

Abstract
Rational protein design is an emerging approach for testing general theories of structure and function. The ability to manipulate function rationally also offers the possibility of creating new proteins of biotechnological value. Here we use the design approach to test the current understanding of the structural principles of allosteric interactions in proteins and demonstrate how a simple allosteric system can form the basis for the construction of a generic biosensor molecular engineering system. We have identified regions in Escherichia coli maltose-binding protein that are predicted to be allosterically linked to its maltose-binding site. Environmentally sensitive fluorophores were covalently attached to unique thiols introduced by cysteine mutations at specific sites within these regions. The fluorescence of such conjugates changes cooperatively with respect to maltose binding, as predicted. Spatial separation of the binding site and reporter groups allows the intrinsic properties of each to be manipulated independently. Provided allosteric linkage is maintained, ligand binding can therefore be altered without affecting transduction of the binding event by fluorescence. To demonstrate applicability to biosensor technology, we have introduced a series of point mutations in the maltose-binding site that lower the affinity of the protein for its ligand. These mutant proteins have been combined in a composite biosensor capable of measuring substrate concentration within 5% accuracy over a concentration range spanning five orders of magnitude.
Keywords: rational protein design, maltose-binding protein, allostery
Three phenomena illustrate the remarkable degree of functional control displayed by some proteins. First, structurally dissimilar (“allosteric”) ligands can influence the activity of one another. Such allosteric interactions are responsible for controlling most metabolic and cellular signal transduction pathways and therefore play a central role in regulating cellular physiology (1). Second, some proteins bind their ligands sigmoidally, which results in a transition between fully bound and ligand-free forms over a relatively short concentration range. This allows exquisite control over ligand loading, as illustrated by the efficient transport by hemoglobin of oxygen between tissues with high partial oxygen pressure to metabolically active, oxygen-starved tissues. Finally, some proteins are capable of exerting action at a distance. For instance, the binding of a hormone to a receptor at one side of a membrane results in a change of receptor activity at the other side. One of the great triumphs of molecular biology has been the unification of these three apparently disparate phenomena into a single theory of cooperative interactions between binding sites (2–4). Here we demonstrate how a rational design strategy can be derived from simple structural principles to introduce a heterotropically cooperative interaction between ligand binding at one site and activity at another site (in this case, the fluorescence of a fluorophore) in a monomeric protein, Escherichia coli maltose-binding protein (MBP). We further show that the spatial separation of the two sites allows modular engineering at the ligand-binding site without destroying allosteric linkage to the fluorophore and how this can be used to create novel biosensors.
In cooperatively interacting ligand-binding sites, the sites are spatially separated, and the properties of one site (binding constant and catalytic activity) depend on the degree of occupancy by a ligand at another. Cooperativity between identical copies of a site (homotropic cooperativity) results in sigmoidal binding. If dissimilar ligand-binding sites are cooperative with respect to each other (heterotropic cooperativity), allosteric interactions result. Heterotropic cooperativity need not involve sigmoidal binding of any of the ligand species. For instance, if each allosteric site is present in only one copy, as would be the case in a monomeric protein with two interacting binding sites, then binding of a given ligand species at its site is hyperbolic.
Cooperativity arises when a protein exists as two or more states in equilibrium with each other and when the intrinsic binding constant or activity of a site differs between states. The structural basis for this lies in the coupling between local conformational changes at a binding site and global conformational states of the protein. Structural analysis has revealed that most natural allosteric proteins are multimers. In such cases, the global conformational states are quaternary in nature and are formed by the relative arrangements of the individual subunits. There are two ways in which coupling between these states and the local conformations in a binding site can be achieved: direct and indirect. Direct coupling occurs when ligand-binding sites are located within subunit interfaces, formed by residues of at least two subunits. Coupling between the global quarternary states and the ligand-binding sites is the direct consequence of the rearrangement of the intersubunit interfaces, which changes the relative dispositions of the residues forming the binding sites. Bacterial phosphofructokinase is a good example of such a case (5). This structural mechanism gives rise to highly concerted transitions (2). Indirect coupling occurs when ligand-binding sites do not form part of the interface but are contained wholly within a monomer, as is the case in hemoglobin (6). Local conformational changes in a binding site are formed by the different conformations of individual monomers. The equilibrium between the monomer conformations dictates the states of the binding sites. This equilibrium is itself dependent on the differential stability of each monomeric conformational state within the multimer. This mechanism does not require concerted transitions (3, 4). Indeed, in hemoglobin, it is now known that there are several combinations of tertiary and quaternary conformational states within the tetramer (7).
