

Abstract
Due to the inhalation of airborne particles containing bacterial lipopolysaccharide (LPS), these molecules might incorporate into the 1,2-dipalmitoylphosphatidylcholine (DPPC)-rich monolayer and interact with surfactant protein A (SP-A), the major surfactant protein component involved in host defense. In this study, epifluorescence microscopy combined with a surface balance was used to examine the interaction of SP-A with mixed monolayers of DPPC/rough LPS (Re-LPS). Binary monolayers of Re-LPS plus DPPC showed negative deviations from ideal behavior of the mean areas in the films consistent with partial miscibility and attractive interaction between the lipids. This interaction resulted in rearrangement and reduction of the size of DPPC-rich solid domains in DPPC/Re-LPS monolayers. The adsorption of SP-A to these monolayers caused expansion in the lipid molecular areas. SP-A interacted strongly with Re-LPS and promoted the formation of DPPC-rich solid domains. Fluorescently labeled Texas red-SP-A accumulated at the fluid-solid boundary regions and formed networks of interconnected filaments in the fluid phase of DPPC/Re-LPS monolayers in a Ca2+-independent manner. These lattice-like structures were also observed when TR-SP-A interacted with lipid A monolayers. These novel results deepen our understanding of the specific interaction of SP-A with the lipid A moiety of bacterial LPS.
INTRODUCTION
Lipopolysaccharide (LPS), the major constituent of the outer membrane of Gram-negative bacteria, is a potent stimulator of the immune system (1). Besides its proinflammatory activities, LPS participates in bacterial membrane functions. LPS is essential for bacterial growth and viability since it contributes to low membrane permeability and enhances resistance toward hydrophobic agents (2). From a structural point of view, LPS consists of a polysaccharide part attached to a lipid (lipid A). Lipid A consists of a diglucosamine backbone to which two phosphates are linked at positions 1 and 4′, and six or seven ester- and amide-linked acyl chains are bound at positions 2, 3, 2′, and 3′ (Fig. 1). The polysaccharide part, which facilitates the solubility of the molecule in water, consists of two parts: 1), the O-specific chain, an oligosaccharide with a composition varying with bacterial species; and 2), a rather invariable core section, which is located between the oligosaccharide and the lipid A. Wild-type enterobacterial species with O-chains are termed “smooth” and their LPS called “smooth LPS” (S-LPS). Mutants producing LPS lacking O-specific chains are termed “rough” (R) and their LPS designated as Ra, Rb, Rc, Rd, and Re in order of decreasing core length (3). The endotoxic molecule with the smallest molecular size and full endotoxic activities is Re-LPS (Fig. 1). Bacteria with Re-LPS phenotypes are more common among pathogens that colonize the upper aerodigestive tract (4).
FIGURE 1.

Chemical structure of DPPC and Re-LPS from S. minnesota R-595. The Re-LPS contains two units of KDO. The lipid A moiety is also indicated. Lipid A consists of a diglucosamine backbone to which two phosphates are linked at positions 1 and 4′, and seven ester- and amide-linked acyl chains at positions 2, 3, 2′, and 3′. R1 and R2 indicate different substitutions of the respective phosphate groups in the lipid A moiety of Re-LPS. R1 can be either H or phosphoethanolamine. R2 can be either H or 4-amino-4-deoxy-L-arabinose (3).
Due to inhalation of airborne particles containing bacteria and LPS, the thin alveolar epithelium is continuously exposed to this potent proinflammatory molecule. When LPS molecules enter the host via airways, they interact with alveolar macrophages in a fluid environment characterized by the presence of pulmonary surfactant, which is involved in reducing the surface tension of the fluid lining the alveoli and in host defense. Several components present in the lipid-rich alveolar fluid, such as surfactant proteins (SP-) A, C, and D (5–8), are involved in the binding and neutralization of LPS and/or downregulation of LPS responses that promote excessive inflammation and compromise gas exchange.
SP-A is a large oligomeric extracellular protein found primarily in the alveolar fluid of mammals. It belongs to the structurally homologous family of innate immune defense proteins known as collectins for their collagen-like and lectin domains (9,10). Unlike other collectins, SP-A is mainly associated with surfactant lipids, especially with DPPC. SP-A's ability to bind lipids 1), improves the adsorption and spreading of surfactant membranes onto an air-liquid interface (11); 2), protects surfactant biophysical activity from the inhibitory action of serum proteins (12); and 3), allows this protein to position and concentrate along with surfactant membranes as an initial defense against inhaled toxins and pathogens. We previously reported that SP-A interacts with DPPC monolayers (13,14) and with gel-like regions of monolayers of lung surfactant lipid extract (15). SP-A is able to bind not only to surfactant membranes but also to pathogen-associated molecular patterns on microorganisms, such as bacterial LPS, which has long been known to bind to SP-A (5,9). We have recently reported a detailed study of the characteristics of the interaction of human SP-A with bacterial Re-LPS (6).
We expect that, given the lipophilic nature of LPS, inhaled LPS might incorporate into the lung surfactant DPPC-rich monolayer and then interact with SP-A at the interface. This would likely make LPS less available for signaling. Thus the first objective of this study was to investigate the miscibility of Re-LPS in DPPC monolayers and the effect of Re-LPS on the lateral lipid organization of these monolayers, determined by epifluorescence microscopy. The second objective was to find out whether SP-A from the hypophase associates with DPPC/Re-LPS-mixed monolayers as well as with DPPC monolayers (13,14) and whether SP-A modifies lipid lateral organization of such monolayers. The third objective was to visualize the arrangement of fluorescently labeled SP-A (TR-SP-A) in association with DPPC, DPPC/Re-LPS, and lipid A monolayers.
