

Abstract
The structure of a membrane-embedded α-helical reference protein, the M13 major coat protein, is characterized under different conditions of hydrophobic mismatch using fluorescence resonance energy transfer in combination with high-throughput mutagenesis. We show that the structure is similar in both thin (14:1) and thick (20:1) phospholipid bilayers, indicating that the protein does not undergo large structural rearrangements in response to conditions of hydrophobic mismatch. We introduce a “helical fingerprint” analysis, showing that amino acid residues 1–9 are unstructured in both phospholipid bilayers. Our findings indicate the presence of π-helical domains in the transmembrane segment of the protein; however, no evidence is found for a structural adaptation to the degree of hydrophobic mismatch. In light of current literature, and based on our data, we conclude that aggregation (at high protein concentration) and adjustment of the tilt angle and the lipid structure are the dominant responses to conditions of hydrophobic mismatch.
INTRODUCTION
The interaction between integral membrane proteins and cell membranes is dominated by hydrophobic forces (1). For this reason, it is energetically favorable for integral membrane proteins to have a transmembrane segment with the same hydrophobic length as the hydrophobic thickness of the lipid bilayer. However, in certain cases the hydrophobic length of the membrane protein and of the surrounding lipids does not match, a phenomenon called “hydrophobic mismatch”. Hydrophobic mismatch has been shown to modulate the function and structure of different membrane proteins, including the BK channel, the M2 proton channel, and various types of ion-transporting ATPases (2–5). However, despite the tremendous progress that has been made in understanding protein-lipid interactions, the detailed molecular mechanisms by which membrane proteins respond to conditions of hydrophobic mismatch remain unknown. Several structural mechanisms have been proposed by which an α-helix can alleviate hydrophobic mismatch. For example, Fourier transform infrared experiments on α-helical polyleucine peptides suggested distortions of the α-helix in the N- and C-termini in response to hydrophobic mismatch (6). However, in the case of polyleucine-alanine (KALP) peptides, similar experiments indicated that the peptide backbone structure is not significantly affected by mismatch, even if the extent of the mismatch is large (7). Recently, it was proposed that transmembrane helices might flex around a well-defined kink in response to hydrophobic stress (8). Moreover, it was suggested that transmembrane α-helices can reduce their hydrophobic length by the formation of a π-helix. Alternatively, an α-helix could increase its hydrophobic length by the formation of a 310-helix (9,10). Recently, low-level quantum mechanical calculations confirmed that an α-helix can undergo such a structural transition to a 310 or π-helix if a force is exerted along the helix axis (11).
To fully grasp the concept of hydrophobic mismatch, new information on the structural response of proteins to conditions of hydrophobic mismatch is crucial. However, because of the difficulty involved in studying the structure of membrane proteins in bilayers, this represents an enormous biophysical challenge. Conventional techniques for structure determination of water-soluble proteins, such as NMR spectroscopy and x-ray crystallography, require meticulously tuned experimental conditions. We believe that a systematic structural study of membrane proteins in bilayers of different hydrophobic thickness using these techniques is currently unfeasible. Alternatively, circular dichroism and infrared spectroscopy have been successfully applied to study the structure of model peptides under conditions of hydrophobic mismatch (6,7). However, although, undeniably, powerful techniques exist for the overall characterization of secondary structure of proteins and peptides (12,13), these approaches are not suitable for obtaining site-specific information.
Here, we present an approach based on high-throughput mutagenesis in combination with site-specific labeling to obtain low-resolution, but site-specific, information on a membrane protein under conditions of hydrophobic mismatch. For our purpose, we produced several cysteine mutants of M13 major coat protein, a 50-residue-long α-helical model protein, and specifically labeled them using the fluorescence probe AEDANS. Following our own approach, described in the literature (14–16), we perform FRET experiments from the natural Trp-26 to AEDANS to monitor the conformation of the coat protein in bilayers of different hydrophobic thickness, i.e., 14:1PC and 20:1PC.