Although most allosteric proteins found in nature are oligomers, there is no fundamental structural reason for cooperativity to arise in multimers only. In general, the simplest, minimal structural requirements for highly concerted, directly coupled cooperative interactions are that two rigid domains in a protein exist in two different conformations relative to each other, that the coupled binding sites are located in the interface between these domains, and that they are formed by residues contributed by both domains. The domains can be either whole protein subunits or rigid subdomains within a monomer that undergoes a large conformational change, such as the hinge-bending motion found in many proteins (8).
MBP is a member of a family of structurally related proteins found in the periplasm of E. coli that is involved in chemotactic response and transport (9, 10). This monomeric protein has a single binding site for maltose and is known to undergo a large conformational change upon ligand binding (11, 12). High-resolution x-ray structures of the apo (open) and bound (closed) forms (13, 14) reveal that this motion involves two domains that undergo a large hinge-twist movement relative to each other. This protein is therefore a good candidate for the introduction of a directly coupled, interdomain allosteric interaction.
Biosensors are analytical tools that harness the remarkable specificity of biomolecular recognition, allowing the determination of the concentration of a single molecular species in a complex mixture (15). Detection occurs at two levels: molecular signal transduction in which the physical properties of a macromolecule change upon analyte binding, and macroscopic signal determination to detect this change. Development of most biosensors involves the identification of a naturally occurring macromolecule (such as an enzyme or antibody) with the required specificity, discovery of a suitable signal, and construction of a detector adapted to the properties of the macromolecule in question. Although effective biosensors have been developed in this way, each device is unique and requires substantial development time and optimization. It would therefore be beneficial to develop a generic molecular engineering system, allowing the facile construction of new biosensors. The molecular signal transduction by an allosteric linkage mechanism described here is well suited for this purpose. It is a homogeneous system that does not require a change in composition for measurement, unlike heterogeneous systems, which consume either the analyte [e.g., glucose detection by glucose oxidase (16, 17)] or a signal transduction component [e.g., displacement of a labeled antigen in a competitive immunoassay (18)]. Spatial separation between an allosterically linked reporter group and ligand-binding site allows the facile isolation of sensor molecules with altered binding properties while retaining a common molecular signal transduction mechanism.
MATERIALS AND METHODS
Molecular Modeling.
Calculations were carried out using the dezymer protein design program (19) on a NeXT computer. Coordinates for the free (13) and maltose-bound (14) forms of MBP were obtained from the Protein Data Bank (references 1OMP and 2MBP, respectively). Molecular drawings were made with molscript (20).
Mutagenesis.
A phagemid containing the malE gene, pMAL-C2 (21), was purchased from New England Biolabs. This vector was modified by oligonucleotide-directed mutagenesis using the uracil method (22) to introduce a BamHI site at the C terminus of the protein, to allow facile cloning of oligonucleotide cassettes in this region. This allowed the reconstruction of the wild-type C-terminal sequence and fusion of a His5 peptide for affinity purification. All subsequent variants are derived from this construct.
Protein Expression and Purification.
The MBP expression constructs lack the wild-type leader sequence and are expressed cytoplasmically at high levels. The C-terminal His5 peptide allows the proteins to be purified using immobilized metal affinity chromatography on an iminodiacetate/zinc column (Pharmacia), so that maltose-free protein is easily prepared in one step (23). Overloaded SDS/polyacrylamide gels stained with Coomassie blue showed only one band. Typical yields were 70 mg of pure protein from 1 liter of fermentation broth.
Fluorophore Coupling.