EXPERIMENTAL PROCEDURES
Materials
1,2-Dipalmitoylphosphatidylcholine (DPPC) and the fluorescent lipid probe 1-palmitoyl-1-{12-[(7-nitro-2-1,3-benzoxadizole-1-yl)amino]dodecanoyl} phosphatidylcholine (NBD-PC) were obtained from Avanti Polar Lipids (Birmingham, AL). Re-LPS and diphosphoryl lipid A from Salmonella minnesota (serotype Re 595) were purchased from Sigma (St. Louis, MO). The fluorescent probe used to chemically label SP-A, sulforhodamine 101 sulfonyl chloride or Texas red (TR), was obtained from Molecular Probes (Eugene, OR). The organic solvents (methanol and chloroform) used to dissolve DPPC, lipid A, and Re-LPS were high performance liquid chromatography grade.
Concentrations of DPPC, NBD-PC, and lipid A were assessed by measuring the phospholipid phosphorus and Re-LPS concentration by quantification of the 2-keto-3-deoxyoctulosonic acid (KDO). Water used in all experiments and analytical procedures was deionized and doubly distilled in glass, the second distillation being from dilute potassium permanganate solution.
Isolation and labeling of SP-A
SP-A was isolated from bronchoalveolar lavage of patients with alveolar proteinosis using a sequential butanol and octylglucoside extraction (11). The purity of SP-A was checked by one-dimensional sodium dodecylsulfate-polyacrylamide gel electrophoresis in 12% acrylamide under reducing conditions and mass spectrometry. The oligomerization state of SP-A was assessed by electrophoresis under nondenaturing conditions and electron microscopy as reported elsewhere (16,17).
Fluorescently labeled SP-A was prepared as described in Ruano et al. (13). Briefly, SP-A in 5 mM Tris-HCl buffer, pH 8.3, was incubated with 1 mM TR (SP-A/TR molar ratio of 6:1) for 90 min in darkness at room temperature. To remove unreacted fluorescent reagent, the mixture was exhaustively dialyzed against 5 mM Tris-HCl, pH 7.4. Activity of labeled TR-SP-A compared to that of native SP-A was assayed by testing its ability to self-associate and to induce aggregation of DPPC and Re-LPS in the presence of calcium at 37°C as described elsewhere (16–19). The effect of TR-SP-A on π-A isotherms of DPPC monolayers was essentially identical (within our ability to measure) to that of nonconjugated SP-A.
Surface pressure-area measurements and epifluorescence microscopy
DPPC, lipid A, Re-LPS, or DPPC mixed with different amounts of Re-LPS were spread from chloroform/methanol 3:1 (v/v) solutions onto a buffer A subphase (150 mM NaCl, 5 mM Tris-HCl, pH 7.4) containing either 150 μM EDTA or 150 μM CaCl2, with or without SP-A, in a thermostated Langmuir-Blodgett trough (302RB Ribbon Barrier Film Balance, NIMA Technologies, Coventry, UK). The concentration of Ca2+ was set at 150 μM because at this concentration there is a molar excess of calcium in the subphase with respect to lipid and protein concentrations. The use of low Ca2+ concentrations prevents the extensive protein self-association that occurs at higher Ca2+ concentrations (18). Extensive SP-A self-aggregation hampers the interaction of SP-A with lipid films (15).
Epifluorescence microscopy measurements were performed on a surface balance whose construction and operation have been described previously (20,21). DPPC, lipid A, Re-LPS, and DPPC/Re-LPS combinations were mixed in chloroform-methanol solutions with 1 mol % NBD-PC (based on the lipid content). Monolayers were formed by spreading onto a buffered saline subphase (buffer A) containing either 150 μM CaCl2 or 150 μM EDTA, with or without 0.08 μg/ml of TR-SP-A. After a 1-h period, which would have allowed for solvent evaporation and penetration of the protein into the gas or gas-liquid expanded coexistence phases, the monolayer was compressed at a slow speed (20 mm2/s or an initial rate of 0.13 Å2/molecule/s) at 25°C. At selected surface pressures, compression was halted, and a video recording was made for a 1-min period of both NBD-PC and TR fluorescence by switching fluorescence filter combinations. As described previously, our apparatus does not allow for instantaneous recording of images at two wavelengths (13–15). Therefore, images at the two wavelengths are not superimposable because of monolayer movement between the acquisitions of the images. The video images were obtained with a charge-coupled device camera that records in black and white. Images were analyzed with digital image processing using JAVA 1.3 software (Jandel Scientific, San Rafael, CA) as discussed elsewhere (21,22).
RESULTS
Re-LPS effects on DPPC monolayers
Fig. 2 A shows the surface pressure area (π-A) isotherms obtained at 25°C for monolayers of DPPC and Re-LPS and mixtures of DPPC/Re-LPS. Pure DPPC gave monolayers that exhibited a transition region between liquid-expanded (LE) and liquid-condensed (LC) phases at surface pressures in the range of 7–12 mN/m on a buffered saline subphase containing 150 μM CaCl2 (solid line). The collapse pressure for this phospholipid was ∼70 mN/m. The presence of 150 μM CaCl2 in the subphase did not appreciably modify the π-A isotherm of DPPC when compared to that obtained in the absence of calcium (23). Fig. 2 A also shows that the π-A isotherms of DPPC/Re-LPS-mixed monolayers were shifted markedly to larger molecular areas relative to the isotherm of pure DPPC (Fig. 2 A). This shifting depended on the molar fraction of Re-LPS, XRe-LPS. The π-A compression isotherm for pure Re-LPS monolayers showed an easily distinguishable collapse at ∼56 mN/m and a kink at 36 mN/m, which might indicate a LC/LE transition (Fig. 2 A, dashed-dotted-dotted line).
FIGURE 2.