M13 major coat protein was selected as a model protein because it has been the subject of a plethora of biophysical studies in bilayers of matching and mismatching thickness, making it ideally suited as a reference protein (for a review, see Stopar et al. (35)). Moreover, under the experimental conditions used for the FRET experiments, the coat protein is a single membrane-spanning monomeric α-helix, which implies that our findings will not be complicated by protein-protein interactions. Depending on the environment, different conformations of the coat protein have been observed. In sodium dodecyl sulfate and dodecylphosphocholine micelles, U-shaped, L-shaped, and extended structures have been found (17). In some of these structures, the transmembrane helix has a strong curvature. In dehydrated oriented bilayers, the protein forms an L-shaped structure. In this structure, the transmembrane helix shows a distinct kink near residue 39 (18). In fully hydrated vesicles, the coat protein forms an almost straight helix except for the N-terminal hydrophilic anchor, which is unstructured (16). No deviation in the transmembrane helix was observed in the latter case. Thus, the coat protein can be considered as a flexible protein that can adapt to a multitude of environments (19), and we therefore expect it to be particularly sensitive to hydrophobic mismatch.
To analyze our FRET data, we use a “helical fingerprint” to identify 310 or π-helical domains in the coat protein under conditions of hydrophobic mismatch. Surprisingly, the conformational features or the coat protein in thin and in thick membranes are similar, indicating that the protein does not undergo large structural rearrangements in response to hydrophobic mismatch.
METHODS
Sample preparation
Single cysteine mutants A3C, A7C, A9C, A10C, F11C, N12C, L14C, Q15C, A16C, S17C, A18C, T19C, Y21C, I22C, G23C, Y24C, A27C, V29C, V30C, V31C, I32C, V33C, A35C, T36CA27S, I37C, G38CA27S, I39C, L41C, F42C, K43C, K44C, A49CA27S, and S50C of the M13 major coat protein were prepared, purified, and labeled with 1,5-I-AEDANS, as described previously (20). Protein reconstitution was carried out as described in previous studies (14,21). The phospholipids 14:1PC and 20:1PC were purchased from Avanti Polar Lipids (Alabaster, AL). For experiments at high (∼1500) L/P ratio, the concentration of protein in all samples was ∼1 μM. In the case of titration experiments, the concentration of labeled protein was kept constant at ∼1 μM. The concentration of optically inert mutant (i.e., acetamide-labeled mutant Y21A/Y24A/W26A/T46C) was varied—0, 0.5, 2.0, 6.5, and 14 μM—and the amounts corresponded to total L/P ratios of 1500, 1000, 500, 200, and 100, respectively. This optically inert mutant exhibited behavior similar to that of the wild-type protein in preparation, isolation, and gel electrophoresis.
Fluorescence measurements
Fluorescence emission and fluorescence excitation spectra were recorded on a Fluorolog 3.22 (Jobin Yvon Spex, Edison, NJ) at 20°C, as described elsewhere (14). The energy transfer efficiency was calculated from the fluorescence intensities in the excitation spectra of AEDANS-labeled mutants, given by (14)
 |
(1) |
In this equation, F(290) and F(340) are the fluorescence intensities in the excitation spectrum at 290 and 340 nm.  and
and  are the extinction coefficients of AEDANS at 290 and 340 nm, respectively, and
are the extinction coefficients of AEDANS at 290 and 340 nm, respectively, and 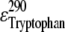 is the extinction coefficient of tryptophan at 290 nm. The values of 1200, 6000, and 4800 M−1 cm−1, respectively, for
is the extinction coefficient of tryptophan at 290 nm. The values of 1200, 6000, and 4800 M−1 cm−1, respectively, for 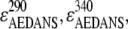 and
and 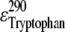 were taken from the literature (22,23).
were taken from the literature (22,23).
Molecular modeling
The energy transfer efficiencies for helices composed of both α-helical and π-helical domains, and for helices composed of both α-helical and 310-helical domains, were calculated based on modifications of analytical expressions for an α-helix, as described in the text using PERL-scripts, which can be obtained from the authors upon request. The energy transfer efficiencies in the case of an unstructured helix were calculated using the computer program FRETsim (15,16,24), which also can be obtained from the authors upon request.
RESULTS AND DISCUSSION
Titration experiments
From the literature, it is known that membrane proteins and peptides can respond to hydrophobic mismatch by aggregation. For example, the β-helical peptide gramicidin A′ aggregates at a high concentration in the membrane under conditions of hydrophobic mismatch (25). It has also been suggested that α-helical polyleucine-alanine peptides (KALP peptides) can form oligomers (10,26). However, bacteriorhodopsin, consisting of multiple transmembrane α-helices, can accommodate large differences between protein and lipid hydrophobic thickness without protein aggregation (27).