Iodoacetamide and chloride derivatives of N-((2-(iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diazole (IANBD), 4-chloro-7-nitrobenz-2-oxa-1,3-diazole (CNBD), and acrylodan were purchased from Molecular Probes. The fluorophores were dissolved in acetonitrile and reacted with freshly purified cysteine mutants of MBP (10:1) in 1 M NaCl/50 mM phosphate buffer, pH 7.0, for 5 hr at room temperature. Unreacted fluorophore was separated from protein by gel filtration. The extent of coupling was measured both by determining the remaining free thiol concentration using Ellman’s reagent (24) and from the ratio of the absorbances of the major protein and fluorophore chromophores (ɛ280(MBP) = 69 mM−1⋅cm−1; ɛ420(CNBD) = 13 mM−1⋅cm−1; ɛ469(IANBD) = 23 mM−1⋅cm−1; ɛ392(acrylodan) = 20 mM−1⋅cm−1). Coupling was always found to be >95%. The conjugates were stable at 4°C for a period of months, as determined by maltose binding assays.
Measurement of Maltose Binding.
Maltose binding was determined by measuring changes in fluorescence on a SLM (Urbana, IL) Aminco–Bowman series 2 fluorimeter at 25 ± 1°C. Maltose (Sigma) was titrated stepwise into a 50 nM conjugated protein solution in 0.1 M NaCl/50 mM phosphate, pH 7.0, which was continuously mixed with a magnetic stirrer. For titrations, the excitation and emission slit widths were set to 4 and 16 nm, respectively (tryptophan, λex = 280 nm, λem = 340 nm; CNBD, λex = 420 nm, λem = 530 nm; IANBD, λex = 469 nm, λem = 540 nm; acrylodan, λex = 392 nm, λem = 520 nm). Under these conditions, the instrument noise was <1% of the fluorescence signal observed in saturating solutions of maltose.
Binding Curve Fits.
Experimentally observed binding curves of single proteins were fit to the standard equation describing binding to a single site:
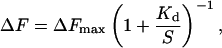 |
1 |
where ΔF is the change in fluorescence, ΔFmax is the fluorescence change at saturating concentrations of maltose, Kd is the binding constant, and S the concentration of maltose. In a composite sensor where n proteins with different binding constants Kd,i but identical maximal fluorescent signal response are mixed together at equimolar concentrations, the total signal is the sum of the individual binding curves:
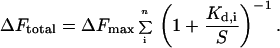 |
2 |
The fractional error in the concentration measurement, δS/S, is related to the first derivative of the binding curve as follows:
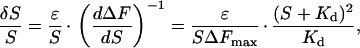 |
3 |
where ɛ is the absolute instrument error in the measurement of a fluorescence signal. The corresponding aggregate error for a composite sensor is given by:
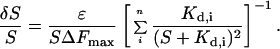 |
4 |
RESULTS AND DISCUSSION
Molecular Modeling.
A difference distance matrix technique (25) was used to analyze the conformational changes in MBP. An N × N distance matrix was calculated of the Cα interatomic distances for the open (Do) and closed (Dc) forms. The difference matrix, ΔD = Do − Dc, records all the changes in distance due to the conformational differences between the two states (Fig. 1, below diagonal line). To extract the local conformational changes, the pairs of interatomic distances were restricted to those that approach within 10 Å of each other in either of the two states (Fig. 1, above diagonal line).
Figure 1.

Dot plot of a double difference distance matrix describing the conformational changes between the open and closed forms of MBP. Below the diagonal line, there is a dot for every pair of Cα atoms that move >4 Å relative to each other. Above the diagonal line, a subset of those pairs, which are within 10 Å of each other in either of the two global conformational states, is shown. This triangle therefore represents the regions of local conformational changes. Groups A–D are spatially separated from the maltose-binding pocket and are predicted to be allosterically linked. Groups 1–4 are either too close to the maltose-binding site (1 and 2) or form part of the partially disordered N terminus (3 and 4).