(A) Surface pressure (π)-area isotherms of DPPC/Re-LPS-mixed monolayers as a function of Re-LPS molar fraction (XRe-LPS): 0.0 (solid line); 0.2 (long dashed line); 0.4 (short dashed line); 0.6 (dotted line); 0.8 (dash-dot-dashed line); and 1.0 (dash-dot-dot-dashed line). (B) Variation of the collapse pressure as a function of XRe-LPS. (C) Mean molecular area of DPPC/Re-LPS-mixed monolayers as a function of XRe-LPS at three surface pressures. The dotted lines are the theoretical variations assuming the additivity rule. The monolayers were compressed at 50 cm2/min on a subphase containing 150 μM Ca2+, 150 mM NaCl, 5 mM Tris-HCl, pH 7.4. The temperature of the subphase was 25.0°C ± 0.1°C. Error bars are within the size of the symbol.
The collapse of all DPPC/Re-LPS-mixed monolayers took place at surface pressures between those of pure DPPC and Re-LPS monolayers (Fig. 2 B). According to the two-dimensional phase rule, if the monolayer components are immiscible in the condensed and collapsed states, the isotherm will show two distinct collapse pressures corresponding to those for the pure components. However, collapse pressures of mixed monolayers composed of two miscible components will vary with composition (24). Thus, the dependence of the collapse pressure on the molar fraction of Re-LPS and the fact that the collapse pressures of the mixed films lie between the collapse pressures of pure components indicate that Re-LPS and DPPC are miscible. To demonstrate more clearly the interaction between these two components of the mixtures, the mean area per molecule was plotted as a function of the mole fraction of Re-LPS at three surface pressures: 11, 32, and 45 mN/m (Fig. 2 C). The dotted lines are the mean molecular areas calculated by assuming ideal mixing. If an ideal mixed monolayer is formed or the two components are completely immiscible in the two-dimensional state, the plot of mean molecular areas as a function of the mole fraction of one of the components at a given surface pressure would be a straight line. The results show that, for all surface pressures studied, negative deviations from ideal behavior were observed, indicating attractive interaction between DPPC and Re-LPS.
To characterize the possible effects of Re-LPS on DPPC lateral organization, epifluorescence microscopy images of DPPC monolayers containing 1 mol % NBD-PC were recorded in the absence and presence of different amounts of Re-LPS (Fig. 3). The fluorescent dye (NBD-PC) concentrated in the LE phase. which appeared bright in the fluorescence images of the monolayers (13–15). In the low pressure regime, DPPC monolayers showed black nonfluorescent domains, the LC phase dispersed very homogeneously in a fluorescent environment (LE). As the monolayer was compressed the relative area occupied by the LC domains increased (Fig. 3). Kidney-shaped solid domains, typical of the LE/LC coexistence region of DPPC monolayers (25), were observed over the range of 7–12 mN/m. At higher surface pressures solid domains grew to occupy most of the monolayer. Comparison of these images with those obtained in the absence of calcium (13,26) indicates that calcium lowers the surface pressure corresponding to LE-LC phase transition. Thus dark probe-depleted condensed domains were first seen at ∼5.4 mN/m in the presence of Ca2+ (data showed in Fig. 5) and at ∼7.4 mN/m in its absence (26).
FIGURE 3.

Typical fluorescent images obtained from pure DPPC and DPPC/Re-LPS-mixed monolayers containing 1 mol % NBD-PC at the indicated surface pressures. The monolayers were spread on a subphase containing 150 μM Ca2+, 150 mM NaCl, 5 mM Tris-HCl, pH 7.4. The bright background indicates the phase containing the fluorescent probe NBD-PC. The scale bar is 25 μm.
FIGURE 5.

Typical images obtained from a DPPC monolayer containing 1 mol % NBD-PC spread onto a buffered saline subphase containing 150 μM Ca2+, with and without 0.08 μg/ml TR-SP-A at the surface pressures indicated. Images were recorded through filters selecting fluorescence coming either from NBD-PC (emission centered at 520 nm) or TR-SP-A (emission centered at 590 nm). The scale bar is 25 μm.
On the other hand, Fig. 3 shows that the presence of Re-LPS in DPPC/Re-LPS-mixed films increased the transition pressure from LE to LC phase. Furthermore, the presence of Re-LPS changed the shape of liquid-condensed domains to trefoil-like forms and reduced their size, causing a relative decrease in the LC phase in the DPPC/Re-LPS films. As the amount of Re-LPS in the monolayer increased, the size of the nonfluorescent DPPC-rich LC domains decreased (Fig. 3). For XRe-LPS ≥ 0.5 no LC domains were observed and the mixed monolayers are in the LE fluid state. Thus, it is likely that DPPC contributes mainly to the LC domains and that Re-LPS mixed with DPPC are in the brilliant LE phase. In contrast to DPPC monolayers, no LE to LC phase transition could be observed for pure Re-LPS monolayers at relevant lateral pressures (7–30 mN/m) (data not shown). This indicates either that there is a single phase or that LE and LC domains have much smaller dimensions than the optical resolution. In this way, Roes et al. (27) recently reported that small LE and LC domains can be observed in pure Re-LPS monolayers by atomic force microscopy, the sizes of which are below the optical resolution of a light microscope.
SP-A interaction with DPPC and DPPC/Re-LPS monolayers in the presence of calcium
Fig. 4 shows the π-area per lipid molecule isotherms of monolayers of DPPC alone and in the presence of XRe-LPS = 0.2 spread over buffered saline subphases containing 150 μM CaCl2 with or without 0.08 μg/ml SP-A. SP-A caused an expansion of either DPPC or DPPC/Re-LPS isotherms, which suggests that the protein was taking up some space in these monolayers or was interacting with the phospholipid monolayer enough to perturb the usual lipid packing. SP-A alone did not adsorb to the interface at the concentration used in the experiments. SP-A needs a lipid monolayer to pull it into the surface. The SP-A-induced displacement of π-A isotherms to larger molecular areas was greater for DPPC/Re-LPS than for DPPC monolayers. For instance, at 20 mN/m, the area increase was ∼3 times larger for DPPC/Re-LPS than for DPPC monolayers. SP-A adsorption to the monolayers could affect the interactions between Re-LPS and DPPC, driving their ideal mixing or complete demixing. Either of these would effectively lead to an increase in the mean molecular area per lipid molecule toward the additive line (see Fig. 2 C). On the other hand, Re-LPS in DPPC/Re-LPS may promote the adsorption of SP-A, which would cause greater increase in the mean area per lipid molecule in DPPC/Re-LPS compared to DPPC monolayers. The presence of large fluorescent clusters of TR-SP-A in the DPPC/Re-LPS films (see Fig. 6) seems to support this explanation.
