

Abstract
Three Rab GTPases, Rab1, Rab2 and Rab6, are involved in protein transport between the endoplasmic reticulum (ER) and the Golgi. Whereas Rab1 regulates the anterograde ER-to-Golgi transport, Rab2 and Rab6 coordinate the retrograde Golgi-to-ER transport. We have previously demonstrated that Rab1 differentially modulates the export trafficking of distinct G protein-coupled receptors (GPCRs). In this report, we determined the role of Rab2 and Rab6 in the cell surface expression and signaling of α2B-adrenergic (α2B-AR), β2-AR and angiotensin II type 1 receptors (AT1R). Expression of the GTP-bound mutant Rab2Q65L significantly attenuated the cell-surface expression of both α2B-AR and β2-AR, whereas the GTP-bound mutant Rab6Q72L selectively inhibited the transport of β2-AR, but not α2B-AR. Similar results were obtained by siRNA-mediated selective knockdown of endogenous Rab2 and Rab6. Consistently, Rab2Q65L and Rab2 siRNA inhibited α2B-AR and β2-AR signaling measured as ERK1/2 activation and cAMP production, respectively, whereas Rab6Q72L and Rab6 siRNA reduced signaling of β2-AR, but not α2B-AR. Similar to the β2-AR, AT1R expression at the cell surface and AT1R-promoted inositol phosphate accumulation were inhibited by Rab6Q72L. Furthermore, the nucleotide-free mutant Rab6N126I selectively attenuated the cell-surface expression of β2-AR and AT1R, but not α2B-AR. These data demonstrate that Rab2 and Rab6 differentially influence anterograde transport and signaling of GPCRs. These data also provide the first evidence indicating that Rab6-coordinated retrograde transport selectively modulates intracellular trafficking and signaling of GPCRs.
1. Introduction
G protein-coupled receptors (GPCRs)1 are a superfamily of cell surface receptors which couple to heterotrimeric G-proteins and regulate multiple downstream effectors such as adenylyl cyclases, phospholipases, protein kinases and ion channels [1, 2]. The magnitude of receptor-elicited cellular response to a given signal is modulated by elaborately regulated intracellular trafficking which dictates the level of the receptor expression at the plasma membrane. GPCRs are synthesized in the endoplasmic reticulum (ER) and properly folded receptors are recruited and packaged into ER-derived COPII transport vesicles. Transport vesicles carrying cargo receptors then migrate from the ER to the ER-Golgi intermediate complex (ERGIC) (also referred to as vesiculotubular clusters or pre-Golgi intermediates), which is the first segregation site, sorting proteins to anterograde or retrograde transport pathways [3]. Receptors are then transported from the ERGIC to the Golgi apparatus and the trans-Golgi network (TGN). During their transport, receptors undergo post-translational modifications (e.g. glycosylation), which is essential for achieving mature status of the receptors. Fully mature receptors then move from the TGN to the plasma membrane. At the plasma membrane GPCRs may undergo internalization upon stimulation by their ligands [4–6]. Internalized receptors are either recycled back to the plasma membrane or targeted to the lysosome for degradation. Therefore, the balance of anterograde transport, endocytosis and degradation dictates the level of receptor at the plasma membrane.
Compared with the extensive studies on the events of the endocytic pathway [4], molecular mechanisms underlying the transport processes of GPCRs from the ER through the Golgi to the cell surface and regulation of receptor signaling by these processes are relatively less well understood. The progress achieved over the past few years indicates that GPCR anterograde targeting to the cell surface is a highly regulated process [7]. First, GPCRs have to be correctly folded and assembled in order to pass through the ER quality control mechanism. It has been demonstrated that export from the ER is a rate-limiting step for maturation of the δ-opioid receptor [8], and accumulation of misfolded receptors in the ER is associated with the pathogenesis of human disease [9, 10]. Second, GPCR export from the ER is directed by highly conserved motifs in the membrane-proximal termini [11–15]. We have recently identified the F(X)6LL motif in the C-termini required for α2B-adrenergic (α2B-AR) and angiotensin II type 1 receptor (AT1R) export from the ER [14, 15] and the YS motif in the N-termini essential for α2-AR export from the Golgi [16]. Third, dimerization of GPCRs also plays an important role in proper receptor folding to achieve a confirmation competent for export, in addition to regulating ligand binding, signal transduction and internalization [17]. Fourth, GPCR export from the ER and transport to the cell surface is modulated by direct interactions with multiple regulatory proteins such as the ER chaperones, accessory proteins and receptor activity modifying proteins, which may stabilize receptor conformation, facilitate receptor maturation and promote receptor delivery to the plasma membrane [7]. Finally, recent studies suggest that GPCRs may form a complex with G proteins and downstream effectors in the ER prior to export from the ER [18, 19].
Rab proteins consist of more than 60 members in mammalian cells and 11 members in yeast that form the largest subfamily of the Raslike small GTPase superfamily. Each Rab GTPase has a distinct subcellular localization and regulates discrete protein transport steps in exocytic and endocytic pathways [20]. Elucidation of the roles for Rab GTPases in GPCR trafficking have mainly been focused on the events involved in internalization and degradation of the receptors [21]. Whereas Rab4 and Rab11 are mainly involved in the recycling of internalized receptor from the endosome back to the plasma membrane, Rab5 regulates the internalization of GPCRs from the plasma membrane to the endosome, and Rab7 participates in the process targeting receptors to the lysosome for degradation [22–32]. Our laboratory has focused efforts on understanding the role of Rab1 GTPase in the export trafficking of GPCRs [33–35]. Rab1 is localized in the ER and the Golgi apparatus and regulates anterograde protein transport specifically from the ER to the Golgi and between the Golgi compartments [36–38]. We have previously demonstrated that Rab1 differentially regulates the anterograde trafficking of distinct GPCRs. Whereas the transport from the ER to the cell surface of α1-AR, β-AR and AT1R is dependent on Rab1, the transport of α2B-AR to the cell surface utilizes a non-classic, Rab1-independent pathway, suggesting multiple pathways mediating the cell-surface targeting of GPCRs [33–35].
In contrast to the function of Rab1 GTPase regulating the ER-to-Golgi traffic, Rab2 and Rab6 GTPases modulate retrograde protein transport from the Golgi to the ER. Rab2 GTPase is mainly localized to the ERGIC and has been proposed to control retrograde transport from the ERGIC to the ER [36, 39, 40]. There are three Rab6 isoforms: Rab6A, Rab6A’ and Rab6B. Rab6A and Rab6A’ differ in only three amino acid residues in regions flanking the GTP-binding domain and are ubiquitously expressed. In contrast, Rab6B is only expressed in neuronal cells. Rab6 is mainly localized to the Golgi and the TGN and are involved in the retrograde transport between Golgi cisternae or from the Golgi to the ER [41–43]. In this work, we have determined the role of Rab2 and Rab6 GTPases in the cell-surface targeting and signaling of α2B-AR, β2-AR and AT1R, three representative GPCRs, which couple to different G proteins and initiate distinct signaling pathways. We have demonstrated that Rab2 and Rab6 differentially modulate export trafficking of distinct GPCRs with similar structural features, and indicate that Rab GTPase-coordinated retrograde protein transport from the Golgi through the ERGIC to the ER may function as a novel site for control of GPCR biosynthesis.
2. Experimental procedures
2.1. Materials
Rat α2B-AR in vector pcDNA3, human β2-AR in vector pBC and rat AT1R in vector pCDM8 were kindly provided by Stephen M. Lanier and John D. Hildebrandt (Department of Pharmacology, Medical University of South Carolina, Charleston, SC) and Kenneth E. Bernstein (Department of Pathology, Emory University, Atlanta, GA), respectively. Antibodies against phospho-ERK1/2, green fluorescent protein (GFP), Rab1, Rab2, Rab6, calregulin and β-actin were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibodies against ERK1/2 were from Cell Signaling Technology. Anti-GM130 antibodies were from Transduction Laboratories (San Diego, CA). Anti-ERGIC53 antibodies were from AXXORA, LLC (San Diego, LA). Anti-giantin antibodies were purchased from Abcam Inc. (Cambridge, MA). Alexa Fluor 594- or 488-labeled antibodies and 4,6-diamidino-2-phenylindole were obtained from Molecular Probes, Inc. (Eugene, OR). Anti-FLAG M2 monoclonal antibody, isoproterenol (ISO) and UK14304 were obtained from Sigma (Saint Louis, MO). Human angiotensin II (Ang II) was purchased from EMD Chemicals Inc. (San Diego, CA). Penicillin/streptomycin, L-glutamine, and trypsin/EDTA were from Invitrogen (Carlsbad, CA). [³H]-CGP12177 (specific activity = 51 Ci/mmol), [³H]-RX821002 (50 Ci/mmol), [³H]-Ang II (50.5 = Ci/mmol) and myo-[³H]Inositol were purchased from Amersham Biosciences/GE Healthcare. All other materials were obtained as described elsewhere [16, 33].
2.2. Plasmid constructions
α2B-AR, β2-AR and AT1R tagged with GFP at their C-termini (α2B-AR-GFP, β2-AR-GFP, AT1R-GFP) were generated as described previously [33]. Rab6 was tagged with the FLAG epitope at its N-terminus by PCR using primer GACTACAAGGACGACGATGACAAG (coding peptide DYKDDDDK) and subcloned into the XhoI and HindIII restriction sites of pcDNA3.1(−) (Invitrogen). Human Rab2 was tagged with the GFP epitope at its N-terminus by releasing Rab2 from the pcDNA3.1(+) vector with KpnI and ApaI followed by ligation into the pEGFP-C1 vector at KpnI and ApaI sites. The GTP-bound constitutively active Rab2Q65L and Rab6Q72L mutants and the dominant-negative Rab6N126I mutant were generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The sequence of each construct used in this study was verified by restriction mapping and nucleotide sequence analysis (Louisiana State University Health Sciences Center DNA Sequence Core).