Eight regions that undergo appreciable local conformational change were identified in this way. Two of these (regions 1 and 2) are very close to the maltose-binding site (at least one of the atoms in the side chains is within 8 Å of at least one of the maltose atoms) and are therefore not sufficiently separated to satisfy spatial independence. Regions 3 and 4 are not located in the interdomain interface and were therefore also not considered further. The remaining four regions, A–D, are located at least 11 Å from the maltose-binding site and involve packing rearrangements between secondary structure elements near the surface of the protein, forming a band defining the interface between the two large subdomains of MBP (Fig. 2). All contain residues contributed by both subdomains. These regions therefore satisfy all conditions that are predicted to give rise to allosteric linkage to the maltose-binding site.
Figure 2.

MBP undergoes a large conformational change upon ligand binding. This involves a relative rearrangement of large rigid subdomains (I and II) best described as a combined hinge-twist movement. The maltose-binding site is formed by the interface between the two subdomains. The shaded spheres (A–D) indicate the regions that are predicted to be allosterically linked to maltose binding.
Allosteric Linkage.
The allosteric coupling between the maltose-binding site and the set of predicted “protoallosteric” sites was tested using a combined mutagenesis and semisynthetic strategy. Environmentally sensitive fluorescent dyes were covalently attached to unique thiols introduced in three of the four sites by constructing cysteine point mutations of residues located in these regions. Any attempt at the prediction of locations with the highest response would require a detailed molecular simulation of the conformational ensembles of the fluorophores in the presence of solvent, a nontrivial proposition. We therefore constructed several different cysteine mutations in a given region to establish empirically which mutation gives the most pronounced changes. In the case of a protoallosteric site composed of many residues, such as site B, we selected mutations based on the largest, maltose binding-induced changes in calculated solvent-accessible areas (26) of the residues. Three different fluorophores were used to probe the sites: two 7-nitrobenz-2-oxa-1,3-diazole derivatives with different linker lengths (IANBD and CNBD) and acrylodan. The prediction is that if a site is allosterically coupled to the maltose-binding site and if the attached fluorophore is appropriately placed within the site so that its environment is sufficiently perturbed by the local conformational changes to give a change in fluorescence, then the fluorescence of the conjugated protein should change heterotropically cooperatively with respect to maltose.
The protoallosteric sites all showed a change in fluorescence of at least one of their conjugates upon ligand binding (Table 1). To establish that this response is due to coupling to maltose binding and not some other effect the change in fluorescence was measured as a function of maltose concentration. This revealed that, in each case, an asymptotic, single-site binding curve for maltose could be determined by following the fluorescence of the conjugated fluorophore (Fig. 3). Furthermore, there was no response in the presence of glucose and sucrose. Together, these results indicate that the sites are heterotropically cooperative with respect to specific binding of maltose as predicted.
Table 1.
Properties of the mutant proteins
| Construct | Mutation | dmin, Å | ΔA, Å2 | R(acrylodan) | R(CNBD) | R(IANBD) | Kd, μM | Kd(IANBD), μM |
|---|---|---|---|---|---|---|---|---|
| Wild-type | — | — | — | — | — | — | 0.9 | — |
| A1 | Phe-92 → Cys | 15 | +51 | 1.5 | 0.92 | 0.84 | 0.6 | 0.3 |
| A2 | Ile-329 → Cys | 11 | +37 | 1.0 | 1.0 | 1.8 | 1.0 | 0.1 |
| B1 | Asn-100 → Cys | 19 | +50 | 0.85 | 1.0 | 1.0 | 1.0 | — |
| B2 | Arg-98 → Cys | 17 | +31 | 1.0 | 1.0 | 1.05 | 1.1 | — |
| B3 | Asp-95 → Cys | 17 | +52 | 0.8 | 1.0 | 4.4 | 1.5 | 1.4 |
| C1 | Ser-233 → Cys | 14 | −48 | 0.85 | 1.4 | 2.0 | 2.0 | 11.0 |
Figure 3.

Change in fluorescence of IANBD conjugated to the B3 variant upon maltose binding. The experimental points of the binding curve have been fit to Eq. 1. (Inset) Changes in the emission spectrum. – – –, No maltose; ——, addition of 1.3 mM maltose.