FIGURE 4.

Pressure-area isotherms of DPPC and DPPC/Re-LPS (XRe-LPS = 0.2) monolayers spread onto a buffered saline subphase containing 150 μM Ca2+ with (solid circles) or without (open circles) 0.08 μg/ml SP-A. The temperature of the subphase was 25°C ± 0.1°C.
FIGURE 6.

Typical images obtained from a DPPC/Re-LPS (XRe-LPS = 0.2) mixed monolayer containing 1 mol % NBD-PC spread onto a buffered saline subphase containing 150 μM Ca2+, with and without 0.08 μg/ml TR-SP-A at the surface pressures indicated. Images were recorded through filters selecting fluorescence coming either from NBD-PC (emission centered at 520 nm) or TR-SP-A (emission centered at 590 nm). Arrows in the central panel show a distinct, brilliant new phase that dissolves more lipophilic dye. The scale bar is 25 μm.
At a surface pressure of ∼50 mN/m the mean area per lipid molecule in the DPPC/Re-LPS films was the same in the absence and presence of SP-A in the subphase (Fig. 4). Therefore, at surface pressures >50 mN/m, SP-A did not occupy space in the monolayers and did not perturb the binary DPPC/Re-LPS monolayers. In contrast, the perturbing influence of SP-A on DPPC monolayers was observed only up to ∼30 mN/m. These observations suggest that specific interactions between Re-LPS and SP-A might facilitate both the adsorption of SP-A to the monolayers and its perturbing effects on the lipid monolayers at higher surface pressures.
Fig. 5 shows images obtained from a monolayer of DPPC containing 1 mol % NBD-PC formed on a buffered saline subphase containing 150 μM Ca2+, with or without 0.08 μg/ml TR-SP-A. Fluorescence coming from either the lipid probe (NBD-PC) or the fluorescently labeled protein (TR-SP-A) was selectively recorded from the same monolayers by switching the filters. NBD fluorescence showed that at low surface pressures (3.9 mN/m) solid domains were formed in the presence but not in the absence of SP-A. Thus, SP-A increased the formation of solid phases. This SP-A effect on DPPC monolayers in the presence of calcium differs from that observed in its absence, where SP-A did not promote solid phase formation (13). Fig. 5 also shows that the fluorescence of the protein (TR-SP-A) was not observed in the monolayers at very low pressures (below 3.9 mN/m) and was only seen when LC domains began to appear. At 3.9 mN/m, intense rings of TR fluorescence surrounding DPPC-condensed domains were apparent. As the pressures increased, the protein fluorescence was evenly distributed in the fluid film and the dark LC domains became fuzzy and were penetrated by fluorescent points, indicating TR-SP-A incorporation in the condensed regions. This behavior differs from that observed in the absence of CaCl2 in the subphase, where SP-A does not directly associate with DPPC in LC phase (13,14).
Fig. 6 shows images obtained from a mixed monolayer of DPPC/Re-LPS (XRe-LPS = 0.2) containing 1 mol % NBD-PC formed over a buffered saline subphase containing 150 μM CaCl2, with or without 0.08 μg/ml TR-SP-A. NBD fluorescence showed that at low surface pressure (6–8 mN/m) solid domains were formed in the presence but not the absence of SP-A. At higher surface pressures (8.4 or 11.3 mN/m), the relative area occupied by the LC domains was higher in the presence of SP-A than in its absence. Thus SP-A promoted the formation of DPPC-rich solid domains. SP-A adsorption to mixed DPPC/Re-LPS monolayers could affect the interactions between Re-LPS and DPPC and lead to decreased miscibility between Re-LPS and DPPC. This would favor the formation of DPPC-rich solid domains under compression. The micrographs in the central panel of Fig. 6 show that up to ∼11.3 mN/m LE and LC phases coexisted in the monolayers. At 15 mN/m a distinct new phase that dissolves more lipophilic dye appeared (see arrows). It is difficult to experimentally assess the composition of this new phase. Since it occurs only in the ternary system DPPC/Re-LPS/SP-A but not in the binary systems DPPC/SP-A (Fig. 5) and DPPC/Re-LPS (Fig. 6, left panel), it seems reasonable to infer that SP-A is inducing this new phase as a consequence of its interaction with Re-LPS. It is possible that at surface pressures ∼15 mN/m, DPPC/Re-LPS/SP-A (but not DPPC/Re-LPS) monolayers were in the LC state. This is consistent with the facts that SP-A induced solid domains and that the pressure-area curves for DPPC/Re-LPS films formed on SP-A had an inflection point at ∼16 mN/m, which might indicate the transition pressure to LC phase (Fig. 4). The brilliantly fluorescent LE phase enriched with NBD probe, visualized in epifluorescence images, might be “frozen” within the LC phase. The latter appeared both dark (the typically dye-depleted LC phase) and bright gray in appearance because it had dissolved some of the NBD probe.
Fig. 6 also shows that TR fluorescence was only visible when condensed domains began to appear at 6 mN/m. However, the distribution of TR-SP-A in DPPC/Re-LPS (XRe-LPS = 0.2) monolayers was different from that observed in monolayers of pure DPPC. At surface pressures >6mN/m, TR-SP-A accumulated at the fluid-solid boundary regions and formed networks of interconnected filaments in the fluid phase of DPPC/Re-LPS monolayers. These reticular structures were also seen in monolayers containing XRe-LPS = 0.3 in which the size of LC domains are reduced with respect to mixed monolayers containing XRe-LPS = 0.2 (data not shown). They were also observed in DPPC/lipid A-mixed monolayers (Xlipid A = 0.2) (data not shown) and in monolayers of pure lipid A formed over a buffered saline subphase containing calcium and TR-SP-A.