2.3. Cell culture and transient transfection
HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum, 100 units/ml penicillin, and 100 µg/ml streptomycin. Transient transfection of the HEK293T cells was carried out using Lipofectamine 2000 reagent (Invitrogen) as previously described [33]. The transfection efficiency was estimated to be greater than 70% based on the GFP fluorescence.
2.4. Radioligand binding
Cell-surface expression of α2B-AR, β2-AR and AT1R in HEK293T cells was measured by ligand binding of intact live cells using [³H]-RX821002, [³H]-CGP12177 and [³H]-Ang II, respectively, as previously described [16, 34, 35]. Briefly, HEK293T cells were cultured in 6-well dishes and transfected with 0.5 µg of the α2B-AR, β2-AR or AT1R plasmid together with 1.5 µg of Rab mutants or empty vectors. After 6 h the cells were split into 12-well plates at a density of 4 × 105 cells/well and cultured for an additional 24 h.
For measurement of α2B-AR expression at the cell surface, the cells were incubated with DMEM plus [³H]-RX821002 at a concentration of 20 nM for 90 min at room temperature. The nonspecific binding was determined in the presence of rauwolscine (10 µM). For measurement of β2-AR expression at the cell surface, the cells were incubated with DMEM containing the ligand [³H]-CGP12177 at a concentration of 20 nM for 90 min at room temperature. Non-specific binding was determined by pre-incubation with alprenolol at a concentration of 20 µM for 30 min followed by incubation with [³H]-CGP12177 (20 nM) in the presence of alprenolol (20 µM). The cells were washed twice with 1 ml of ice-cold phosphate-buffered saline (PBS), and the cell surface-bound ligands were extracted by 1M NaOH treatment for 2 h. The radioactivity was counted by liquid scintillation spectrometry in 3.5 ml of Ecoscint A scintillation solution (National Diagnostics, Inc., Atlanta, GA). For measurement of AT1R expression at the cell surface, the cells were incubated with DMEM containing the ligand [³H]-Ang II at a concentration of 10 nM at 4 °C (to limit AT1R internalization induced by ligand Ang II during the binding) overnight with constant shaking. The nonspecific binding was determined in the presence of nonradioactive Ang II (10 µM). The cells were washed twice with 1 ml of ice-cold PBS, and the cell surface-bound [³H]-Ang II was extracted by mild acid treatment (2 × 5 min with 0.5 ml of buffer containing 50 mM glycine, pH 3, and 125 mM NaCl). The radioactivity was counted by liquid scintillation spectrometry in 6 ml of Ecoscint A scintillation solution.
Radioligand binding of membrane preparations was carried out essentially as described [14]. Briefly, HEK293T cells were cultured on 100-mm dishes and transiently transfected with α2B-AR and Rab mutants as described above. The cells were washed twice with PBS, harvested and homogenized in 3 ml of buffer containing 5 mM Tris-HCl, pH 7.4, 5 mM EGTA and 5 mM EDTA supplemented with Complete Mini protease inhibitor cocktail (Roche Applied Science, Mannheim, Germany). After centrifugation at 100,000 × g for 30 min, the pellet was resuspended in 300 µl of membrane buffer containing 50 mM Tris-HCl, pH 7.4, 0.6 mM EDTA and 5 mM MgCl2. Our previous data demonstrated that the membrane preparation does not contain the ER and the Golgi [23]. The membrane suspension (15 µg membrane protein) was incubated with increasing concentrations of [³H]-RX821002 (5 – 160 nM) in a total volume of 100 µl. Non-specific binding was determined in the presence of the selective α2B-AR antagonist rauwolscine (10 µM). Duplicate samples were incubated for 30 min at room temperature with constant shaking, and the reaction was terminated by vacuum filtration. After washing with 100 mM Tris-HCl, pH 7.4 (4 × 4 ml), the retained radioactivity was measured by liquid scintillation spectrometry.
2.5. Flow cytometric analysis of receptor expression
For measurement of total receptor expression, HEK293T cells transfected with individual GFP-tagged receptor together with Rab mutants or siRNA were collected and resuspended in PBS containing 1% fetal calf serum at a density of 8 × 106 cells/ml. Total receptor expression was determined by measuring total GFP fluorescence on a flow cytometer (BD Biosciences FACSCalibur) as described [16].
2.6. Immunofluorescence microscopy
For fluorescence microscopic analysis of receptor subcellular localization, HEK293T cells were grown on coverslips pre-coated with poly-L-lysine in 6-well plates and transfected with 100 ng of GFP-tagged receptors with or without co-transfection together with 400 ng of Rab mutants. The cells were fixed with 4% paraformaldehyde-4% sucrose mixture in PBS for 15 min and stained with 4, 6-diamidino-2-phenylindole for 5 min. The coverslips were mounted, and fluorescence was detected with a Leica DMRA2 epifluorescent microscope as described previously [33]. Images were deconvolved using SlideBook software and the nearest neighbors deconvolution algorithm (Intelligent Imaging Innovations, Denver, CO). For co-localization studies involving immunostaining, HEK293T cells were permeabilized with PBS containing 0.2% Triton X-100 for 5 min, and blocked with 5% normal donkey serum for 1 h. The cells were then incubated with primary antibodies for 1 h. After washing with PBS (3 × 5 min), the cells were incubated with Alexa Fluor 594-or 488-labeled secondary antibody (1:2000 dilution) for 1 h at room temperature, and the fluorescence was analyzed as described above.
2.7. siRNA-mediated knockdown of Rab2 and Rab6
Stealth™ RNAi duplex targeting Rab2 and Rab6 GTPases were purchased from Invitrogen. For Rab2 GTPase, a sense (5'-GCGUACGCCUAUCUCUUCAAGUACA-3’) and an antisence (5’–UGUACUUGAAGAGAUAGGCGUACGC-3’) targeted on the sequence at positions 212–236 relative to the start codon of human Rab2 (accession number NM_002865) and the RNAi duplex (sense: 5’-GCGGCCCAUCUCUUUACGAUAUACA-3’ and antisense: 5’-UGUAUAUCGUAAAGAGAUGGGCCGC-3’) was used as a control. For Rab6 GTPase, a sense (5'-GGAAGUGAUGUUAUCAUCAUGCUAG-3’) and an antisence (5’–CUAGCAUGAUGAUAACAUCACUUCC-3’) targeted on the sequence at positions 246–370 relative to the start codon of Rab6 (accession number AF498939) and the RNAi duplex (sense: 5’-ACUGUCACGGUUUGAGUUGUCCUUA-3’ and antisense: 5’-UAAGGACAACUCAAACCGUGACAGU-3’) was used as a control. HEK293T cells were plated on 6-well dishes at a density of 4 × 105 cells/well for 12–16 h before transfection. The plasmid encoding the selected receptor was mixed with siRNA and simultaneously transfected by using Lipofectamine 2000 reagent. Briefly, 7.5 µl of Lipofectamine 2000 and 6 µl of 20 µM RNAi plus 0.5 µg of individual receptor plasmids were added separately into 100 µl of Opti-MEM medium. After 5 min incubation, both solutions were mixed and incubated for 20 min at room temperature. The transfection mixture was added to culture dishes containing 0.8 ml fresh DMEM without antibiotics. After incubation at 37°C for at 6–8 h, culture medium was changed to full DMEM medium. All the experiments were performed at 48 h after RNAi transfection.
2.8. Measurement of ERK1/2 activation
HEK293T cells were cultured in 6-well dishes and transfected with 0.5 µg of α2B-AR with or without co-transfection with 1.5 µg of Rab mutants or 6 µl of 20 µM Rab siRNA as described above. At 6–8 h after transfection, the cells were split into 6-well dishes and cultured for additional 36 h. The cells were starved for at least 3 h and then stimulated with 1 µM UK14304 for 5 min. Stimulation was terminated by addition of 1 × SDS gel loading buffer. After solubilizing the cells, 20 µl of total cell lysates was separated by 12% SDS-PAGE. ERK1/2 activation was determined by measuring the levels of phosphorylation of ERK1/2 with phospho-specific ERK1/2 antibodies by immunoblotting [33].
2.9. Measurement of cAMP production
cAMP production in response to stimulation with ISO was measured by using cAMP enzymeimmunoassay system (Biotrak, Amersham Pharmacia Biotech, Piscataway, NJ) as described previously [23]. HEK293T cells were cultured in 100-mm dishes and transfected with 3 µg of β2-AR. After 6 h, the cells were split into 12-well plates and cultured for 12 h. The cells were then starved for 24 h before stimulation with ISO at a concentration of 10 µM for 10 min at room temperature. The reactions were stopped by aspirating the medium and then the cells were lysed using 200 µl of dodecyltrimethylammonium (2.5%). One hundred µl of cell lysate was transferred into microtitre plates and incubated with anti-cAMP antiserum, followed by the incubation with cAMP-peroxidase. After washing and addition of substrate, peroxidase activity was measured by spectrometry. cAMP concentrations were calculated based on the competition of cAMP in samples with a fixed quantity of peroxidase-labeled cAMP.