At low lipid/protein ratios, M13 coat protein is known to aggregate in bilayers of mismatching lipids (28). To assay possible protein aggregation of the coat protein embedded in lipid bilayers, the intermolecular energy transfer of AEDANS-labeled mutant A3C was investigated upon titration with an acetamide-labeled mutant Y21A/Y24A/W26A/T46C. This mutant is an optically inert coat protein because of the absence of tryptophan and tyrosine residues, and will not contribute to the energy transfer processes. M13 coat protein mutant A3C was selected, because the AEDANS label is expected to be far away from the lipid-water interface, thus avoiding possible specific interactions with the lipid membrane. The energy transfer efficiencies for different L/P ratios were determined using Eq. 1 and are depicted in Fig. 1. At high L/P ratios (L/P = 1000 and 1500), the energy transfer efficiencies level off at a value of ∼0.3 in the case of the thin 14:1PC lipids, and ∼0.2 for the thick 20:1PC lipids. This suggests that the resulting energy transfer is related to intramolecular effects and that the protein is essentially monomeric and randomly distributed under these conditions.
FIGURE 1.

Energy transfer efficiencies of AEDANS-labeled M13 mutant coat protein A3C at different lipid/protein ratios for 14:1PC (⋄) and 20:1PC membranes (•). The concentration of AEDANS-labeled mutant protein was kept constant at 1 μM. Error bars were calculated based on an uncertainty of ±200 M−1 cm−1 in the extinction coefficient.
However, upon decreasing the L/P ratio below these values, the energy transfer efficiency increases for both thick and thin lipids. Because the protein that is titrated in is optically inactive, this increase can only arise from a decrease of protein-protein distances, indicating that oligomers are being formed. This conclusion is in good qualitative agreement with results from previous studies (28), although slightly different lipids were used in the work described here. Based on this analysis, all further FRET experiments were carried out at L/P 1500, where the intermolecular energy transfer is negligible. This avoids complications in the interpretation of the energy transfer efficiencies in terms of intramolecular structural effects.
Intramolecular energy transfer efficiencies
To monitor the conformation of membrane-embedded M13 coat protein under conditions of mismatch, the intramolecular energy transfer efficiencies for different mutant positions at L/P 1500 in 14:1PC and 20:1PC bilayers were determined from the excitation spectra of AEDANS-labeled mutants (14) (see Fig. 2). Overall, the energy transfer efficiencies in thin 14:1PC and thick 20:1PC membranes are similar. For both lipid systems, on going from mutant position 1 to the tryptophan residue at position 26, the energy transfer efficiency gradually increases. As expected, the energy transfer decreases gradually again on going from mutant position 27 to position 50, i.e., when the distance from the AEDANS acceptor label to the tryptophan donor at position 26 is increased. In both lipid systems, some local fluctuations are visible, which could arise from a helical protein conformation. For reference, we display the theoretical energy transfer efficiencies in the case of a straight α-helix calculated as described in our previous work (14,16). Especially for mutant position 3 in the N-terminal domain and position 37 in the transmembrane domain, relatively large deviations from a straight α-helix are observed. These deviations are consistently observed in multiple experiments.
FIGURE 2.

Energy transfer efficiencies of various AEDANS-labeled M13 mutant coat proteins incorporated in 14:1PC (⋄) and 20:1PC membranes (•). For reference, the theoretical efficiencies in the case of an α-helix are indicated by a black line that interconnects the theoretical efficiencies per mutant. The shaded line represents the theoretical efficiencies in the case of an α-helix with a π-helical segment at residues 38–50. Error bars were calculated based on an uncertainty of ±200 M−1 cm−1 in the extinction coefficient.