SP-A interaction with lipid A monolayers in the presence of calcium
Fig. 7 (left panel) shows the π-A compression isotherms for pure lipid A monolayers in the absence and presence of 0.08 μg/ml SP-A. The isotherms showed a collapse at ∼60 mN/m and an inflection point at ∼35 mN/m. The presence of SP-A in the subphase led to an increase in the film area of lipid A monolayers, suggesting that SP-A also incorporated into these monolayers. Epifluorescence microscopy images indicated that bright fluorescent reticular structures of TR-SP-A appeared in lipid A monolayers at surface pressures ≥9 mN/m at which LE-LC phases coexist (27) (Fig. 7, right panel). These lattice-like structures appear to be specific for the interaction of SP-A with the lipid A moiety of Re-LPS.
FIGURE 7.

(Left panel) Isotherms of monolayers of lipid A spread onto a buffered saline subphase containing 150 μM Ca2+ in the absence (○) and presence (•) of 0.08 μg/ml SP-A at 25°C. (Right panel) Typical epifluorescence microscopic images recorded through a filter that selects fluorescence coming from TR-SP-A (emission centered at 590 nm) at different surface pressures. The scale bar is 25 μm.
SP-A interaction with DPPC/Re-LPS and lipid A monolayers in the presence of EDTA
Since SP-A self-associates (18,19) and induces LPS aggregation in the presence of calcium (6,16,17), experiments were performed in the presence of 150 μM EDTA to determine whether the effects of SP-A on DPPC/Re-LPS and lipid A films were calcium dependent. Fig. 8 (left panel) shows typical π-A isotherms of DPPC/Re-LPS (XRe-LPS = 0.2) monolayers spread onto a buffered saline subphase containing EDTA, with or without 0.08 μg/ml SP-A. SP-A expanded the interfacial DPPC/Re-LPS film in the presence of EDTA (solid line). However, the perturbing influence of SP-A on DPPC/Re-LPS-mixed monolayers was observed over a greater range of surface pressure in the presence of calcium (∼6–50 mN/m) than in its absence (∼10–30 mN/m) (Figs. 4 and 8). NBD-PC fluorescence (Fig. 8, right panel) showed that, in the absence of calcium, SP-A hardly increased the formation of solid domains. With respect to microscopic images obtained with the TR filter (Fig. 8, right panel), LE-LC phase coexistence was also required for the appearance of fluorescent SP-A around liquid-condensed domains in the presence of EDTA. Once solid domains were formed, TR-SP-A was seen forming a network of interconnected filaments (Fig. 8) as had been observed in the presence of calcium (Fig. 6). At a surface pressure of 30 mN/m no protein fluorescence was detected, consistent with protein exclusion from the interface or less SP-A binding to the film.
FIGURE 8.

(Left panel) Isotherms of DPPC/Re-LPS (XRe-LPS = 0.2) monolayers spread onto a buffered saline subphase containing 150 μM EDTA in the absence (○) and presence (•) of 0.08 μg/ml SP-A at 25°C. (Right panel): Typical fluorescent images recorded through filters selecting fluorescence coming either from NBD-PC or TR-SP-A at different surface pressures. The scale bar is 25 μm.
Fig. 9 (left panel) shows the π-A compression isotherm for pure lipid A monolayers formed over a buffered saline subphase containing 150 μM EDTA, with and without 0.08 μg/ml SP-A. SP-A also expanded lipid A interfacial films in the presence of EDTA. TR epifluorescence microscopy images (Fig. 9, right panel) indicated that bright fluorescent reticular structures of TR-SP-A appeared in lipid A monolayers at surface pressures ≥9 mN/m . These results indicate that these lattice-like structures formed by SP-A in either lipid A or DPPC/Re-LPS films are independent of calcium and seem to depend on the specific interaction of SP-A with the lipid A part of bacterial LPS.
FIGURE 9.

(Left panel) Isotherms of monolayers of lipid A spread onto a buffered saline subphase containing 150 μM EDTA in the absence (○) and presence (•) of 0.08 μg/ml SP-A at 25°C. (Right panel) Typical epifluorescence microscopic images recorded through a filter that selects fluorescence coming from TR-SP-A at different surface pressures. The scale bar is 25 μm.
DISCUSSION
In this study, epifluorescence microscopy combined with a surface balance was used to examine the interaction of SP-A with mixed monolayers of DPPC/Re-LPS. The rationale for this study is based on the fact that DPPC/LPS-mixed monolayers might be formed in the lung as a consequence of inhaled airborne particles containing bacterial LPS. Given the lipophilic nature of LPS, these molecules might incorporate into the DPPC-rich monolayer.
Using surface pressure area isotherms, we show that DPPC and Re-LPS were miscible and that there was an attractive interaction between the lipids (Fig. 2). The fact that the collapse pressures of DPPC/Re-LPS-mixed films were between the collapse pressures of pure components (70 mN/m for DPPC and 56 mN/m for Re-LPS) indicates miscibility between both lipids. In addition, binary monolayers of Re-LPS plus DPPC showed negative deviations from ideal behavior of the mean areas of the films, which is consistent with partial miscibility and attractive interaction between the lipids. Epifluorescence microscopy shows that Re-LPS decreased the relative area occupied by the probe-depleted LC domains (Fig. 3) and increased the surface pressure corresponding to the LE/LC transition: solid domains began to appear at higher surface pressures in DPPC/Re-LPS films than in DPPC films. Furthermore, the presence of Re-LPS in DPPC films reduced the size of solid domains. Given that the size of LC domains of DPPC-Re-LPS monolayers decreased with increasing Re-LPS mol % in the monolayer, it is likely that LC domains of DPPC/Re-LPS monolayers consisted mainly of DPPC. The dissolution, rearrangement, and decrease in the size of DPPC-rich solid domains induced by Re-LPS are consistent with miscibility and attractive interaction between these two lipids.