2.10. Measurement of inositol phosphate (IP) production
IP accumulation was measured as described [44] with modifications [33]. Briefly, HEK293T cells were cultured on 100-mm dishes and transfected. At 8–12 h after transfection, the transfected cells were split into 60-mm culture dishes. At 24 h after transfection, the cells were labeled with fresh medium supplemented with myo-[³H]inositol (2 µCi/ml) for 24 h. The cells were washed three times with 2 ml of ice-cold Hanks' balanced salt solution containing 1.26 mM Ca2+ and 0.89 mM Mg2+ and incubated in 2 ml of Hanks' balanced salt solution containing 10 mM LiCl at room temperature for 20 min. The cells were then stimulated with Ang II at 1 µM for 45 min at 37 °C. The reactions were terminated with 1 ml of ice-cold 10% trichloroacetic acid. The cells were frozen at −80 °C for 10 min, collected, and mixed with 0.5 ml of 0.72 M KOH and 0.6 M KHCO3. After centrifugation at 3000 rpm for 5 min to remove the precipitated proteins, the supernatants were applied to Dowex AG 1-X8 anion exchange columns (1.0 ml) equilibrated with 0.1 M formic acid. The columns were washed with 0.1 M formic acid (4 × 4 ml) to remove free myo-[³H]inositol, and IPs were eluted with 0.1 M formic acid (3 × 2.0 ml) containing 1 M ammonium formate and counted by liquid scintillation spectrometry in 18 ml of Ecoscint A scintillation solution.
2.11. Immunoblotting
Western blot analysis of protein expression was carried out as previously described [33]. HEK293T cell lysates were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes. The signal was detected using ECL Plus (PerkinElmer Life Sciences) and a Fuji Film luminescent image analyzer (LAS-1000 Plus) and quantitated using the Image Gauge program (Version 3.4).
2.12. Statistical analysis
Differences were evaluated using Student's t test, and p < 0.05 was considered as statistically significant. Data are expressed as the mean ± S.E.
3. Results
3.1. Regulation of the cell-surface expression of α2B-AR and β2-AR by Rab2 and Rab6 GTPases
To distinguish the endogenous Rab GTPases and exogenously transfected Rab GTPase mutants and to facilitate the subcellular localization of the Rab GTPases, the constitutively active GTP-bound Rab2Q65L and Rab6Q72L mutants were tagged with GFP and FLAG at their N-termini, respectively. Rab2Q65L and Rab6Q72L were transiently expressed in HEK293T cells and their expression were determined by Western blotting using Rab isoform-specific antibodies and antibodies against the epitope GFP (Fig. 1A) or FLAG (Fig. 1B). Subcellular localization showed that Rab2Q65L and Rab6Q72L were localized to the perinuclear regions of the transfected cells (Fig. 1C). These data are consistent with the localization and function of Rab2 and Rab6 GTPases in modulating protein transport [36–43], suggesting that the GFP and FLAG epitop tagging did not influence the subcellular localization of Rab2 and Rab6 GTPases.
Fig. 1.

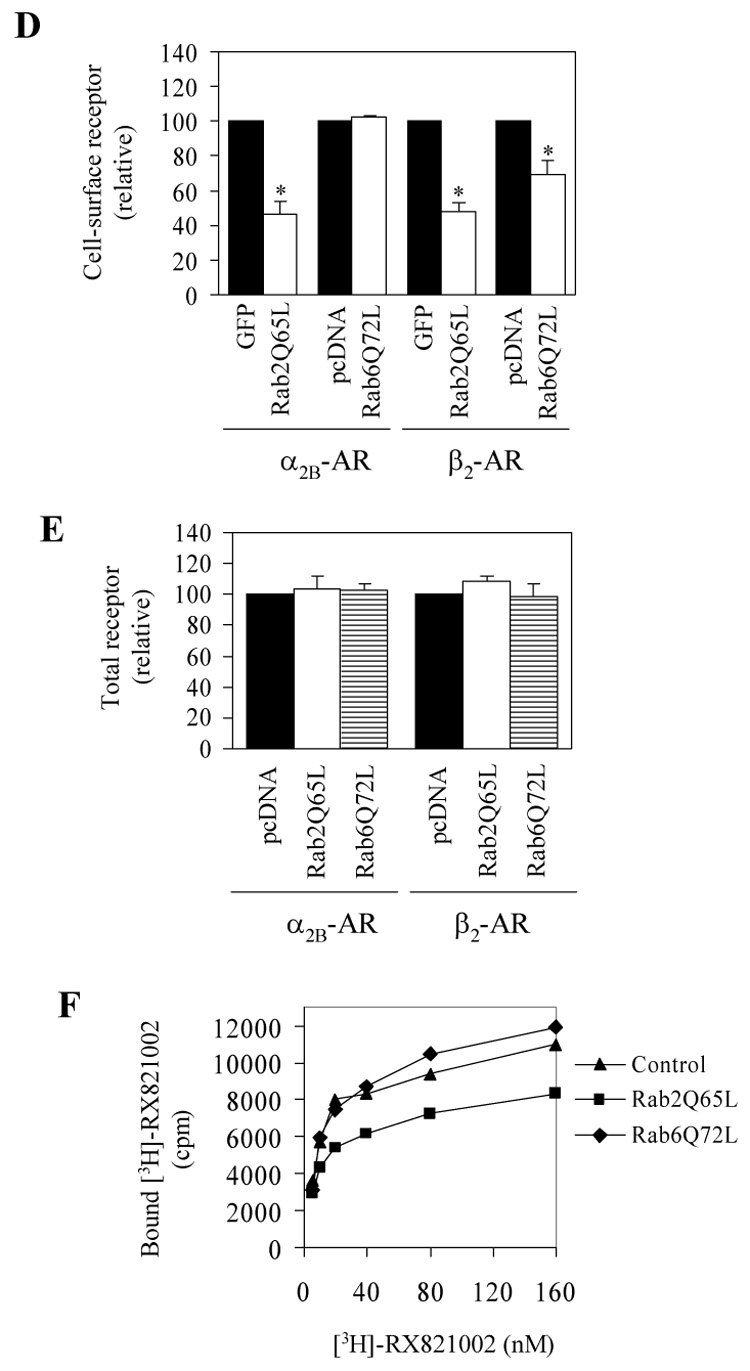
Effect of transient expression of Rab2Q65L and Rab6Q72L on the cell-surface expression of α2B-AR and β2-AR. A. Western blot analysis of GFP-tagged Rab2Q65L expression. HEK293T cells cultured on 6-well plates were transfected with the pEGFP-C1 vector (control) or GFP-Rab2Q65L mutant. Cell homogenates were separated by 12% SDS-PAGE and expression of Rab2Q65L was detected by Western blotting using anti-Rab2 (upper panel) and anti-GFP antibodies (middle panel). B. Western blot analysis of FLAG-tagged Rab6Q72L expression. HEK293T cells cultured on 6-well plates were transfected with the pcDNA3 vector (control) or FLAG-Rab2Q72L mutant and expression of Rab672L was detected by Western blotting using anti-Rab6 (upper panel) and anti-FLAG M2 antibodies (middle panel). β-actin expression is shown in lower panels as a loading control. C. The subcellular distribution of GFP-Rab2Q65L and FLAG-Rab6Q72L. HEK293T cells cultured on coverslips were transfected with GFP-conjugated Rab2Q65L or FLAG-Rab6Q72L. The subcellular distribution of GFP-Rab2Q65L was revealed by fluorescence microscopy detecting GFP and FLAG-Rab6Q72L by immunostaining with anti-FLAG antibodies as described under “Experimental procedures.” Scale bars, 10 µm. D. Inhibition of the cell-surface expression of α2B-AR and β2-AR by Rab2Q65L and Rab6Q72L. HEK293T cells were transfected with GFP-conjugated α2B-AR or β2-AR together with the pEGFP-C1 vector (GFP), GFP-tagged Rab2Q65L, the pcDNA3 vector or FLAG-Rab6Q72L. The expression of α2B-AR or β2-AR at the cell surface was determined by intact cell ligand binding using [³H]-RX821002 and [³H]-CGP12177, respectively, as described in the “Experimental procedures.” The mean values of specific ligand binding were 43371 ± 6368, 44247 ± 3979, 49014 ± 8433 and 51385 ± 2879 cpm (n = 3, each in triplicate) from cells transfected with α2B-AR with pEGFP-C1, α2B-AR with pcDNA3, β2-AR with pEGFP-C1 or β2-AR with pcDNA3, respectively. The data shown are percentages of the mean value obtained from cells transfected with individual receptor and the pcDNA3 or pEGFP-C1 vector and are presented as the mean ± S.E. of three experiments. *, p < 0.05 versus the cells transfected with respective receptor and the pcDNA3 or pEGFP-C1 vector. E. Effect of Rab2Q65L and Rab6Q72L on total expression of α2B-AR and β2-AR. HEK293T cells were transfected with GFP-conjugated α2B-AR or β2-AR together with the pcDNA3 vector, FLAG-Rab6Q72L or Rab2Q65L in the pcDNA3 vector. The overall receptor expression was determined by measuring GFP fluorescence using a flow cytometer as described in the “Experimental procedures.” F. Specific [³H]-RX821002 binding to membrane fractions prepared from the cells transfected with α2B-AR and Rab2Q65L or Rab6Q72L. HEK293T cells were transiently transfected with α2B-AR together with the pcDNA3 vector, FLAG-Rab6Q72L or Rab2Q65L. The membrane preparation (15 µg of protein) was incubated with increasing concentrations of [³H]-RX821002 (5 – 160 nM) for 30 min. Specific binding was determined in duplicate, and nonspecific binding was determined in the presence of 10 µM rauwolscine as described under "Experimental procedures." The data shown are representative of at least three separate experiments, each with similar results.
