR experiments. Finally, to confirm that the perturbation effect is caused by Sap C and not by any residual TFE used to initially dissolve the protein, another control sample was prepared. In this second procedure, the phospholipids (dissolved in chloroform) were mixed with ∼50 μl of TFE. Then, the sample was dried using a steady steam of N2 gas, placed in a vacuum desiccator overnight, and resuspended in HEPES buffer (pH = 7.0) as described previously. The pure MLVs prepared in the absence or presence of TFE (as described above) showed similar static 31P NMR spectra (Supplementary Material, Fig. 5S).
Sap C-POPS complex resuspended in a neutral buffer
Sap C not only facilitates the phospholipids lysosomal digestion but also participates in lipid trafficking as a transfer protein (2,9). However, whether the Sap C-lipid complex is capable of transferring lipids to their targets through different pH environments (without the dissociation back to lipids and protein) is still unclear in the literature. In a previous report, de Alba and co-workers utilized solution NMR spectroscopy to study the association of Sap C with vesicles prepared with L-α-phosphatidylecholine from egg and L-α-phosphatidyleserine (using a high protein/lipid molar ratio (1:1) or 0.5:0.5 mM protein/total lipid mixture) (13). When the pH of this system (pH = 5.4) was increased back to the original state (pH = 6.8), the authors noticed that the NMR signal intensity recovers to 91% of its original value implying that the majority of the protein gets released. The authors concluded that the binding of Sap C to phospholipid vesicles is a pH-controlled reversible process under the conditions described in their report (13). Moreover, this conclusion was based upon the assumption that vesicles that have a molecular mass larger than 100 kDa tumble very slow, and therefore, the NMR signal that was observed in the presence of vesicles was considered to belong only to the Sap C that is free in solution (13).
Conversely, in this study, we probed the effect of Sap C insertion on both the headgroups' mobility and the acyl chains disorder of the POPC and POPS phospholipids. Using this approach, the data cannot confirm directly whether or not Sap C was partially released from the Sap C-POPS complex after its resuspension in a neutral buffer and whether or not the binding process is reversible. However, our data can clearly confirm using several solid-state NMR experiments that a significant protein-lipid perturbation effect was observed after the exposure of protonated Sap-C/POPS complex to a neutral pH under the current experimental conditions (low protein/lipid molar ratio (1:50); or 2 mol % protein with respect to lipid). A similar observation has been made in the case of the binding of Sap C to PS liposomal vesicles. Salt concentrations have been known to affect the binding of Sap C to PS membranes (52). The insertion of Sap C into PS membranes is blocked at 0.4 M sodium chloride (NaCl). However, the Sap C-PS complex is very stable at salt concentrations up to 1 M (52).
CONCLUSIONS
This study indicates that 2H order parameter profiles, 31P CSA widths, and 31P T1 relaxation times has been successfully used to probe the interaction of the protonated Sap C with neutral (POPC) and negatively charged (POPS) phospholipids at neutral pH conditions. Additionally, oriented phospholipids on glass plates were used to investigate the perturbation effect of Sap C inserted into both POPS and POPC using 31P solid-state NMR spectroscopy. The Sap C, protonated in a weak acid, was unable to produce well-oriented POPS bilayers. From the prospective of the lipids' acyl chain and headgroup dynamics, our new solid-state NMR spectroscopic spectra indicate that Sap C (initially protonated in a weak acid) perturbs the negatively charged (POPS) and not the neutrally charged lipid headgroups (POPC). Unexpectedly, the protonated Sap C was able to conserve its perturbation effect on the POPS membrane even after the Sap C-POPS system was resuspended in neutral buffer. This suggests that Sap C inserted into POPS bilayers initially form a stable complex that lasts even after its resuspension in neutral buffer. Similar complexes have been reported to be stable even at high salt concentrations up to 1 M (52). Finally, as expected, unprotonated Sap C is unable to directly associate with the POPS membrane under neutral pH conditions.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health grants DK 57690 (to X.Q.) and GM080542 (to G.A.L.) and National Science Foundation award CHE-0645709 (to G.A.L.). The 500-MHz wide-bore spectrometer was obtained from a National Science Foundation grant (10116333 to G.A.L.).
Editor: Anne S. Ulrich.