The adsorption of SP-A to DPPC/Re-LPS monolayers caused expansion in the lipid molecular areas in both the presence and absence of calcium, which indicates an intercalation of SP-A molecules into the DPPC/Re-LPS monolayer (Figs. 4 and 8). The lack of Ca2+ requirement for the interaction of SP-A with DPPC/Re-LPS films is consistent with previous results that indicate that SP-A binds to Re-LPS and DPPC in the absence of calcium (6,13,14,28,29). However, it would be expected that the presence of Ca2+ influences the interaction of SP-A with DPPC/Re-LPS monolayers since calcium causes conformational changes in the globular domain of SP-A, as detected by fluorescence spectroscopy (16,18,30) and transmission electron microscopy (31). This conformational change enhances lipid binding and allows SP-A-SP-A self-association and SP-A-mediated lipid aggregation (18,30). In addition, Ca2+ binds tightly to the KDO moieties of Re-LPS (with an apparent dissociation constant of 14 μM) (32), so that Ca2+ could neutralize these negatively charged moieties. Here we show that the presence of calcium influenced the effect of SP-A on the lipid lateral organization of DPPC/Re-LPS monolayers as determined by epifluorescence microscopy. The presence of calcium promoted SP-A-induced formation of solid domains in these monolayers, decreasing the surface pressure corresponding to the LE/LC transition (NBD epifluorescence images of Figs. 6 and 8). In addition, we found that calcium makes DPPC/Re-LPS monolayers more sensitive to SP-A over a larger surface pressure range (Figs. 4 and 8).
The use of fluorescent TR-SP-A allows for the analysis of SP-A interaction with domains or regions of DPPC/Re-LPS monolayers. We found that TR fluorescence was only visible when LC domains began to appear, which occurred at ∼6 and 9.6 mN/m in the presence and absence of calcium, respectively. These results are consistent with the concept that SP-A recognizes the lipid in the gel phase and penetrates the membrane interface through lipid packing defects at liquid-condensed-liquid expanded boundaries (13). Once solid domains are formed, TR-SP-A was observed forming a network of interconnected filaments in the fluid phase of DPPC/Re-LPS monolayers, possibly connecting solid domains in which TR-SP-A seems to accumulate. These protein reticular structures were formed when there was a relatively low SP-A concentration in the subphase (0.08 μg/ml) in both the presence and absence of calcium (Figs. 6 and 8). However, in the absence of this cation no protein fluorescence was detected at surface pressures ≥30 mN/m, which is consistent with protein exclusion from the interface or less protein binding at such surface pressures in the absence of calcium. Given that this protein network was also seen in DPPC/lipid A-mixed monolayers (Xlipid A = 0.2) and in monolayers of pure lipid A (in both the presence and absence of calcium) (Figs. 7 and 9) but not in DPPC monolayers (Fig. 5), we conclude that the formation of extensive lattice-like structures at the interface depends on the specific interaction of SP-A with the lipid A part of bacterial LPS.
These SP-A lattice-like arrays seemed to form through extensive SP-A-SP-A association, stabilized by the binding of the protein to the lipid film. SP-A binds to Re-LPS in solution with high affinity (KD = 0.028 μM) in a Ca2+-independent manner (6), and the formation of this SP-A network on DPPC/Re-LPS monolayers was calcium independent. However, self-association of SP-A in solution requires Ca2+, with the calcium activation constant (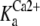 ) for human SP-A self-association at physiological ionic strength 12 ± 1.8 μM (18). Our results suggest that the adsorption of SP-A to DPPC/Re-LPS monolayers facilitated cooperative assembling of SP-A molecules in a lattice-supraquaternary structure even in the absence of calcium. This type of supraquaternary organization of SP-A and cooperative interaction with DPPC/Re-LPS monolayers might play a physiological role. For instance, SP-A could induce aggregation of mixed Re-LPS/phospholipid membranes that are squeezed out from the monolayer on expiration in situ.
) for human SP-A self-association at physiological ionic strength 12 ± 1.8 μM (18). Our results suggest that the adsorption of SP-A to DPPC/Re-LPS monolayers facilitated cooperative assembling of SP-A molecules in a lattice-supraquaternary structure even in the absence of calcium. This type of supraquaternary organization of SP-A and cooperative interaction with DPPC/Re-LPS monolayers might play a physiological role. For instance, SP-A could induce aggregation of mixed Re-LPS/phospholipid membranes that are squeezed out from the monolayer on expiration in situ.
Aggregation of membranes containing Re-LPS could be important to reduce LPS toxicity (6) and facilitate phagocytosis of these aggregates by alveolar macrophages. This would prevent the binding of LPS to its receptor complex on alveolar epithelial and immune cells, which would launch an inflammatory response. On the other hand, the formation of a lattice of protein aggregates on pure lipid A monolayers suggests that this might be the initial step in lesion formation in the outer membranes of Gram-negative bacteria induced by SP-A. It was recently reported that SP-A causes increased Gram-negative bacteria permeability and killing (33), presumably by direct effect of SP-A on the properties of the microbial cell membrane (34).
It is important to mention that tubular myelin and multilamellar vesicles from native surfactant also contain arrays of SP-A (35,36). These arrays seem to be formed as a result of SP-A's capability to self-associate and bind to membranes. SP-A arrays remain intact when the lipid is partially removed with acetone (35,36), and their spacing is comparable to the size of SP-A. Interconnected SP-A molecules seem to form the skeleton of these structures. The formation of this type of SP-A supraquaternary organization adsorbed to surfactant membranes must be of physiological importance. It is thought that these protein arrays may stabilize large surfactant aggregates and decrease surfactant inactivation in the presence of serum protein inhibitors (10).