To define the role of Rab2 and Rab6 GTPases in GPCR targeting to the cell surface, GFP-Rab2Q65L and FLAG-Rab6Q72L were co-expressed with α2B-AR or β2-AR in HEK293T cells and their effects on the cell-surface expression of the receptors was quantified by ligand binding in intact live cells. The cell-surface numbers of α2B-AR and β2-AR were significantly inhibited by 54 and 52%, respectively, by co-expression with Rab2Q65L as compared with cells expressing receptor alone (Fig. 1D). Interestingly, in contrast to Rab2Q65L, transient expression of Rab6Q72L differentially modulated the cell-surface expression of α2B-AR and β2-AR. Whereas Rab6Q72L inhibited β2-AR expression at the plasma membrane by 30%, the cell-surface number of α2B-AR was not influenced by expression of Rab6Q72L (Fig. 1D). In contrast, the overall expression of α2B-AR and β2-AR was not significantly altered by Rab2Q65L and Rab6Q72L (Fig. 1E). These data indicate that, whereas Rab2 is involved in the cell-surface targeting of both of α2B-AR and β2-AR, Rab6 selectively modulates the export trafficking of β2-AR, but not α2B-AR.
To further confirm that Rab2 and Rab6 GTPase were capable of differentially modulating the transport of α2B-AR to the cell surface as measured by intact cell ligand binding, the plasma membrane fractions were prepared from cells transfected with α2B-AR and Rab2Q65L or Rab6Q72L and the receptor number was measured by radioligand binding. The radioligand [³H]-RX821002 binding to the membrane fraction prepared from the cells transfected with α2B-AR alone was similar to the membrane preparations from cells expressing α2B-AR and Rab6Q72L, but was clearly higher than that from cells expressing α2B-AR and Rab2Q65L (Fig. 1F). These data further indicate that the transport of α2B-AR to the cell surface is independent of Rab6 GTPase.
3.2. Effect of Rab2 and Rab6 GTPases on the subcellular distribution of α2B-AR and β2-AR
We then determined the effects of Rab2 and Rab6 on the subcellular distribution of α2B-AR and β2-AR. Rab2Q65L and FLAG-Rab6Q72L in the pcDNA3.1 vector were coexpressed with α2B-AR-GFP or β2-AR-GFP. The subcellular localization of GFP-conjugated receptors at steady state was revealed by fluorescence microscopy analysis. As anticipated, the majority of α2B-AR and β2-AR was expressed at the cell surface, which was confirmed by co-localization with tetramethylrhodamine-conjugated concanavalin A, a plasma membrane marker (data not shown). Consistent with the marked reduction in the cell-surface expression as measured by radioligand binding, β2-AR lost its cell-surface expression pattern in cells transfected with Rab2Q65L or Rab6Q72L and was arrested in the perinuclear region. In contrast, α2B-AR export to the cell surface was selectively blocked in cells expressing Rab2Q65L (Fig. 2A) and retained its ability to target to the cell surface in cells expressing Rab6Q72L. These data further indicate that Rab2 is involved in the transport of both α2B-AR and β2-AR, whereas Rab6 selectively influences intracellular trafficking of β2-AR, but not α2B-AR.
Fig. 2.
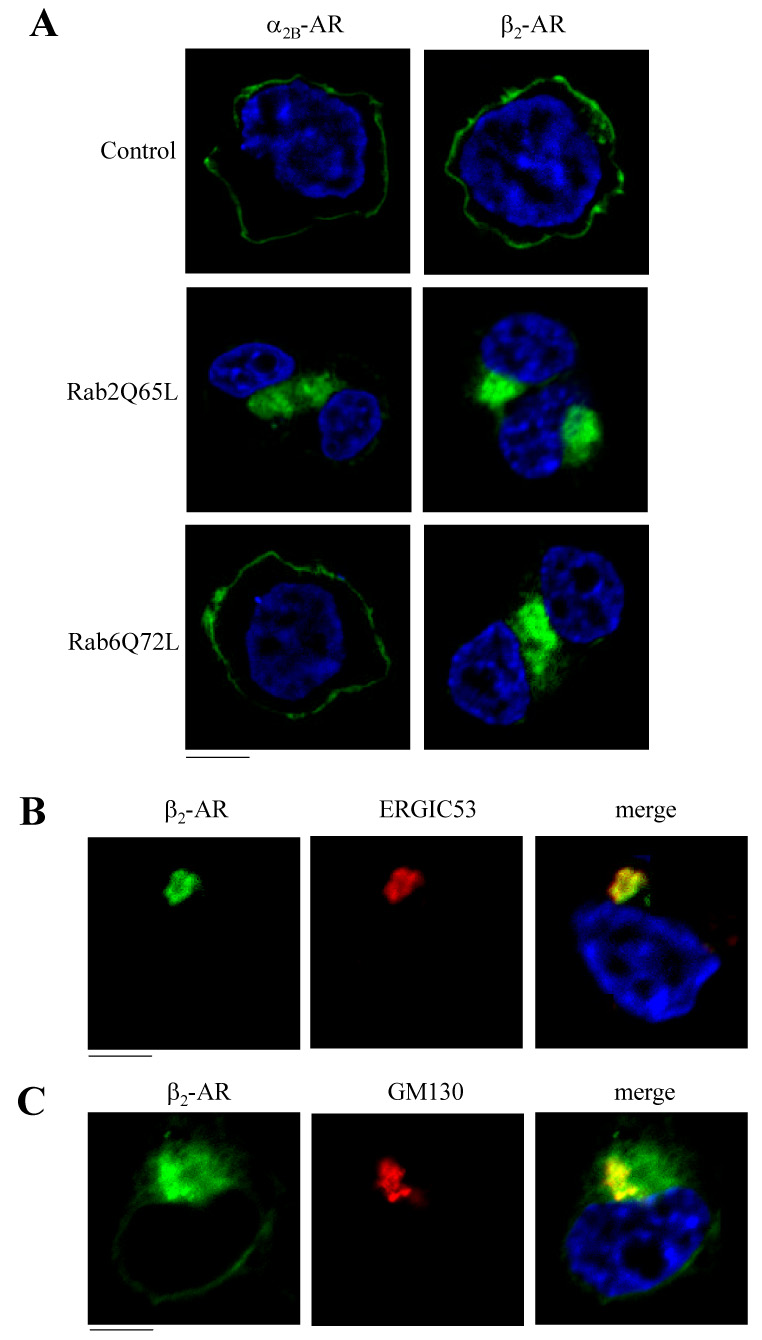
Effect of Rab2Q65L and Rab6Q72L on the subcellular distribution of α2B-AR and β2-AR. A. HEK293T cells cultured on coverslips were transfected with α2B-AR-GFP or β2-AR-GFP (100 ng) together with the pcDNA3 vector (control), Rab2Q65L or Rab6Q72L (400 ng). The subcellular distribution of the receptors was revealed by detecting GFP fluorescence as described under “Experimental procedures.” B. Co-localization of β2-AR-GFP with ERGIC53. HEK293T cells cultured on coverslips were transfected with β2-AR-GFP and Rab2Q65L and stained with anti-ERGIC53 antibodies (1:200 dilution). C. Co-localization of β2-AR-GFP with GM130. HEK293T cells cultured on coverslips were transfected with β2-AR-GFP and Rab6Q72L and stained with anti-GM130 antibodies (1:200 dilution). Green, β2-AR-GFP; red, ERGIC53 (B) and GM130 (C); blue, DNA staining by 4,6-diamidino-2-phenylindole (nucleus); yellow, co-localization of β2-AR with ERGIC53 (B) and GM130 (C). The data shown are representative images of at least three independent experiments. Scale bars, 10 µm.
To further define the intracellular compartments that the receptors are possibly arrested by expression of Rab2Q65L and Rab6Q72L, we co-localized β2-AR with the ER marker calregulin, the ERGIC marker ERGIC53 and the cis-Golgi marker GM130. GFP-tagged β2-AR was co-expressed with Rab2Q65L or FLAG-Rab6Q72L in the pcDNA3 vector in HEK293T cells and the co-localization of the receptor with the markers was visualized by microscopy analysis following staining with antibodies against calregulin, ERGIC53 or GM130. β2-AR was partially co-localized with ERGIC53 (Fig. 2B), but not calregulin (data not shown), in cells expressing Rab2Q65L. These data suggest that β2-AR retained its ability to exit from the ER in cells expressing Rab2Q65L, but was trapped in the ERGIC compartment. These data are consistent with the effect of Rab2Q65L on the transport of other plasma membrane proteins from the ERGIC compartment [40]. When co-expressed with FLAG-Rab6Q72L, β2-AR was partially co-localized with GM130 (Fig. 2C), suggesting that β2-AR was retained in the cis-Golgi compartment by Rab6Q72L.
3.3. Effect of siRNA-mediated knockdown of Rab2 and Rab6 GTPases on the cell-surface expression of α2B-AR and β2-AR
To further define the function of Rab2 and Rab6 in regulating receptor anterograde transport, we determined the effect of siRNA-mediated knockdown of endogenous Rab2 and Rab6 expression on the cell-surface targeting of α2B-AR and β2-AR. Introduction of Rab2 and Rab6 siRNA into HEK293T cells specifically reduced Rab2 and Rab6 expression by 62 and 68%, respectively, when compared with cells transfected with control siRNA (Fig. 3A and 3B). Rab2 siRNA transfection did not produce a significant effect on the expression of the closely related small GTPases Rab1 and Rab6 nor Rab6 siRNA trasnfection produce a significant effect on Rab1 and Rab2 expression (Fig. 3A). Similar to transient expression of their constitutively active GTP-bound mutants, depletion of endogenous Rab2 and Rab6 by siRNA transfection differentially modulated the cell-surface expression of α2B-AR and β2-AR. Rab2 siRNA inhibited the transport to the cell surface of both α2B-AR and β2-AR, whereas Rab6 siRNA selectively attenuated the transport of β2-AR, but not α2B-AR (Fig. 3C). Inhibition on receptor expression at the cell surface induced by transient expression of Rab2 and Rab6 siRNA was specific, since the control siRNA did not have a clear effect on the receptor expression on the cell surface when compared with cells without siRNA transfection (data not shown). Furthermore, both Rab2 and Rab6 siRNA had no influence on the overall expression levels of both α2B-AR and β2-AR (Fig. 3D). These data further support differential regulation of export trafficking of α2B-AR and β2-AR by Rab2 and Rab6 GTPases.