In a previous study, an increase in energy transfer efficiency in the N-terminus was attributed to the presence of an unstructured domain arising from the N-terminal hydrophilic anchor of the protein (16). The set of experimental energy transfer efficiencies in Fig. 2 provides an opportunity to test this idea. To monitor the effect of an unstructured domain in the N-terminus, we define a FRET quality parameter Q1–15, given by
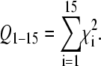 |
(2) |
Here, i runs over the mutants, and 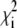 is defined as
is defined as
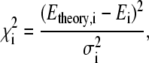 |
(3) |
where Ei is the experimental efficiency and 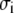 is the standard deviation calculated based on an uncertainty of ±200 M−1 cm−1 in the extinction coefficient (14). With this definition of parameter Q1–15, a low value implies that the structure is in good agreement with our FRET data. The theoretical intramolecular energy transfer efficiency for each mutant, Etheory,i, is calculated from the distance between the donor and acceptor:
is the standard deviation calculated based on an uncertainty of ±200 M−1 cm−1 in the extinction coefficient (14). With this definition of parameter Q1–15, a low value implies that the structure is in good agreement with our FRET data. The theoretical intramolecular energy transfer efficiency for each mutant, Etheory,i, is calculated from the distance between the donor and acceptor:
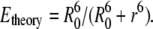 |
(4) |
In this equation, r is the distance between the donor (tryptophan at position 26) and acceptor (AEDANS). 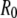 is the Förster radius, which was determined in previous work to be 24 ± 1 Å, assuming a dynamic averaging of the donor and acceptor within the lifetime of the donor excited state for all mutant positions (14). The distance r between donor and acceptor in the molecular model is readily calculated using a formalism that was derived previously, taking the size of the donor and acceptor labels as 6.5 Å and 8.0 Å, respectively (29).
is the Förster radius, which was determined in previous work to be 24 ± 1 Å, assuming a dynamic averaging of the donor and acceptor within the lifetime of the donor excited state for all mutant positions (14). The distance r between donor and acceptor in the molecular model is readily calculated using a formalism that was derived previously, taking the size of the donor and acceptor labels as 6.5 Å and 8.0 Å, respectively (29).
The quality parameter Q1–15 will be specifically sensitive to structural changes of the N-terminal protein domain. We define the unstructured region of the protein as domain U. This domain starts at residue 1 and ends at residue nU. In this analysis, the remaining part of the protein is assumed to be an α-helix. So, for a value of nU = 6, residues 1–6 are unstructured and residues 7–50 are α-helical. For our analysis, we use the previously described computer program FRETsim to calculate the efficiencies in the case of an unstructured domain (16). This computer program models the unstructured region as a chain of vectors joining the Cα atoms. To calculate the efficiency of each mutant, a structure was randomly generated. Structures with clashing Cα atoms (i.e., with an interatom distance of <1.54 Å, twice the Van der Waals radius of the carbon atom) were rejected, and a new random structure was generated until 1000 iterations were reached. This procedure was repeated for increasing lengths of the unstructured domain, for values of nU = 1–15. The resulting quality parameters Q1–15 are depicted in Fig. 3. Clearly the introduction of an unstructured domain in the N-terminus leads to a reduction in the quality parameter Q1–15. In both 14:1PC and 20:1PC bilayers, the value Q1–15 is minimal for an unstructured domain of nine residues. We therefore conclude that in the N-terminal domain, amino acid residues 1–9 are unstructured. This is in excellent agreement with previous findings (16,19).
FIGURE 3.

Quality parameters Q1–15 for an increasing unstructured region in the N-terminal domain of AEDANS-labeled M13 mutant coat protein for 14:1PC (⋄) and 20:1PC (•).
The deviating efficiency of residue 37 of the coat protein in Fig. 2 suggests that the α-helix (Fig. 2, black line) is distorted in this region, for instance via the formation of a kink or the formation of a 310- or π-helix. Previous work showed no evidence for a kink in the transmembrane protein domain (15,29), and therefore we do not take into account flexing of the α-helix in our further analysis. Instead, we allow the α-helix to adapt its helical structure to a 310- or π-helix. To this end, we introduce a protein model of a mixed helix, composed of an α-helical domain and a 310- or π-helical domain (see Fig. 4). Hydrogen bonding in an α-helix is strongly cooperative (30), and for this reason a helix with alternating short stretches of α and π-helix, or with short stretches of α and 310-helix is expected to be energetically unfavorable. We therefore assume that only a single, continuous 310- or π-helical domain is formed, and that the remaining part of the protein forms an α-helix. To describe the parameters for the different helical regions, we use known values for the rotation and rise/residue (31). Because the first nine amino acid residues are unstructured, this analysis is performed on the remaining amino acid residues 10–50.