The responses of different sites within a protoallosteric region and different fluorophores coupled at a particular site vary widely, ranging from a 4.4-fold increase in fluorescence of IANBD attached at position B3 to a complete lack of response of all fluorophores at position B2. The degree of coupling between changes in local conformation and changes in fluorescence is therefore highly dependent on the mechanism of environmental sensitivity. Acrylodan is sensitive to polarity of the environment (27). The 7-nitrobenz-2-oxa-1,3-diazole derivatives are sensitive to quenching by solvent (28) and differ from each other in the length of their linker. However, in each of the three regions, one site could be identified that gave a good response with IANBD (A2, B3, and C1). The best response (4.4-fold at B3) was found in a region with the most extensive number of interfacial contacts between the two domains, which would therefore be expected to be the most extensively coupled region, based on the simple steric model of allosteric behavior.
In allosterically linked sites, the binding behavior of a ligand at one site should be dependent on the degrees of occupancy at the other. We have already established that this is true for the fluorophore site. The converse also holds: comparison of the maltose binding constants in the presence and absence of conjugated fluorophore shows that, in each case, the Kd for maltose depends on the fluorophore (Table 1). Most interestingly, in three out of the four cases, the Kd decreases, indicating that the protein has a stronger affinity for maltose in the presence of the fluorophore (in one instance, IANBD coupled to A2, by one order of magnitude), which in turn implies that the fluorophore stabilizes the closed form of the protein. The one exception to this is conjugation of a fluorophore to the C1 site, which results in a marked increase in the Kd for maltose, suggesting that the reporter group sterically interferes with maltose binding.
Spatial separation between the binding sites also implies that the intrinsic binding properties of each should be independent of the other when unoccupied. Independence of the maltose-binding site from the engineered fluorophore sites is revealed by the effect of the single cysteine mutations on the Kd of maltose (Table 1), measured in the absence of conjugated fluorophore. The largest change is a 2-fold increase (C1); three out of five show no change within the error of the measurement; and two show a 1.5-fold decrease (A2) or increase (B3). This indicates that the intrinsic binding of maltose is independent of most of the engineered sites, with the possible exception of the C1 site.
Taken together, these results demonstrate that the intrinsic properties of the maltose-binding pocket are sterically independent of sites A and B, but not site C. The locations of these three sites relative to the maltose-binding pocket provides a possible explanation for this observation. The maltose-binding pocket and the A and B sites are located on opposite sides of the two β-strands, which form the hinge region connecting the two protein domains. The hinge therefore forms a physical barrier between these sites, ensuring total steric independence. In contrast, the C site is located on the same side of the hinge, at the tip of the “jaws” that form the maltose-binding pocket. It is therefore much more likely to interfere directly with maltose binding. This direct interaction has been used to measure ligand binding by a quasicompetitive, rather than allosteric, linkage method. Cysteine mutations have been constructed in the jaw region of both MBP (29) and its structural homologue, phosphate-binding protein (30). Environmentally sensitive fluorophores conjugated to these mutants respond to ligand but also increase the substrate Kd substantially, indicating steric interference.
Modular Manipulation of the Maltose-Binding Site.
A further consequence of the allosteric coupling mechanism is that the intrinsic binding affinity and specificity of the maltose-binding pocket should be independently manipulable without destroying linkage with the fluorophore. To test this, three tryptophans in the binding pocket (at positions 62, 230, and 340) that form van der Waals contacts with maltose were mutated singly to alanine in the B3 background, the site which showed the largest fluorescence signal in response to maltose binding. These mutations were chosen because they had been shown previously to decrease the affinity for maltose (31). The IANBD conjugates of all three variants retained the same level of fluorescent response as the original B3 protein within experimental error. The binding constants of the mutants were determined by following the IANBD fluorescence (Fig. 4A) and were found to have increased as expected. The mutants do not respond to glucose or sucrose and are therefore still specific for maltose. This demonstrates that the maltose-binding pocket can be manipulated independently without destroying allosteric linkage to the B3 site, as predicted.
Figure 4.