Fig. 3.

Effect of siRNA-mediated knockdown of Rab2 and Rab6 on the cell surface expression of α2B-AR and β2-AR. A. HEK293T cells cultured on 6-well dishes were transiently transfected with control siRNA, Rab2 or Rab6 siRNA as described under "Experimental procedures." At 48 hours after transfection, total homogenate protein was separated by 12% SDS-PAGE, and expression of Rab2 (upper panel), Rab6 (middle panel) and Rab1 (lower panel) was detected by Western blotting using isoform-specific antibodies. B. Effect of Rab2 and Rab6 siRNA on their expression. The data are expressed as percentages of Rab protein expression in cells transfected with control siRNA and presented as the means ± S.E. of three individual experiments. *, p < 0.05 versus their respective control siRNA. C. Effect of siRNA-mediated depletion of Rab2 and Rab6 on the cell-surface expression of α2B-AR and β2-AR. HEK293T cells were transfected with GFP-conjugated α2B-AR or β2-AR together with the control or Rab siRNA as described in the “Experimental procedures.” The cell-surface expression of the receptors was determined by intact cell ligand binding as described in the legend of figure 1. The data shown are percentages of the mean value obtained from cells transfected with individual receptor with control siRNA and are presented as the mean ± S.E. of three experiments. *, p < 0.05 versus the cells transfected with respective receptor and control siRNA. D. Effect of Rab2 and Rab6 siRNA on total expression of α2B-AR and β2-AR by measuring GFP fluorescence as described in the legend of figure 1.
3.4. Regulation of α2B-AR and β2-AR signaling by Rab2 and Rab6 GTPases
We then determined if attenuation of cell-surface expression of α2B-AR and β2-AR induced by transient expression of the active Rab2Q65L and Rab6Q72L mutants and siRNA-mediated knockdown of Rab2 and Rab6 expression could influence receptor signaling propagation. α2B-AR-mediated signaling was measured by activation of the ERK1/2 MAPK in response to stimulation with UK14304 and β2-AR-mediated signaling was measured by cAMP production in response to stimulation with ISO. ERK1/2 activation by UK14304 was markedly compromised in cells co-transfected with α2B-AR and Rab2Q65L as compared with cells expressing α2B-AR alone (Fig. 4A and 4B). Similarly, transient expression of Rab2 siRNA significantly attenuated ERK1/2 activation by UK14304 in cells expressing α2B-AR as compared with cells transfected with control siRNA (Fig. 4A and 4B). In contrast, expression of Rab6Q72L and Rab6 siRNA had no obvious inhibitory effect on UK14304-stimulated ERK1/2 activation. cAMP production in response to stimulation with ISO was markedly inhibited by 67, 72, 48 and 74% by transient expression of Rab2Q65L, Rab6Q72L, Rab2 siRNA and Rab6 siRNA, respectively (Fig. 4C). These data are strongly consistent with the effect of Rab2 and Rab6 GTPases on the cell-surface expression of these receptors, and further indicate that Rab2 is required for the cell-surface targeting and signaling of α2B-AR and β2-AR, whereas Rab6 selectively modulates the trafficking and signaling of β2-AR, but not α2B-AR.
Fig. 4.

Effect of Rab2 and Rab6 on signaling of α2B-AR and β2-AR. A. Effect of Rab2 and Rab6 on ERK1/2 activation by α2B-AR. HEK293T cells were transfected with GFP-tagged α2B-AR together the pEGFP-C1 vector (GFP), GFP-Rab2Q65L, the pcDNA3 vector, FLAG-Rab6Q72L, Rab2 control siRNA, Rab2 siRNA, Rab6 control siRNA or Rab6 siRNA as described under “Experimental procedures”. The cells were stimulated with UK14304 at a concentration of 1 µM for 5 minutes at 37 °C. ERK1/2 activation was determined by Western blot analysis using phospho-specific ERK1/2 antibodies (ERK1/2-P). upper panel, representative blots of ERK1/2 activation; lower panel, total ERK1/2 expression. B. Quantitative data expressed as percentages of ERK1/2 activation obtained from cells transfected with α2B-AR and a separate control and stimulated with 1 µM UK14304 and presented as the mean ± S.E. of three experiments. C. Effect of Rab2 and Rab6 on cAMP production by the β2-AR agonist ISO. HEK293T cells were cultured in 100-mm plates and transfected with β2-AR as described in (A) for α2B-AR and then stimulated with ISO (10 µM) for 10 minutes. cAMP concentrations were determined by using cAMP enzymeimmunoassay system as described under “Experimental procedures.” The data are presented as fold increase in response to ISO stimulation over the basal values and as the mean ± S.E. of three experiments. *, p < 0.05 versus their respective controls.
3.5. Effect of Rab6Q72L on the cell-surface expression, subcellular localization and signaling of AT1R
We then sought to determine if Rab6 is involved in the regulation of cell-surface expression, signaling and subcellular distribution of AT1R. Expression of Rab6Q72L dramatically blocked AT1R transport to the cell surface by 56% as measured by intact cell ligand binding (Fig. 5A) and AT1R was trapped in the perinuclear region of the transfected cells (Fig. 5B). Consistently, co-expression of Rab6Q72L with AT1R profoundly inhibited IP production in response to Ang II stimulation. IP production in response to Ang II stimulation in HEK293T cells transfected with AT1R plus the empty pcDNA3.1 vector or Rab6Q72L was increased by 5.4 ± 0.3− and 1.5 ± 0.1-fold (p < 0.05), respectively (Fig. 5C). These data indicate that, similar to β2-AR, AT1R targeting to the cell surface and signaling is dependent on the normal function of Rab6 GTPase.
Fig. 5.
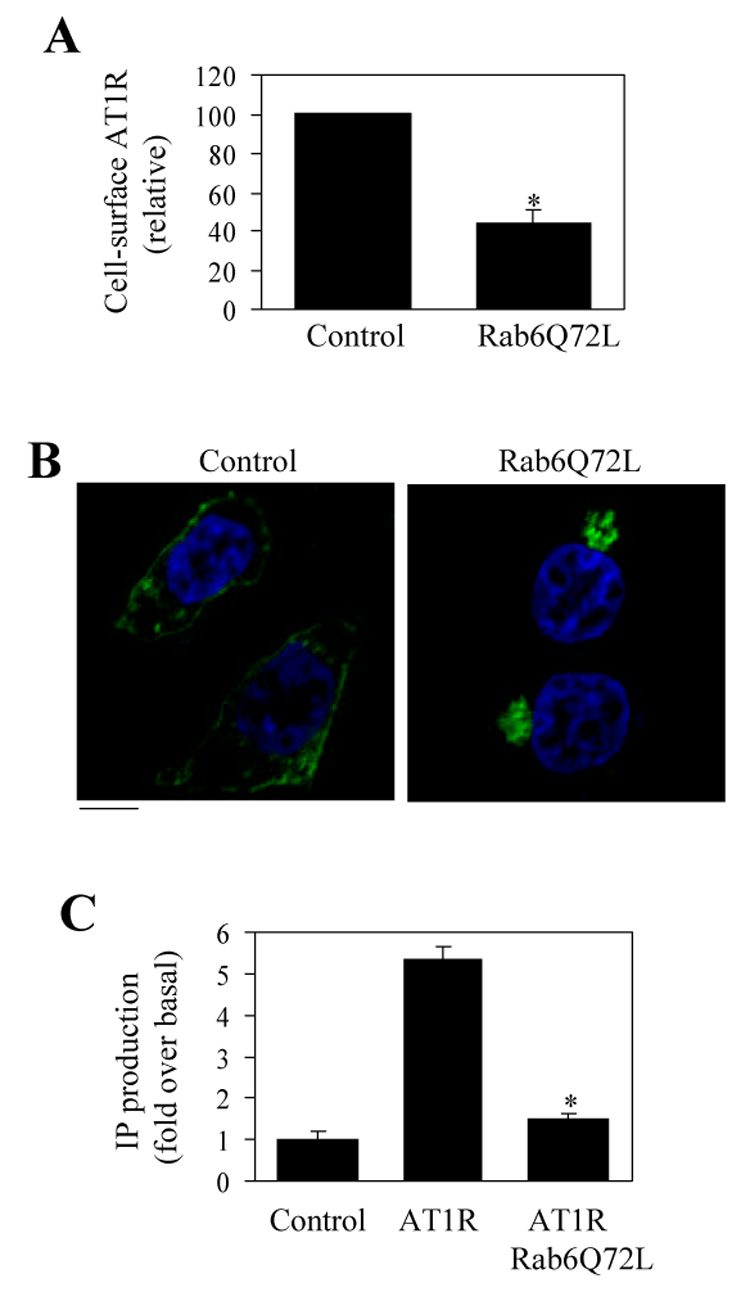
Effect of Rab6Q72L on the cell-surface expression, subcellular localization and signaling of AT1R. A. Effect of Rab6Q72L on AT1R transport to the cell surface. HEK293T cells were cultured and transfected with the pcDNA3 vector or FLAG-Rab6Q72L and the cell surface expression of AT1R was determined by ligand [³H]-Ang II binding as described under "Experimental procedures." Nonspecific binding was obtained in the presence of 10 µM nonradioactive Ang II. The mean values of specific ligand binding were 12730 ± 1562 cpm (n = 3, each in triplicate) from cells transfected with AT1R with the pcDNA3 vector. The data shown are the percentage of the mean value obtained from cells expressing AT1R alone and are presented as the means ± S.E. *, p < 0.05 versus control. B. Effect of Rab6Q72L on the subcellular localization of AT1R. HEK293T cells were transfected with AT1R-GFP and the pcDNA3 vector (control) or FLAG-Rab6Q72L and the subcellular localization of AT1R was revealed by fluorescence microscopy detecting GFP. The data shown are representative images of at least three independent experiments. Scale bar, 10 µm. C. Effect of Rab6Q72L on AT1R-mediated IP accumulation. HEK293T cells were transfected with AT1R-GFP and the pcDNA3.1 vector or FLAG-Rab6Q72L. The transfected cells were split into 60-mm culture dishes, incubated with myo-[³H]inositol, and stimulated with Ang II at 1 µM. IP production was measured as described under "Experimental procedures." The basal levels of IP production in untransfected HEK293T cells and in HEK293T cells transfected with AT1R-GFP and pcDNA3.1 or FLAG-Rab6Q72L were 2643 ± 312, 2544 ± 221 and 1805 ± 342 cpm, respectively. The data are shown as the -fold increase over the respective basal levels of IP production in response to Ang II stimulation and represent the means ± S.E. of three experiments. *, p < 0.05 versus cells transfected with AT1R-GFP alone.