References
- 1.Kishimoto, Y., M. Hiraiwa, and J. S. Obrien. 1992. Saposins: structure, function, distribution, and molecular genetics. J. Lipid Res. 33:1255–1267. [PubMed] [Google Scholar]
- 2.Kolter, T., F. Winau, U. Schaible, M. Lieppe, and K. Sandhoff. 2005. Lipid-binding proteins in membrane digestion, antigen presentation, and antimicrobial defense. J. Biol. Chem. 280:41125–41128. [DOI] [PubMed] [Google Scholar]
- 3.Sandhoff, K., T. Kolter, and G. Van Echten-Deckert. 1998. Sphingolipid metabolism. Sphingiod analogs, sphingolipid activator proteins, and the pathology of the cell. Ann. N. Y. Acad. Sci. 845:139–151. [DOI] [PubMed] [Google Scholar]
- 4.Vaccaro, A. M., M. Tatti, F. Ciaffoni, R. Salvioli, B. Maras, and A. Barca. 1993. Function of saposin C in the reconsititution of glycoceramidase by phosphatidylserine liposomes. FEBS Lett. 336:159–162. [DOI] [PubMed] [Google Scholar]
- 5.Vaccaro, A. M., M. Tatti, F. Ciaffoni, R. Salvioli, A. Serafino, and A. Barca. 1994. Saposin-C induces pH-dependent destabilization and fusion of phosphatidylserine-containing vesicles. FEBS Lett. 349:181–186. [DOI] [PubMed] [Google Scholar]
- 6.Wang, Y., G. A. Grabowski, and X. Y. Qi. 2003. Phospholipid vesicle fusion induced by saposin C. Arch. Biochem. Biophys. 415:43–53. [DOI] [PubMed] [Google Scholar]
- 7.Poste, G., and G. Nicolson. 1978. Cell Surface Reviews, Vol. 5. In Membrane Fusion. G. Poste and G. Nicolson, editors. North-Holland Publishing, Amsterdam, NY.
- 8.Vaccaro, A. M., F. Ciaffoni, M. Tatti, R. Salvioli, A. Barca, D. Tognozzi, and C. Scerch. 1995. pH-dependent conformational properties of saposins and their interaction with phospholipid membranes. J. Biol. Chem. 270:30576–30580. [DOI] [PubMed] [Google Scholar]
- 9.Holthuis, J. C. M., and T. P. Levine. 2005. Lipid traffic: floppy drives and superhighway. Nat. Rev. Mol. Cell Biol. 6:209–220. [DOI] [PubMed] [Google Scholar]
- 10.Rafi, M. A., G. Degala, X. L. Zhang, and D. A. Wenger. 1993. Mutational analysis in a patient with a variant form of Gaucher disease caused by Sap-2 deficiency. Somat. Cell Molec. Gen. 19:1–7. [DOI] [PubMed] [Google Scholar]
- 11.Liou, B., A. Kazimierczuk, M. Zhang, C. R. Scott, R. S. Hegde, and G. A. Grabowski. 2006. Analyses of variant acid beta-glucosidases: effects of Gaucher disease mutations. J. Biol. Chem. 281:4242–4253. [DOI] [PubMed] [Google Scholar]
- 12.Ahn, V. E., P. Leyko, J. R. Alattia, L. Chen, and G. G. Prive. 2006. Crystal structures of saposins A and C. Protein Sci. 15:1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Alba, E., S. Weiler, and N. Tjandra. 2003. Solution structure of human saposin C: pH-dependent interaction with phospholipid vesicles. Biochemistry. 42:14729–14740. [DOI] [PubMed] [Google Scholar]
- 14.Hawkins, C. A., E. de Alba, and N. Tjandra. 2005. Solution structure of human saposin C in a detergent environment. J. Mol. Biol. 346:1381–1392. [DOI] [PubMed] [Google Scholar]
- 15.Abu-Baker, S., X. Qi, J. Newstadt, and G. A. Lorigan. 2005. Structural changes in a binary mixed phospholipid bilayer of DOPG and DOPS upon Sap C interaction at acidic pH utilizing 31P and 2H solid-state NMR spectroscopy. Biochim. Biophys. Acta. 1717:58–66. [DOI] [PubMed] [Google Scholar]
- 16.Qi, X., and G. A. Grabowski. 2001. Differential membrane interactions of saposins A and C. Implication for the functional specificity. J. Biol. Chem. 276:27010–27017. [DOI] [PubMed] [Google Scholar]
- 17.Ohkuma, S., Y. Moriyama, and T. Takano. 1982. Identification and characterization of a proton pump on lysosomes by fluorecien-isothiocyanate-dextran fluorescence. Proc. Natl. Acad. Sci. USA. 79:2758–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aronson, N. N. J., and M. J. Kuranda. 1989. Lysosomal degradation of Asn-linked glycoproteins. FASEB. 3:2615–2622. [DOI] [PubMed] [Google Scholar]
- 19.Bond, J. S., and P. E. Butler. 1987. Intercellular proteases. Annu. Rev. Biochem. 56:333–364. [DOI] [PubMed] [Google Scholar]
- 20.Higa, H. H., A. Manzi, and A. Varki. 1989. O-Acetylation and de-O-acetylation of sialicacids. Purification, characterization and properties of a glycosylated rat liver estrase specific for 9-O-acetyled sialic acid. J. Biol. Chem. 264:19435–19442. [PubMed] [Google Scholar]