In summary, this study shows that Re-LPS is miscible in the DPPC film. There is an attractive interaction between Re-LPS and DPPC, which results in reduction of the size of DPPC-rich solid domains in DPPC/Re-LPS monolayers. SP-A, the major surfactant protein component involved in host defense, incorporates into DPPC/Re-LPS monolayers and produces expansion in the lipid molecular areas. The perturbing influence of SP-A in DPPC/Re-LPS monolayers is seen over a greater range of surface pressure than in DPPC monolayers in the presence of calcium. With respect to the differential partitioning of fluorescently labeled TR-SP-A into regions of DPPC-Re-LPS monolayers, we found that fluorescent TR-SP-A accumulates at the fluid-solid boundary regions of these monolayers and forms networks of interconnected filaments in the fluid phase in a Ca2+-independent manner. Such protein reticular structures are also observed when TR-SP-A interacts with mixed DPPC/lipid A and pure lipid A monolayers. These novel results deepen our understanding of the specific interaction of SP-A with the lipid A moiety of LPS and may explain how SP-A behaves as an effective LPS neutralizing agent in the alveolus, making LPS less available to interact with components of the innate immune system in the airways and thus contributing to maintaining the lung in a noninflamed state.
Acknowledgments
This work was supported by the Ministerio de Educación y Ciencia (SAF2006-04434), Instituto de Salud Carlos III (cibeRES-CB06/06/0002), CAM (S-BIO-0260-2006), Fundación Médica MM, and the Canadian Institutes of Health Research to K.M.W.K. (CIHR MT-9361).
I. García-Verdugo and O. Cañadas contributed equally to this work.
Editor: Thomas J. McIntosh.
References
- 1.Raetz, C. R., R. J. Ulevitch, S. D. Wright, C. H. Sibley, A. Ding, and C. F. Nathan. 1991. Gram-negative endotoxin: an extraordinary lipid with profound effects on eukaryotic signal transduction. FASEB J. 5:2653–2660. [DOI] [PubMed] [Google Scholar]
- 2.Nikaido, H., and M. Vaara. 1987. Outer membrane. In Escherichia coli and Salmonella typhimurium. Cellular and Molecular Biology. C. Neidhardt, J. L. Ingraham, K. Brooks Low, B. Magasanik, M. Schaechter, and H. E. Umbarger, editors. American Society for Microbiology, Washington, DC. 7–22.
- 3.Seydel, U., A. B. Schormm, R. Blunck, and K. Brandenburg. 2000. Chemical structure, molecular conformation and bioreactivity of endotoxins. In Chemical Immunology: CD14 in the Inflammatory Response, vol. 74. R. S. Jack, editor. Karger, Basel, Switzerland. 5–24. [DOI] [PubMed]
- 4.Alexander, C., and E. T. Rietschel. 2001. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 7:197–202. [PubMed] [Google Scholar]
- 5.Van Iwaarden, J. F., J. C. Pikaar, J. Storm, E. Brouwer, J. Verhoef, R. S. Oosting, L. M. van Golde, and J. A. van Strijp. 1994. Binding of surfactant protein A to the lipid A moiety of bacterial lipopolysaccharides. Biochem. J. 303:407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.García-Verdugo, I., F. Sánchez-Barbero, K. Soldau, P. S. Tobias, and C. Casals. 2005. Interaction of SP-A (surfactant protein A) with bacterial rough lipopolysaccharide (Re-LPS), and effects of SP-A on the binding of Re-LPS to CD14 and LPS-binding protein. Biochem. J. 391:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Augusto, L. A., J. Li, M. Synguelakis, J. Johansson, and R. Chaby. 2002. Structural basis for interactions between lung surfactant protein C and bacterial lipopolysaccharide. J. Biol. Chem. 277:23484–23492. [DOI] [PubMed] [Google Scholar]
- 8.Kuan, S. F., K. Rust, and E. Crouch. 1992. Interactions of surfactant protein D with bacterial lipopolysaccharides. Surfactant protein D is an Escherichia coli-binding protein in bronchoalveolar lavage. J. Clin. Invest. 90:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCormack, F. X. 1998. Structure, processing and properties of surfactant protein A. Biochim. Biophys. Acta. 1408:109–131. [DOI] [PubMed] [Google Scholar]
- 10.Casals, C., and I. García-Verdugo. 2005. Molecular and functional properties of surfactant protein A. In Developments in Lung Surfactant Dysfunction in Lung Biology in Health and Disease. K. Nag, editor. Marcel Dekker, New York. 55–84.