3.6. Effect of the dominant-negative mutant Rab6N126I on the cell-surface expression of α2B-AR, β2-AR and AT1R
To further characterize differential regulation by Rab6 GTPase of anterograde transport of GPCRs, we generated the nucleotide-free mutant Rab6N126I and determined its effect on the cell-surface expression of α2B-AR, β2-AR and AT1R. Similar to Rab6Q72L, Rab6N126I was also tagged with the FLAG epitope at its N-terminus and its expression in HEK293T cells was determined by Western blotting using antibodies against Rab6 and FLAG (Fig. 6A). In contrast to Rab6Q72L localized to the perinuclear regions of the transfected cells, FLAG-Rab6N126I mutants displayed a cytosolic distribution pattern (Fig. 6B). Similar to the constitutively active mutant Rab6N72L, transient expression of the dominant-negative mutant Rab6N126I selectively modulated the cell surface expression of three GPCRs as measured by intact cell ligand binding. The cell surface numbers of β2-AR and AT1R was moderately, but significantly attenuated by Rab6N126I (Fig. 6C). In contrast, Rab6N126I did not significantly influence the cell-surface expression of α2B-AR (Fig. 6C). The overall expression of α2B-AR and β2-AR was also not influenced by Rab6N126I (data not shown). These data further indicate that the transport of different GPCRs may be differentially modulated by Rab6 GTPase.
Fig. 6.
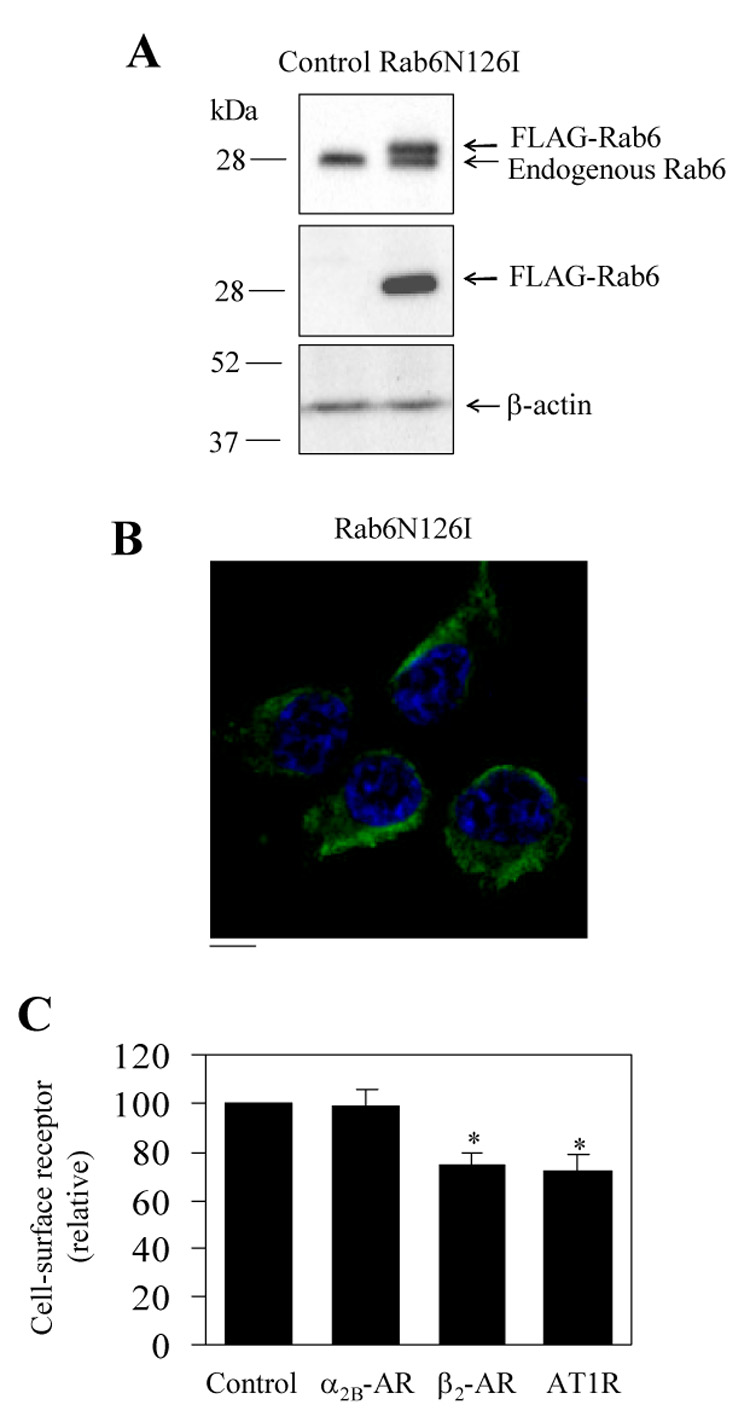
Effect of the dominant-negative mutant Rab6N126I on the cell-surface expression of α2B-AR, β2-AR and AT1R. A and B. Western blot analysis of expression (A) and the subcellular distribution of FLAG-Rab6N126I (B) as described in the legend of figure 1 for FLAG-Rab6Q72L. C. Effect of FLAG-Rab6N126I on cell surface expression of α2B-AR, β2-AR and AT1R. HEK293T cells were transfected with GFP-conjugated α2B-AR, β2-AR or AT1R together with the pcDNA3 vector (control) or FLAG-Rab6N126I. The expression of α2B-AR and β2-AR at the cell surface was determined as described in the legend of figure 1 and the cell-surface AT1R measured as in figure 5. The data shown are percentages of the mean value obtained from cells transfected with individual receptor and the pcDNA3 vector and are presented as the mean ± S.E. of three experiments. *, p < 0.05 versus control.
4. Discussion
There are at least three Rab GTPases, Rab1, Rab2 and Rab6, which coordinate anterograde and retrograde transport between the ER and the Golgi apparatus. Whereas Rab1 GTPase mediates anterograde transport of newly synthesized proteins from the ER to the Golgi, Rab2 and Rab6 GTPases modulate retrograde Golgi-to-ER transport, which is essential for retrieval of ER or Golgi resident proteins to maintain the organelle homeostasis as well as recycling of the components of the transport machinery required for anterograde transport [36 – 43]. We recently demonstrated that Rab1 GTPase differentially regulates the cell-surface targeting of distinct GPCRs [33–35]. Our data indicate that the transport from the ER through to the Golgi to the cell surface of α1-AR, β-AR and AT1R is dependent on Rab1. In contrast, the transport of α2B-AR to the cell surface is independent of Rab1, suggesting that multiple routes mediate anterograde transport of different GPCRs from the ER to the cell surface. However, the role of Rab GTPases involved in retrograde transport in the biosynthesis of the GPCR superfamily in mammalian cells remains undefined. To address this issue, in this report we investigated the function of Rab2 and Rab6 GTPases in regulating the cell-surface targeting, subcellular distribution and signaling of α2B-AR, β2-AR and AT1R.
Rab2 GTPase is predominantly localized to the ERGIC compartment which plays a critical role in separating proteins for anterograde or retrograde transport in the early secretory pathway [3] and has been proposed to modulate retrograde protein transport specifically from the ERGIC to the ER [39, 40]. The Rab2-mediated retrograde protein transport has also been shown to be important for anterograde transport of plasma membrane proteins. For example, expression of GTP-bound Rab2Q65L inhibits the maturation of vesicular stomatitis viral glycoprotein (VSVG) [36]. Interestingly, the trafficking of the cystic fibrosis transmembrane conductance regulator (CFTR) through the early secretory pathway is independent of Rab2 function [45]. Our data demonstrated that transient expression of Rab2Q65L significantly inhibited the cell-surface expression of α2B-AR and β2-AR as determined by ligand binding of intact cells and membrane preparations. Subcellular localization showed that the receptors were unable to transport to the cell surface and accumulated in the ERGIC subcellular compartment in cells expressing Rab2Q65L, which is consistent with an accumulation of VSVG in the ERGIC compartment in the presence of Rab2Q65L [40]. These data support the function of Rab2 in regulating protein transport from the ERGIC. Consistent with attenuated receptor expression at the cell surface, receptor-mediated signaling as measured by ERK1/2 activation and cAMP production was also blocked by Rab2Q65L. Similar to transient expression of Rab2Q65L, siRNA-mediated depletion of endogenous Rab2 GTPase blocked cell-surface expression and signaling of both α2B-AR and β2-AR. These data strongly indicate that Rab2 GTPase-coordinated protein transport plays an essential role in the anterograde transport of α2B-AR and β2-AR to the plasma membrane.