Maltose binding curves for the B3 protein and three point mutants determined by measuring IANBD fluorescence. (A) Binding curves for each individual curve (fit to Eq. 1: B3, •; B3/W230A, ▪; B3/W62A, ♦; B3/W340A, ▴). (B) Response of a composite sensor with all four proteins mixed in equimolar proportions (total protein concentration, 2 μM; fit to Eq. 2). (C) Error analysis (dashed line indicates 5% error level) of the individual proteins (lightface lines, simulated with Eq. 3) compared with the aggregate error of the composite sensor (boldface line, simulated with Eq. 4) using the same total protein concentration.
Construction of a High Dynamic Range “Composite” Biosensor.
The binding constants of the original B3 variant and the three single tryptophan mutants are spaced at approximately one order of magnitude intervals (B3, 1.4 μM; B3/W230A, 22 μM; W62A, 154 μM; W340A, 2800 μM). This allows the accurate determination of the concentration of maltose over a large continuous range of concentrations by mixing these four proteins together in equimolar proportions in a single cuvette. The resulting composite biosensor is capable of determining the concentration of maltose to within 5% accuracy over a five orders of magnitude (Fig. 4 B and C), spanning a concentration range of 0.1–20 mM.
CONCLUSION
Generalization of the observed structural mechanisms of cooperative interactions results in the prediction that in any protein in which there are several rigid units (monomers in a multimer or domains in a monomer) that rearrange relative to each other, it should be possible to establish allosteric linkage between two sites, provided that both sites are located within the interface between two such units and that residues forming a site are contributed by both units. Here we have used a rational design approach to test this prediction by constructing a novel allosteric interaction in MBP, a monomeric protein known to undergo a large conformational change upon ligand binding, resulting in the rearrangement of two rigid domains. We have used the x-ray structures of both forms of MBP to identify regions located in the interdomain interface and spatially separated from the maltose-binding site, which undergo local conformational changes and which are comprised of residues contributed by both domains. We predict such regions to be allosterically linked to maltose binding. We have experimentally demonstrated the reliability of these predictions by showing that the fluorescence of fluorophores covalently attached in the predicted sites is heterotropically cooperative with respect to maltose binding.
One of the consequences of spatially separated, allosterically linked sites is that it should be possible to manipulate the sites independently. We have demonstrated this by constructing a series of mutants in the maltose-binding site that had lowered affinity but retained allosteric coupling. These mutants can be combined to construct a composite biosensor capable of determining the concentration of maltose over a range spanning five orders of magnitude. The next challenge is to change the specificity of MBP without affecting allosteric signal transduction. This could allow the construction of a generic biosensor engineering system in which specificity for different analytes can be systematically changed without requiring development of new detection technologies.
The relative movement of two rigid subdomains separated by a hinge region is a common mechanism for conformational change in a number of enzymes (8). Antibodies undergo a similar conformational change upon ligand binding, in which the variable domain moves relative to the constant domain (32). Under favorable circumstances, it should therefore be possible to create similar allosterically linked sites in antibodies, which could be used either to monitor or modulate their binding properties.
Acknowledgments
We thank Dr. C. A. Fierke for generously allowing us to use her fluorimeter and Dr. T. G. Oas for stimulating discussions. This work was supported by a grant from the Whitaker Foundation to H.W.H. J.S.M. acknowledges partial support by National Institutes of Health Training Grant GM08558.