FIGURE 4.

Membrane-embedded M13 coat protein model used to calculate the energy transfer efficiencies in a mixed helix. In the N-terminus, amino acid residues 1–9 are unstructured. The α-helix is indicated as a dark shaded ribbon. The 310- or π-helical domain is depicted as a black rectangle. The starting and ending positions of this domain vary between 10 and 50 (see Fig. 5).
For this purpose, we define a FRET quality parameter Q10–50 similar to that given by Eq. 2. The resulting quality parameters are plotted in Fig. 5. The plots in Fig. 5 can be seen as helical fingerprints, i.e., given by the shape of the colored domains. Each pixel represents a unique structure, with the starting and ending position of the 310- or π-helix indicated on the y and x axes, respectively. The helical fingerprint indicates the ability of an α-helix to form domains of 310- or π-helices. The diagonal in the panels of Fig. 5 gives the values for a full α-helix from residues 10–50. The value Q10–50 for a full α-helix is 185 in the case of thin 14:1PC bilayers, and 249 in the case of thick 20:1PC bilayers. In the case of thin 14:1PC bilayers, the introduction of a π-helical fragment leads to a reduction in the value Q10–50 from 185 in the case of a full α-helix to 123 in the case of an α-helix with a π-helical domain in the transmembrane helix from residues 38–50. For thick 20:1PC bilayers, the introduction of a π-helix also leads to a reduction in Q10–50 from 244 for a full α-helix to 190 for an α-helix with a π-helical domain in the transmembrane helix from residues 38–50. The introduction of a 310-helix does not lead to a reduction in the value Q10–50 with respect to a full α-helix in the case of thin 14:1PC bilayers. In the case of thick 20:1PC bilayers, the introduction of a 310-helix does lead to a reduction in the value Q10–50, from 244 for a full α-helix to 228 for an α-helix with a 310-helical segment from residue 19 to residue 24. Clearly, the largest decrease in the value Q10–50 is seen on introducing π-helical domains in both thin and thick bilayers. It is therefore tempting to conclude from the helical fingerprint analysis that the coat protein forms an α-helix with a π-helical domain in the transmembrane helix at residues 38–50 in both thin and thick bilayers. For reference, the theoretical energy transfer efficiencies in the case of an α-helix with a π-helical segment for residues 38–50 is also displayed in Fig. 2 (shaded line). We note, however, that the model of an α-helix with a π-helical segment and an unstructured N-terminus performs only slightly better than the model of a full α-helix with unstructured N-terminus. For instance, in the case of thin 14:1PC bilayers, the introduction of an unstructured domain leads to a decrease in the value Q1–15 of 85, as compared to a decrease in value Q10–50 of 62. The effect of the introduction of an unstructured domain is much larger than the effect of the π-helix given the fact that Q1–15 only runs over eight mutants whereas Q10–50 runs over 26 mutants. Likewise, the introduction of an unstructured domain in the case of thick 20:1PC bilayers leads to a decrease in value Q1–15 of 52, compared to a decrease of 54 for the value Q10–50.
FIGURE 5.

Quality parameters Q10–50 for an α-helix with a 310- or π-helical segment of variable length in 14:1PC and 20:1PC membranes. The color coding indicates the level of the quality parameter. (A) π-helical segment in 14:1PC. (B) 310-helical segment in 14:1PC. (C) π-helical segment in 20:1PC. (D) 310-helical segment in 20:1PC.
Although the appearance of π-helical domains in transmembrane helices is rare, it has been reported in both theoretical (32) and experimental (33) studies. It has been noted previously that the presence of a metastable π-helix indicates that the system has a high propensity for conformational transitions (34). In the case of the M13 major coat protein, a π-helix between residues 38 and 50 could also indicate that this part of the protein has a high propensity for helical deformation. In an earlier work, it was shown that the phenylalanine residues at positions 42 and 45 and the lysine residues at positions 40, 43, and 44 are implicated in the anchoring of the coat protein on the C-terminal interface (35). It is possible that the ability of the protein backbone to undergo small helical deformations in this region allows efficient incorporation into the phage particle when these residues are detached from the C-terminal interface.