In contrast to Rab2 function in coordinating the ERGIC-to-ER transport, Rab6 may be involved in transport from the late to the early Golgi cisternae or from the Golgi to the ER of proteins such as β-1, 4-galactosyltransferase and protein toxins [41–43]. It has also been demonstrated that manipulation of Rab6 function through expressing its GTP-bound, GDP-bound or guanidine nucleotide deficient mutants inhibits transport of the plasma membrane proteins hemagglutinin protein and CFTR in mammalian cells and rhodopsin in Drosophila [42, 45, 46]. Consistent with the role of Rab6 in the plasma membrane targeting, our data indicate that Rab6 is required for the transport to the cell surface of some GPCRs. Interestingly, in contrast to Rab2 modulating the trafficking of both α2B-AR and β2-AR, Rab6 selectively regulated the cell-surface targeting of β2-AR and AT1R, but not α2B-AR. First, transient expression of Rab6Q72L and Rab6N126I selectively attenuated β2-AR and AT1R expression at the cell-surface without significantly altering α2B-AR transport. Second, consistent with Rab6 function in modulating retrograde protein transport in intra-Golgi transport, our data demonstrated that β2-AR was partially co-localized with the cis-Golgi marker GM130 in cells expressing Rab6Q72L, suggesting that Rab6Q72L blocked β2-AR exit from the cis-Golgi. In contrast, α2B-AR normally transported to the plasma membrane in cells expressing Rab6Q72L. Third, similar to transient expression of Rab6Q72L, selective knockdown of endogenous Rab6 expression by siRNA specifically attenuated the cell-surface expression of β2-AR, but not α2B-AR. Finally, ISO-promoted cAMP production and Ang II-mediated IP production, but not UK14304-mediated ERK1/2 activation, were blocked by Rab6Q72L. These data provide strong evidence indicating that Rab6 GTPase is capable of selectively modulating anterograde trafficking of GPCRs.
The function of Rab GTPases in coordinating vesicular transport is mediated through their GTP/GDP exchange cycle, which superimposes with Rab protein association with, and dissociation from, subcellular organelle membranes. The inactive, GDP-bound conformation of Rab GTPases is maintained in the cytosol through an association with GDP dissociation inhibitors (GDI), which function as chaperones and mediate Rab translocation from cytosol to membrane. Membrane-associated GDP-bound Rab-GDI complexes undergo GDP exchange for GTP. GTP-bound Rab GTPases recruit their effector proteins, which mediate the migration, docking, and fusion of transport vesicles to acceptor membrane [47]. GTP-bound Rab is then hydrolyzed to GDP-bound Rab for recycling back to donor membrane [20]. Based on this model, GDP-bound Rab mutants, which are unable to undergo exchange for GTP, and GTP-bound Rab mutants, which prevent the recycling of Rab to be reused, function as negative regulators in protein transport. Therefore, the inhibitory effect of the GTP-bound mutants Rab2Q65L and Rab6Q72L and the nucleotide-free mutant Rab6N126I on GPCR export to the plasma membrane could be explained through preventing the retrieval of the ER or Golgi resident proteins and the recycling of transport machinery, which will disrupt the normal anterograde transport. However, it has been demonstrated that expression of GTP-bound Rab6 induces the relocation of medial to trans-Golgi resident proteins to the ER by presumably up-regulating the rate of Golgi-to-ER trafficking [41] and the GTP-bound Rab2 facilitates the formation of vesicles from the ERGIC [40]. It is also possible that the inhibitory effect of Rab2Q65L and Rab6Q72L on GPCR transport to the cell surface is mediated through facilitating the flow of retrograde protein transport, which will ultimately disrupt the anterograde protein transport pathway.
There are several possibilities regarding selective regulation of the transport of α2B-AR, β2-AR and AT1R by Rab6 GTPase. First, the transport of α2B-AR, β2-AR and AT1R from the ER to the Golgi may be mediated through distinct transport pathways, which have different sensitivities to the Rab6-coordinated retrograde transport. This possibility is strongly supported by our previous data demonstrating that there is a non-conventional, Rab1-independent transport pathway mediating α2B-AR export to the plasma membrane [33]. Second, the N-linked post-translational glycosylation of these three receptors may play an important role in their differential regulation by Rab6. Rab6-regulated β2-AR and AT1R have putative N-linked glycosylation sites at positions 6, 15, and 187 and positions 4, 176, and 188, respectively and the glycosylation of GPCRs occurs during their transport through the Golgi apparatus. In contrast, the α2B-AR, which is not regulated by Rab6, does not contain glycosylation signals. Consistent with this hypothesis, it has been demonstrated that Rab6-dependent retrograde transport within the Golgi complex mediates recycling of glycosyltransferases [41]. However, we were unable to test if the N-linked glycosylation is one of the determinants for the Rab6-mediated trafficking of β2-AR and AT1R, as mutation of these glycosylation sites in both receptors dramatically disrupted their overall synthesis and transport to the cell surface [data not shown, 48–51]. Third, the Golgi structure may be differentially required for the transport of α2B-AR, β2-AR and AT1R. It is possible that the transport to the cell surface of α2B-AR, which does not require post-translational glycosylation in the Golgi, bypasses the Golgi. Consistent with this possibility, Golgi-independent transport has been reported [52–54]. However, our previous data have demonstrated that BFA treatment, which induces Golgi collapse, blocked α2B-AR transport to the cell surface [16], suggesting a Golgi-dependent transport pathway for α2B-AR.
Selective export trafficking of different GPCRs with similar structural features may be achieved at discrete intracellular levels and are modulated by multiple regulatory molecules. We have demonstrated that α2B-AR, β2-AR and AT1R use the same F(X)6LL motif in the membrane-proximal C-termini to export from the ER, implying that these receptors may be sorted from other GPCRs with different ER exit motifs [14]. The YS motif in the membrane-proximal N-termini may dictate the α2-AR export from the Golgi, providing a means for the segregation of GPCRs at the level of the Golgi [16]. Our recent studies have showed that the GTP hydrolysis of Sar1 GTPase, which plays an essential role in the formation of COPII vesicle from the ER membrane, differentially modulates the recruitment and package of β2-AR, α2B-AR and AT1R into ER exit sites [55], suggesting that GPCRs could also be sorted at the level of ER exit sites on the ER membrane. Our previous works have also showed that biosynthesis of distinct GPCRs may be differentially modulated by Rab1 GTPase. The data presented in the current report suggest that, similar to Rab1-mediated anterograde transport, Rab2- and Rab6-corrdinated retrograde transport from the Golgi through the ERGIC to the ER may control the specificity/selectivity of GPCR targeting to the plasma membrane.
Acknowledgements
This work was supported by the National Institutes of Health Grant GM076167 (to G.W.) and the American Heart Association, Southeast Affiliate Postdoctoral Fellowship (to C.D.). We thank Dr. Catalin M. Filipeanu for measuring cAMP accumulation and Fuguo Zhou for superb technical assistance. We are grateful to Drs. Stephen M. Lanier, John D. Hildebrandt and Kenneth E. Bernstein for plasmids.