- 11.Hawgood, S., B. J. Benson, J. Schilling, D. Damm, J. A. Clements, and R. T. White. 1987. Nucleotide and amino acid sequences of pulmonary surfactant protein SP 18 and evidence for cooperation between SP 18 and SP 28–36 in surfactant lipid adsorption. Proc. Natl. Acad. Sci. USA. 84:66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cockshutt, A. M., J. Weitz, and F. Possmayer. 1990. Pulmonary surfactant-associated protein A enhances the surface activity of lipid extract surfactant and reverses inhibition by blood proteins in vitro. Biochemistry. 29:8424–8429. [DOI] [PubMed] [Google Scholar]
- 13.Ruano, M. L. F., K. Nag, L. A. Worthman, C. Casals, J. Pérez-Gil, and K. M. W. Keough. 1998. Differential partitioning of pulmonary surfactant protein SP-A into regions of monolayers of dipalmitoylphosphatidylcholine and dipalmitoylphosphatidylcholine/dipalmitoylphosphatidylglycerol. Biophys. J. 74:1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruano, M. L., K. Nag, C. Casals, J. Pérez-Gil, and K. M. Keough. 1999. Interactions of pulmonary surfactant protein A with phospholipid monolayers change with pH. Biophys. J. 77:1469–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Worthman, L. A., K. Nag, N. Rich, M. L. Ruano, C. Casals, J. Pérez-Gil, and K. M. Keough. 2000. Pulmonary surfactant protein A interacts with gel-like regions in monolayers of pulmonary surfactant lipid extract. Biophys. J. 79:2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.García-Verdugo, I., F. Sánchez-Barbero, F. U. Bosch, W. Steinhilber, and C. Casals. 2003. Effect of hydroxylation and N187-linked glycosylation on molecular and functional properties of recombinant human surfactant protein A. Biochemistry. 42:9532–9542. [DOI] [PubMed] [Google Scholar]
- 17.Sánchez-Barbero, F., J. Strassner, R. García-Cañero, W. Steinhilber, and C. Casals. 2005. Role of the degree of oligomerization in the structure and function of human surfactant protein A. J. Biol. Chem. 580:7659–7670. [DOI] [PubMed] [Google Scholar]
- 18.Ruano, M. L., I. García-Verdugo, E. Miguel, J. Pérez-Gil, and C. Casals. 2000. Self-aggregation of surfactant protein A. Biochemistry. 39:6529–6537. [DOI] [PubMed] [Google Scholar]
- 19.Ruano, M. L., E. Miguel, J. Pérez-Gil, and C. Casals. 1996. Comparison of lipid aggregation and self-aggregation activities of pulmonary surfactant-associated protein A. Biochem. J. 313:683–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nag, K., N. H. Rich, C. Boland, and K. M. W. Keough. 1990. Design and construction of an epifluorescence microscopic surface balance for the study of lipid monolayer phase transitions. Rev. Sci. Instrum. 61:3425–3430. [Google Scholar]
- 21.Nag, K., C. Boland, N. Rich, and K. M. Keough. 1991. Epifluorescence microscopic observation of monolayers of dipalmitoylphosphatidylcholine: dependence of domain size on compression rates. Biochim. Biophys. Acta. 1068:157–160. [DOI] [PubMed] [Google Scholar]
- 22.Pérez-Gil, J., K. Nag, S. Taneva, and K. M. W. Keough. 1992. Pulmonary surfactant protein SP-C causes packing rearrangements of dipalmitoylphosphatidylcholine in spread monolayers. Biophys. J. 63:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taneva, S., T. McEachren, J. Stewart, and K. M. Keough. 1995. Pulmonary surfactant protein SP-A with phospholipids in spread monolayers at the air-water interface. Biochemistry. 34:10279–10289. [DOI] [PubMed] [Google Scholar]
- 24.Wu, S., and J. R. Huntsberger. 1969. Intermolecular interaction in some mixed polymer monolayers at the air water interface. J. Colloid Interface Sci. 29:138–147. [Google Scholar]
- 25.McColongue, C. W., and T. K. Vanderlick. 1997. A close look at domain formation in DPPC monolayers. Langmuir. 13:7158–7164. [Google Scholar]
- 26.Plasencia, I., K. M. W. Keough, and J. Pérez-Gil. 2005. Interaction of the N-terminal segment of pulmonary surfactant protein SP-C with interfacial phospholipid films. Biochim. Biophys. Acta. 1713:118–128. [DOI] [PubMed] [Google Scholar]
- 27.Roes, S., U. Seydel, and T. Gutsmann. 2005. Probing the properties of lipopolysaccharide monolayers and their interaction with the antimicrobial peptide polymyxin B by atomic force microscopy. Langmuir. 21:6970–6978. [DOI] [PubMed] [Google Scholar]
- 28.King, R. J., M. C. Phillips, P. M. Horowitz, and S. C. Dang. 1986. Interaction between the 35 kDa apolipoprotein of pulmonary surfactant and saturated phosphatidylcholines. Effects of temperature. Biochim. Biophys. Acta. 879:1–13. [DOI] [PubMed] [Google Scholar]
- 29.Casals, C., E. Miguel, and J. Pérez-Gil. 1993. Tryptophan fluorescence study on the interaction of pulmonary surfactant protein A with phospholipid vesicles. Biochem. J. 296:585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haagsman, H. P., T. Sargeant, P. V. Hauschka, B. J. Benson, and S. Hawgood. 1990. Binding of calcium to SP-A, a surfactant-associated protein. Biochemistry. 29:8894–8900. [DOI] [PubMed] [Google Scholar]
- 31.Ridsdale, R. A., N. Palaniyar, C. E. Holterman, K. Inchley, F. Possmayer, and G. Harauz. 1999. Cation-mediated conformational variants of surfactant protein A. Biochim. Biophys. Acta. 1453:23–34. [DOI] [PubMed] [Google Scholar]
- 32.Schindler, M., and M. J. Osborn. 1979. Interaction of divalent cations and polymyxin B with lipopolysaccharide. Biochemistry. 18:4425–4430. [DOI] [PubMed] [Google Scholar]
- 33.Wu, H., A. Kuzmenko, S. Wan, L. Schaffer, A. Weiss, J. H. Fisher, K. S. Kim, and F. X. McCormack. 2003. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J. Clin. Invest. 111:1589–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuzmenko, A. I., H. Wu, and F. X. McCormack. 2006. Pulmonary collectins selectively permeabilize model bacterial membranes containing rough lipopolysaccharide. Biochemistry. 45:2679–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nag, K., J. G. Munro, S. A. Hearn, J. Rasmusson, N. O. Petersen, and F. X. Possmayer. 1999. Correlated atomic force and transmission electron microscopy of nanotubular structures in pulmonary surfactant. J. Struct. Biol. 126:1–15. [DOI] [PubMed] [Google Scholar]
- 36.Palaniyar, N., R. A. Ridsdale, S. A. Hearn, F. Possmayer, and G. Harauz. 1999. Formation of membrane lattice structures and their specific interactions with surfactant protein A. Am. J. Physiol. 276:L642–L649. [DOI] [PubMed] [Google Scholar]