In summary, the presence of a π-helix slightly increases the performance of our model. However, both the model of an α-helix with an unstructured end and the model of an α-helix with an unstructured end and a small π-helical fragment between residues 38 and 50 are acceptable based on the helical fingerprint analysis. Our analysis shows that the general characteristics of the protein are the same in both thin and thick bilayers, but the values of the quality parameters Q10–50 in thin 14:1PC bilayers are slightly lower than those in thick 20:1PC bilayers. This could indicate that there are still small structural differences between the coat protein in thin and thick bilayers that are not accounted for in our models.
Our results show that the overall conformation of the M13 coat protein does not respond to hydrophobic stress. We do not find any evidence for a disturbance of the helical structure in the termini in response to hydrophobic stress. Furthermore, no evidence is found for a structural adaptation of the backbone to a 310- or π-helix due to interaction with the bilayer or with the lipid-water interface. Clearly, the uniaxial mismatch force is not large enough to induce structural transitions between different types of helices, i.e., from an α-helix to a 310- or a π-helix, or from a combined α-helix/π-helix to an α-helix or 310-helix. The question arises why the uniaxial force is insufficient to induce structural transitions of the transmembrane helix. The uniaxial force depends on multiple factors, such as the anchoring of the helix in both lipid-water interfaces, the ability of the protein to tilt, the rigidity of the helix, and the magnitude of the forces due to hydrophobic mismatch. The anchoring of the coat protein is different from that of other model peptides, such as lysine-flanked polyleucine peptides, in that the anchoring is not symmetric. In the C-terminal interface, the coat protein is strongly anchored by a combination of phenylalanine and lysine residues, whereas it is more weakly anchored in the N-terminal interface. Because of the weak anchoring in the N-terminal interface, the coat protein can adapt its tilt angle with relative ease, as was shown in previous work on fluorescent-labeled coat protein (29). In this way, the forces due to hydrophobic mismatch are exerted partially along the helix axis, and partially along the membrane normal, exerting a force on the surrounding phospholipids. Possibly, this force deforms the lipid bilayer, analogous to the “mattress model” of Mouritsen and Bloom (36). The rigidity of the α-helix does not seem to play a crucial role, as the coat protein and other α-helical model peptides behave alike under conditions of hydrophobic stress. Apparently, in general, α-helices have an intrinsic rigidity along the helix axis, even if they are interrupted by a more compressed π-helical structure. This is consistent with findings from theoretical calculations, showing that an α-helix can support a large uniaxial load without yielding (11).
CONCLUDING REMARKS
In conclusion, membrane-embedded M13 major coat protein forms an almost uniform α-helical structure when incorporated into lipid membranes of varying hydrophobic thickness, with a few unstructured residues in the N-terminus and a small tendency to form π-helical domains in the transmembrane domain. The conformational features of the coat protein in thin and thick membranes are similar, indicating that the protein does not undergo large structural rearrangements in response to hydrophobic mismatch. Most likely, aggregation (at high protein concentration) (28) and adjustment of the tilt angle (29) are the main responses of a transmembrane helix to conditions of hydrophobic mismatch.
Acknowledgments
The authors thank Thomas Huber and Antoinette Killian for fruitful discussions.
This work was supported by contract No. QLG-CT-2000-01801 from the European Commission (Molecular Inhibitors of Vacuolar ATPase: New Therapeutic Approaches to Osteoporosis—Targeting the Osteoclast V-ATPase).
Abbreviations used: AEDANS, N-(acetylaminoethyl)-5-naphthylamine-1-sulfonic acid; FRET, Förster (or fluorescence) resonance energy transfer; 14:1PC, 1,2-dimyristoleoyl-sn-glycero-3-phosphocholine; 20:1PC, 1,2-dieicosenoyl-sn-glycero-3-phosphocholine; L/P, lipid/protein molar ratio.
Editor: Peter Tieleman.