- 21.Mononen, I., K. J. Fisher, V. Kaartinrn, and N. N. J. Aronson. 1993. Aspartyleglycosaminuria: protein chemistry and molecular biology of the most common lysosomal storage disorder of glycoprotein degradation. FASEB. 7:1247–1256. [DOI] [PubMed] [Google Scholar]
- 22.Butor, C., G. Griffiths, N. N. J. Aronson, and A. Varki. 1995. Co-localization of hydrolytic enzymes with widely disparate pH optima: implication for the regulation of lysosomal pH. J. Cell Sci. 108:2213–2219. [DOI] [PubMed] [Google Scholar]
- 23.Oglobina, T. A., L. L. Litinskaia, and A. M. Veksler. 1987. Spatial and temporal heterogeneity of pH values in lysosomes from different types of cells cultured in situ. Vopr. Med. Khim. 5:56–59. [PubMed] [Google Scholar]
- 24.Bach, G., C. Chen, and R. E. Pagano. 1999. Elevated lysosomal pH in Mucolipidosis type IV cells. Clinica Chimica Acta. 280:173–179. [DOI] [PubMed] [Google Scholar]
- 25.Holopainen, J., J. Saarikoski, P. K. J. Kinnunen, and I. Jarvela. 2001. Elevated lysosomal pH in neutral ceroid lipofuscinoses. Eur. J. Biochem. 268:5851–5856. [DOI] [PubMed] [Google Scholar]
- 26.Ahn, V. E., K. F. Faull, J. P. Whitelegge, A. L. Fluharty, and G. G. Prive. 2004. Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc. Natl. Acad. Sci. USA. 100:38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou, D., C. Cantu III, Y. Sagiv, N. Schrantz, A. B. Kulkarni, X. Qi, D. J. Mahuran, C. R. Moraales, G. A. Grabowski, K. Benlagha, P. Savage, A. Bendelac, and L. Teyton. 2004. Editing of CD1d-bound lipid antigens by endosomal lipid transfer proteins. Science. 303:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi, X. Y., T. Leonova, and G. A. Grabowski. 1994. Functional human saposins expressed in Escherichia coli: evidence for binding and activation properties of saposin-C with acid beta-glucosidase. J. Biol. Chem. 269:16746–16753. [PubMed] [Google Scholar]
- 29.Qi, X. Y., and G. A. Grabowski. 1998. Acid beta-glucosidase: intrinsic fluorescence and conformational changes induced by phospholipids and saposin C. Biochemistry. 37:11544–11554. [DOI] [PubMed] [Google Scholar]
- 30.Dave, P. C., E. K. Tiburu, K. Damodaran, and G. A. Lorigan. 2004. Investigating structural changes in the lipid bilayer upon insertion of the transmembrane domain of the membrane-bound protein phospholamban utilizing 31P and 2H solid-state NMR spectroscopy. Biophys. J. 86:1564–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Llinas, M., and M. P. Klein. 1975. Charge relay at peptide bond. A proton magnetic resonance of solvation effects on the amide electron density distribution. J. Am. Chem. Soc. 97:4731–4737. [Google Scholar]
- 32.Davis, J. H., K. R. Jeffrey, M. Bloom, and M. I. Valic. 1976. Quadrupolar echo deuteron magnetic resonance spectroscopy in ordered hydrocarbon chains. Chem. Phys. Lett. 42:390–394. [Google Scholar]
- 33.Lu, J., K. Damodaran, J. Blazyk, and G. A. Lorigan. 2005. Solid-state nuclear magnetic resonance relaxation studies of the interaction mechanism of antimicrobial peptides with phospholipid bilayer membranes. Biochemistry. 44:10208–10217. [DOI] [PubMed] [Google Scholar]
- 34.Abu-Baker, S., and G. A. Lorigan. 2006. Phospholamban and its phosphorylated form interact differently with lipid bilayers: A 31P, 2H, and 13C solid-state NMR spectroscopic study. Biochemistry. 45:13312–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Massiot, D., F. Fayon, M. Capron, I. King, S. Le Calve, B. Alonso, J. O. Durand, B. Bujoli, Z. Gan, and G. Hoatson. 2002. Modelling one- and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 40:70–76. [Google Scholar]
- 36.You, H. X., X. Qi, and L. Yu. 2004. Direct AFM observation of saposin C-induced membrane domains in lipid bilayers: from simple to complex lipid mixtures. Chem. Phys. Lipids. 132:15–22. [DOI] [PubMed] [Google Scholar]
- 37.Marassi, F. M., and K. J. Crowell. 2003. Hydration-optimized oriented phospholipid bilayer samples for solid-state NMR structural studies of membrane proteins. J. Magn. Res. 161:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith, I. C. P., and I. H. Ekiel. 1984. Phosphorus-31 NMR of phospholipids in membranes, Chapter 5. In Phosphorus-31 NMR. D. G. Gorenstein, editor. Academic Press, NY. 447–475.