ABBREVIATIONS
- MBP
maltose-binding protein
- IANBD
N-((2-(iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diazole
- CNBD
4-chloro-7-nitrobenz-2-oxa-1,3-diazole
References
- 1.Perutz M F. Mechanisms of Cooperativity and Allosteric Regulation in Proteins. Cambridge, U.K.: Cambridge Univ. Press; 1990. [DOI] [PubMed] [Google Scholar]
- 2.Monod J, Wyman J, Changeux J P. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 3.Koshland D E, Nemethy G, Filmer D. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 4.Hammes G G, Wu C-W. Science. 1971;172:1205–1211. doi: 10.1126/science.172.3989.1205. [DOI] [PubMed] [Google Scholar]
- 5.Schirmer T, Evans P R. Nature (London) 1990;343:140–145. doi: 10.1038/343140a0. [DOI] [PubMed] [Google Scholar]
- 6.Perutz M F, Fermi G, Luisi B, Shaanan B, Liddington R C. Acc Chem Res. 1987;20:309–321. doi: 10.1101/sqb.1987.052.01.063. [DOI] [PubMed] [Google Scholar]
- 7.Ackers G K, Doyle M L, Myers D, Daugherty M W. Science. 1992;255:54–63. doi: 10.1126/science.1553532. [DOI] [PubMed] [Google Scholar]
- 8.Gerstein M, Lesk A M, Chothia C. Biochemistry. 1994;33:6739–6749. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 9.Higgins C F. Annu Rev Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- 10.Ames G F-L. Annu Rev Biochem. 1986;55:397–425. doi: 10.1146/annurev.bi.55.070186.002145. [DOI] [PubMed] [Google Scholar]
- 11.Zukin R S. Biochemistry. 1979;18:2139–2145. doi: 10.1021/bi00578a001. [DOI] [PubMed] [Google Scholar]
- 12.Zukin R S, Hartig P R, Koshland D E. Proc Natl Acad Sci USA. 1977;74:1932–1936. doi: 10.1073/pnas.74.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharff A J, Rodseth L E, Spurlino J C, Quiocho F A. Biochemistry. 1992;31:10657–10663. doi: 10.1021/bi00159a003. [DOI] [PubMed] [Google Scholar]
- 14.Spurlino J C, Lu G-Y, Quiocho F A. J Biol Chem. 1991;266:5202–5219. doi: 10.2210/pdb1mbp/pdb. [DOI] [PubMed] [Google Scholar]
- 15.Hall E E H. Biosensors. Englewood Cliffs, NJ: Prentice–Hall; 1991. [Google Scholar]
- 16.Teller J D. 130th Meeting of the American Chemical Society. Washington, DC: Am. Chem. Soc.; 1956. [Google Scholar]
- 17.Keston A S. 129th Meeting of the American Chemical Society. Washington, DC: Am. Chem. Soc.; 1956. [Google Scholar]
- 18.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 19.Hellinga H W, Richards F M. J Mol Biol. 1991;222:763–785. doi: 10.1016/0022-2836(91)90510-d. [DOI] [PubMed] [Google Scholar]
- 20.Krollis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 21.Maina C V, Riggs P D, Grandea A G, Slatko B E, Moran L S, Tagemonte J A, McReynolds L A, Guan C D. Gene. 1988;74:365–373. doi: 10.1016/0378-1119(88)90170-9. [DOI] [PubMed] [Google Scholar]
- 22.Kunkel T A. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hochuli E, Bannwarth W, Döbeli H, Gentz R, Stuber D. Bio/Technology. 1988;6:1321–1325. [Google Scholar]
- 24.Ellman G L. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 25.Nishikawa K, Ooi T, Ysogai Y, Saito N. J Phys Soc Jpn. 1972;32:1331–1337. [Google Scholar]
- 26.Lee B, Richards F M. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 27.Prendergast F G, Meyer M M, Carlson G L, Iida S, Potter J D. J Biol Chem. 1983;259:7541–7544. [PubMed] [Google Scholar]
- 28.Ghosh P B, Whitehouse M W. Biochem J. 1968;108:155–156. doi: 10.1042/bj1080155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilardi G, Zhou L Q, Hibbert L, Cass A E G. Anal Chem. 1994;66:3840–3847. doi: 10.1021/ac00093a047. [DOI] [PubMed] [Google Scholar]
- 30.Brune M, Hunter J L, Corrie J E T, Webb M R. Biochemistry. 1994;33:8262–8271. doi: 10.1021/bi00193a013. [DOI] [PubMed] [Google Scholar]
- 31.Martineau P, Szmelcman S, Spurlino J C, Quiocho F A, Hofnung M. J Mol Biol. 1990;214:337–352. doi: 10.1016/0022-2836(90)90165-I. [DOI] [PubMed] [Google Scholar]
- 32.Lesk A M, Chothia C. Nature (London) 1988;335:188–190. doi: 10.1038/335188a0. [DOI] [PubMed] [Google Scholar]