References
- 1.Mitra, K., I. Ubarretxena-Belandia, T. Taguchi, G. Warren, and D. M. Engelman. 2004. Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc. Natl. Acad. Sci. USA. 101:4083–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yuan, C., R. J. O'Connell, P. L. Feinberg-Zadek, L. J. Johnston, and S. N. Treistman. 2004. Bilayer thickness modulates the conductance of the BK channel in model membranes. Biophys. J. 86:3620–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duong-Ly, K. C., V. Nanda, W. F. DeGrado, and K. P. Howard. 2005. The conformation of the pore region of the M2 proton channel depends on the lipid bilayer environment. Protein Sci. 14:856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cornelius, F. 2001. Modulation of Na,K-ATPase and Na-ATPase activity by phopholipids and cholesterol. I. Steady-state kinetics. Biochemistry. 40:8842–8851. [DOI] [PubMed] [Google Scholar]
- 5.Lee, A. G. 1998. How lipids interact with an intrinsic membrane protein: the case of the calcium pump. Biochim. Biophys. Acta. 1376:381–390. [DOI] [PubMed] [Google Scholar]
- 6.Liu, F., R. N. A. H. Lewis, R. S. Hodges, and R. N. McElhaney. 2002. Effects of variations in the structure of a polyleucine-based α-helical transmembrane peptide on its interaction with phosphatidylcholine bilayers. Biochemistry. 41:9197–9207. [DOI] [PubMed] [Google Scholar]
- 7.De Planque, M. R. R., E. Goormaghtigh, D. V. Greathouse, R. E. Koeppe, J. A. W. Kruijtzer, R. M. J. Liskamp, B. De Kruijff, and J. A. Killian. 2001. Sensitivity of single membrane-spanning α-helical peptides to hydrophobic mismatch with a lipid bilayer: effects on backbone structure, orientation, and extent of membrane incorporation. Biochemistry. 40:5000–5010. [DOI] [PubMed] [Google Scholar]
- 8.Yeagle, P. L., M. Bennett, V. Lemaître, and A. Watts. 2006. Transmembrane helices of membrane proteins may flex to satisfy hydrophobic mismatch. Biochim. Biophys. Acta. 1768:530–537. [DOI] [PubMed] [Google Scholar]
- 9.Killian, J. A. 1998. Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta. 1376:401–416. [DOI] [PubMed] [Google Scholar]
- 10.Kandasamy, S. K., and R. G. Larson. 2006. Molecular dynamics simulations of model trans-membrane peptides in lipid bilayers: a systematic investigation of hydrophobic mismatch. Biophys. J. 90:2326–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ireta, J., J. Neugebauer, M. Scheffler, A. Rojo, and M. Galván. 2005. Structural transitions in the polyalanine α-helix under uniaxial strain. J. Am. Chem. Soc. 127:17241–17244. [DOI] [PubMed] [Google Scholar]
- 12.Pelton, J. T. 2001. Secondary considerations. Science. 16:2175–2176. [Google Scholar]
- 13.Goormaghtigh, E., J.-M. Ruysschaert, and V. Raussens. 2006. Evaluation of the information content in infrared spectra of protein secondary structure information. Biophys. J. 90:2946–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vos, W. L., R. B. M. Koehorst, R. B. Spruijt, and M. A. Hemminga. 2005. Membrane-bound conformation of M13 major coat protein: A structure validation through FRET-derived constraints. J. Biol. Chem. 280:38522–38527. [DOI] [PubMed] [Google Scholar]
- 15.Nazarov, P. V., R. B. M. Koehorst, W. L. Vos, V. V. Apanasovich, and M. A. Hemminga. 2006. FRET study of membrane proteins: simulation-based fitting for analysis of membrane protein embedment and association. Biophys. J. 91:454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nazarov, P. V., R. B. M. Koehorst, W. L. Vos, V. V. Apanasovich, and M. A. Hemminga. 2007. FRET study of membrane proteins: determination of the tilt and orientation of the N-terminal domain of M13 major coat protein. Biophys. J. 92:1296–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papavoine, C. H. M., B. E. C. Christiaans, R. H. A. Folmer, R. N. H. Konings, and C. W. Hilbers. 1998. Solution structure of the M13 major coat protein in detergent micelles: a basis for a model of phage assembly involving specific residues. J. Mol. Biol. 282:401–419. [DOI] [PubMed] [Google Scholar]