Footnotes
The abbreviations used are: GPCR, G protein-coupled receptors; AR, adrenergic receptor; AT1R, angiotensin II type 1 receptor; ER, endoplasmic reticulum; ERGIC, ER-Golgi intermediate complex; TGN, trans-Golgi network; GFP, green fluorescent protein; VSVG, vesicular stomatitis viral glycoprotein; CFTR, cystic fibrosis transmembrane conductance regulator; GDI, GDP dissociation inhibitors; PBS, phosphate-buffered saline; DMEM, Dulbecco's modified Eagle's medium; ISO, isoproterenol; Ang II, angiotensin II; ERK, extracellular signal-regulated kinase; IP, inositol phosphate.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wess J. Pharmacol. Ther. 1998;80:231–264. doi: 10.1016/s0163-7258(98)00030-8. [DOI] [PubMed] [Google Scholar]
- 2.Pierce KL, Premont RT, Lefkowitz RJ. Nat. Rev. Mol. Cell Biol. 2002:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 3.Appenzeller-Herzog C, Hauri HP. J. Cell Sci. 2002;119:2173–2183. doi: 10.1242/jcs.03019. [DOI] [PubMed] [Google Scholar]
- 4.Von Zastrow M. Life Sci. 2002;74:217–224. doi: 10.1016/j.lfs.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Wu G, Krupnick JG, Benovic JL, Lanier SM. J. Biol. Chem. 1997;272:17836–17842. doi: 10.1074/jbc.272.28.17836. [DOI] [PubMed] [Google Scholar]
- 6.Wu G, Benovic JL, Hildebrandt JD, Lanier SM. J. Biol. Chem. 1998;273:7197–7200. doi: 10.1074/jbc.273.13.7197. [DOI] [PubMed] [Google Scholar]
- 7.Dong C, Filipeanu CM, Duvernay MT, Wu G. Biochim. Biophys. Acta. 2007;1768:853–870. doi: 10.1016/j.bbamem.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petaja-Repo UE, Hogue M, Laperriere A, Walker P, Bouvier M. J. Biol. Chem. 2000;275:13727–13736. doi: 10.1074/jbc.275.18.13727. [DOI] [PubMed] [Google Scholar]
- 9.Stojanovic A, Hwa J. Receptors Channels. 2002;8:33–50. [PubMed] [Google Scholar]
- 10.Morello JP, Bichet DG. Annu. Rev. Physiol. 2001;63:607–630. doi: 10.1146/annurev.physiol.63.1.607. [DOI] [PubMed] [Google Scholar]
- 11.Bermak JC, Li M, Bullock C, Zhou QY. Nat. Cell Biol. 2001;3:492–498. doi: 10.1038/35074561. [DOI] [PubMed] [Google Scholar]
- 12.Robert J, Clauser E, Petit PX, Ventura MA. J. Biol. Chem. 2005;280:2300–2308. doi: 10.1074/jbc.M410655200. [DOI] [PubMed] [Google Scholar]
- 13.Schulein R, Hermosilla R, Oksche A, Dehe M, Wiesner B, Krause G, Rosenthal W. Mol. Pharmacol. 1998;54:525–535. doi: 10.1124/mol.54.3.525. [DOI] [PubMed] [Google Scholar]
- 14.Duvernay MT, Zhou F, Wu G. J. Biol. Chem. 2004;279:30741–30750. doi: 10.1074/jbc.M313881200. [DOI] [PubMed] [Google Scholar]
- 15.Zhou F, Filipeanu CM, Duvernay MT, Wu G. Cell. Signal. 2006;18:318–327. doi: 10.1016/j.cellsig.2005.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong C, Wu G. J. Biol. Chem. 2006;281:38543–38554. doi: 10.1074/jbc.M605734200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bulenger S, Marullo S, Bouvier M. Trends Pharmacol. Sci. 2005;26:131–137. doi: 10.1016/j.tips.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Dupre DJ, Robitaille M, Ethier N, Villeneuve LR, Mamarbachi AM, Hebert TE. J. Biol. Chem. 2006;281:34561–34573. doi: 10.1074/jbc.M605012200. [DOI] [PubMed] [Google Scholar]
- 19.Dupre DJ, Baragli A, Rebois RV, Ethier N, Hebert TE. Cell. Signal. 2007;19:481–489. doi: 10.1016/j.cellsig.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 20.Martinez O, Goud B. Biochim. Biophys. Acta. 1998;1404:101–112. doi: 10.1016/s0167-4889(98)00050-0. [DOI] [PubMed] [Google Scholar]
- 21.Duvernay MT, Filipeanu CM, Wu G. Cell. Signal. 2005;17:1457–1465. doi: 10.1016/j.cellsig.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 22.Seachrist JL, Anborgh PH, Ferguson SS. J. Biol. Chem. 2000;275:27221–27228. doi: 10.1074/jbc.M003657200. [DOI] [PubMed] [Google Scholar]
- 23.Filipeanu CM, Zhou F, Lam ML, Kerut KE, Claycomb WC, Wu G. J. Biol. Chem. 2006;281:11097–11103. doi: 10.1074/jbc.M511460200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore RH, Millman EE, Alpizar-Foster E, Dai W, Knoll BJ. J. Cell Sci. 2004;117:3107–3117. doi: 10.1242/jcs.01168. [DOI] [PubMed] [Google Scholar]
- 25.Fan GH, Lapierre LA, Goldenring JR, Richmond A. Blood. 2003;101:2115–2124. doi: 10.1182/blood-2002-07-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seachrist JL, Laporte SA, Dale LB, Babwah AV, Caron MG, Anborgh PH, Ferguson SS. J. Biol. Chem. 2002;277:679–685. doi: 10.1074/jbc.M109022200. [DOI] [PubMed] [Google Scholar]
- 27.Volpicelli LA, Lah JJ, Fang G, Goldenring JR, Levey AI. J. Neurosci. 2002;22:9776–9784. doi: 10.1523/JNEUROSCI.22-22-09776.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roosterman D, Schmidlin F, Bunnett NW. Am. J. Physiol. Cell Physiol. 2003;284:C1319–C1329. doi: 10.1152/ajpcell.00540.2002. [DOI] [PubMed] [Google Scholar]
- 29.Dale LB, Seachrist JL, Babwah AV, Ferguson SS. J. Biol. Chem. 2004;279:13110–13118. doi: 10.1074/jbc.M313333200. [DOI] [PubMed] [Google Scholar]
- 30.Iwata K, Ito K, Fukuzaki A, Inaki K, Haga T. Eur. J. Biochem. 1999;263:596–602. doi: 10.1046/j.1432-1327.1999.00549.x. [DOI] [PubMed] [Google Scholar]
- 31.Murph MM, Scaccia LA, Volpicelli LA, Radhakrishna H. J. Cell. Sci. 2003;116:1969–1980. doi: 10.1242/jcs.00397. [DOI] [PubMed] [Google Scholar]
- 32.Li JG, Benovic JL, Liu-Chen LY. Mol. Pharmacol. 2000;58:795–801. [PubMed] [Google Scholar]
- 33.Wu G, Zhao G, He Y. J. Biol. Chem. 278;2003:47062–47069. doi: 10.1074/jbc.M305707200. [DOI] [PubMed] [Google Scholar]
- 34.Filipeanu CM, Zhou F, Claycomb WC, Wu G. J. Biol. Chem. 2004;279:41077–41084. doi: 10.1074/jbc.M405988200. [DOI] [PubMed] [Google Scholar]
- 35.Filipeanu CM, Zhou F, Fugetta EK, Wu G. Mol. Pharmacol. 2006;69:1571–1578. doi: 10.1124/mol.105.019984. [DOI] [PubMed] [Google Scholar]
- 36.Tisdale EJ, Bourne JR, Khosravi-Far R, Der CJ, Balch WE. J. Cell Biol. 1992;119:749–761. doi: 10.1083/jcb.119.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plutner H, Cox AD, Pind S, Khosravi-Far R, Bourne JR, Schwaninger R, Der CJ, Balch WE. J. Cell Biol. 1991;115:31–43. doi: 10.1083/jcb.115.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allan BB, Moyer BD, Balch WE. Science. 2000;289:444–448. doi: 10.1126/science.289.5478.444. [DOI] [PubMed] [Google Scholar]
- 39.Tisdale EJ, Balch WE. J. Biol. Chem. 1996;271:29372–29379. doi: 10.1074/jbc.271.46.29372. [DOI] [PubMed] [Google Scholar]
- 40.Tisdale EJ. Mol. Biol. Cell. 1997;10:1837–1849. doi: 10.1091/mbc.10.6.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez O, Antony C, Pehau-Arnaudet G, Berger EG, Salamero J, Goud B. Proc. Natl. Acad. Sci. U. S. A. 1997;94:1828–1833. doi: 10.1073/pnas.94.5.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez O, Schmidt A, Salamero J, Hoflack B, Roa M, Goud B. J. Cell Biol. 1994;127:1575–1588. doi: 10.1083/jcb.127.6.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White J, Johannes L, Mallard F, Girod A, Grill S, Reinsch S, Keller P, Tzschaschel B, Echard A, Goud B, Stelzer EH. J. Cell Biol. 1999;147:743–760. doi: 10.1083/jcb.147.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Downes CP, Michell RH. Biochem. J. 1981;198:133–140. doi: 10.1042/bj1980133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoo JS, Moyer BD, Bannykh S, Yoo HM, Riordan JR, Balch WE. J. Biol. Chem. 2002;277:11401–11409. doi: 10.1074/jbc.M110263200. [DOI] [PubMed] [Google Scholar]
- 46.Shetty KM, Kurada P, O'Tousa JE. J. Biol. Chem. 1998;273:20425–20430. doi: 10.1074/jbc.273.32.20425. [DOI] [PubMed] [Google Scholar]
- 47.Grosshans BL, Ortiz D, Novick P. Proc. Natl. Acad. Sci. U. S. A. 2006;103:11821–11827. doi: 10.1073/pnas.0601617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lanctot PM, Leclerc PC, Escher E, Leduc R, Guillemette G. Biochemistry. 1999;38:8621–8627. doi: 10.1021/bi9830516. [DOI] [PubMed] [Google Scholar]
- 49.Montiel M, Jimenez E. J. Mol. Endocrinol. 1998;20:299–304. doi: 10.1677/jme.0.0200299. [DOI] [PubMed] [Google Scholar]
- 50.Dohlman HG, Bouvier M, Benovic JL, Caron MG, Lefkowitz RJ. J. Biol. Chem. 1987;262:14282–14288. [PubMed] [Google Scholar]
- 51.Hughes RJ, Pasillas M, Saiz J, Jasper J, Insel PA. Biochim. Biophys. Acta. 1997;1356:281–291. doi: 10.1016/s0167-4889(97)00005-0. [DOI] [PubMed] [Google Scholar]
- 52.Urbani L, Simoni RD. J. Biol. Chem. 1990;265:1919–1923. [PubMed] [Google Scholar]
- 53.Jourdan N, Maurice M, Delautier D, Quero AM, Servin AL, Trugnan G. J. Virol. 1997;71:8268–8278. doi: 10.1128/jvi.71.11.8268-8278.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toyooka K, Okamoto T, Minamikawa T. J. Cell Biol. 2000;148:453–464. doi: 10.1083/jcb.148.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong C, Zhou F, Filipeanu CM, Wu G. (manuscript submitted) [Google Scholar]