- 39.Ramamorthy, A., S. Thennarau, A. Tan, D. Lee, C. Clayberger, and A. M. Krensky. 2006. Cell selectivity correlates with membrane-specific interactions: a case study on the antimicrobial peptide G15 derived from granulysin. Biochim. Biophys. Acta. 1758:145–163. [DOI] [PubMed] [Google Scholar]
- 40.Lu, J.-X., J. Blazyk, and G. A. Lorigan. 2006. Exploring membrane selectivity of the antimicrobila peptide KIGAKI using solid-state NMR spectroscopy. Biochim. Biophys. Acta. 1303–1313. [DOI] [PubMed]
- 41.Watts, A. 1998. Solid-state NMR approaches for studying the interaction of peptides and proteins with membranes. Biochim. Biophys. Acta. 1376:297–318. [DOI] [PubMed] [Google Scholar]
- 42.Pinheiro, T. J. T., and A. Watts. 1994. Resolution of individual lipids in mixed phospholipid-membranes and specific lipid cytochrome-c interactions by magic-angle-spinning solid state P-31 NMR. Biochemistry. 33:2459–2467. [DOI] [PubMed] [Google Scholar]
- 43.Pinheiro, T. J. T., and A. Watts. 1994. Lipid specificity in the interaction of cytochrome-C with anionic phospholipid-bilayers revealed by solid-state 31P NMR. Biochemistry. 33:2451–2458. [DOI] [PubMed] [Google Scholar]
- 44.Opella, S. J., A. Nevzorov, M. F. Mesleh, and F. M. Marassi. 2002. Structure determination of membrane proteins by NMR spectroscopy. Biochemistry and Cell Biology-Biochimie Et Biologie Cellulaire. 80:597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mani, R., A. J. Warning, R. I. Lehnrer, and M. Hong. 2005. Membrane-disruptive abilities of B-hairpin antimicrobial peptides correlate with conformation and activity: A 31P and 1H NMR study. Biophys. Biochem. Acta. 1716:11–18. [DOI] [PubMed] [Google Scholar]
- 46.Kovacs, F. A., J. K. Denny, J. R. Song, J. R. Quine, and T. A. Cross. 2000. Helix tilt of the M2 transmembrane peptide from influenza A virus: an intrinsic property. J. Mol. Biol. 295:117–125. [DOI] [PubMed] [Google Scholar]
- 47.Yamaguchi, S., and M. Hong. 2002. Determination of membrane peptide orientation by 1H-detected 2H NMR spectroscopy. J. Magn. Reson. 155:244–250. [DOI] [PubMed] [Google Scholar]
- 48.Santos, J. S., D. K. Lee, and A. Ramamoorthy. 2004. Effects of antidepressants on the conformation of phospholipid headgroups studied by solid-state NMR. Magn. Reson. Chem. 42:105–114. [DOI] [PubMed] [Google Scholar]
- 49.Hallock, K. J., D. K. Lee, and A. Ramamoorthy. 2003. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid structure via positive curvature strain. Biophys. J. 84:3052–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thennarasu, S., D. K. Lee, A. Tan, U. P. Kari, and A. Ramamoorthy. 2005. Antimicrobial activity and membrane selective interactions of a synthetic lipopeptide MSI-843. Biochim. Biophys. Acta. 1711:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hori, Y., M. Demura, T. Niidome, H. Aoyagi, and T. Asakura. 1999. Orientational behavior of phospholipid membranes with mastoparan studied by 31P solid-state NMR. FEBS Lett. 455:228–232. [DOI] [PubMed] [Google Scholar]
- 52.Liu, A., N. Wenzel, and X. Qi. 2005. Role of lysine residues in membrane anchoring of Saposin C. Arch. Biochem. Biophys. 443:101–112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.