- 18.Marassi, F. M. M., and S. J. Opella. 2003. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci. 12:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stopar, D., J. Štrancar, R. B. Spruijt, and M. A. Hemminga. 2006. Motional restrictions of membrane proteins: a site-directed spin labeling study. Biophys. J. 91:3341–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spruijt, R. B., A. B. Meijer, C. J. A. M. Wolfs, and M. A. Hemminga. 2000. Localization and rearrangement modulation of the N-terminal arm of the membrane-bound major coat protein of bacteriophage M13. Biochim. Biophys. Acta. 1509:311–323. [DOI] [PubMed] [Google Scholar]
- 21.Spruijt, R. B., C. J. A. M. Wolfs, and M. A. Hemminga. 1989. Aggregation-related conformational change of the membrane-associated coat protein of bacteriophage M13. Biochemistry. 28:9158–9165. [DOI] [PubMed] [Google Scholar]
- 22.Hudson, E. N., and G. Weber. 1973. Synthesis and characterization of two fluorescent sulfhydryl reagents. Biochemistry. 12:4154–4161. [DOI] [PubMed] [Google Scholar]
- 23.Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 6:1948–1954. [DOI] [PubMed] [Google Scholar]
- 24.Nazarov, P. V., V. V. Apanasovich, V. M. Lutkovski, M. M. Yatskou, R. B. M. Koehorst, and M. A. Hemminga. 2004. Artificial neural network modification of simulation-based fitting: application to a protein-lipid system. J. Chem. Inf. Comput. Sci. 44:568–574. [DOI] [PubMed] [Google Scholar]
- 25.Ge, M., and J. H. Freed. 1999. Electron-spin resonance study of aggregation of gramicidin in dipalmitoylphosphatidylcholine bilayers and hydrophobic mismatch. Biophys. J. 76:264–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mall, S., R. Broadbridge, R. P. Sharma, J. M. East, and A. G. Lee. 2001. Self-association of model transmembrane α-helices is modulated by lipid structure. Biochemistry. 40:12379–12386. [DOI] [PubMed] [Google Scholar]
- 27.Lewis, B. A., and D. M. Engelman. 1983. Bacteriorhodopsin remains dispersed in fluid phospholipid bilayers over a wide range of bilayer thicknesses. J. Mol. Biol. 166:203–210. [DOI] [PubMed] [Google Scholar]
- 28.Fernandes, F., L. M. S. Loura, M. Prieto, R. B. M. Koehorst, R. B. Spruijt, and M. A. Hemminga. 2003. Dependence of M13 major coat protein oligomerization and the lateral segregation on bilayer composition. Biophys. J. 85:2430–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koehorst, R. B. M., R. B. Spruijt, F. J. Vergeldt, and M. A. Hemminga. 2004. Lipid bilayer topology of the transmembrane α-helix of M13 major coat protein and bilayer polarity profile by site-directed fluorescence spectroscopy. Biophys. J. 87:1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ireta, J., J. Neugebauer, M. Scheffler, A. Rojo, and M. Galváni. 2003. Density functional theory study of the cooperativity of hydrogen bonds in finite and infinite α-helix. J. Phys. Chem. B. 107:1432–1437. [Google Scholar]
- 31.Creighton, T. E. 1983. Proteins. Freeman, New York.
- 32.Caballero-Herrera, A., K. Nordstrand, K. D. Berndt, and L. Nilsson. 2005. Effect of urea on peptide conformation in water: molecular dynamics and experimental characterization. Biophys. J. 89:842–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi, G., J. Landin, J. F. Galan, R. R. Birge, A. D. Albert, and P. L. Yeagle. 2002. Structural studies of metarhodopsin II, the activated form of the G-protein coupled receptor, rhodopsin. Biochemistry. 41:7318–7324. [DOI] [PubMed] [Google Scholar]
- 34.Zbilut, J. P., A. Colosimo, F. Conti, M. Colafranceschi, C. Manetti, M. Valerio, C. L. Webber, Jr., and A. Giuliani. 2003. Protein aggregation/folding: the role of deterministic singularities of sequence hydrophobicity as determined by nonlinear signal analysis of acylphosphatase and Aβ(1–40). Biophys. J. 85:3544–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stopar, D., R. B. Spruijt, and M. A. Hemminga. 2006. Anchoring mechanisms of membrane-associated M13 major coat protein. Chem. Phys. Lipids. 141:83–93. [DOI] [PubMed] [Google Scholar]
- 36.Mouritsen, O. G., and M. Bloom. 1984. Mattress model of lipid-protein interactions in membranes. Biophys. J. 1984:141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]