

Summary
Non-specific, extracellular nucleases have received enhanced attention recently as a consequence of the critical role that these enzymes can play in infectivity by overcoming the host neutrophil defense system. The activity of the cyanobacterial nuclease NucA, a member of the ββα Me superfamily, is controlled by the specific nuclease inhibitor NuiA. Here we report the 2.3 Å resolution crystal structure of the NucA:NuiA complex, showing that NucA inhibition by NuiA involves an unusual divalent metal ion bridge that connects the nuclease with its inhibitor. The C-terminal Thr135NuiA hydroxyl oxygen is directly coordinated with the catalytic Mg2+ of the nuclease active site, and Glu24NuiA also extends into the active site, mimicking the charge of a scissile phosphate. NuiA residues Asp75 and Trp76 form a second interaction site, contributing to the strength and specificity of the interaction. The crystallographically-defined interface is shown to be consistent with results of studies using site-directed NuiA mutants. This mode of inhibition differs dramatically from the exosite mechanism of inhibition seen with the DNase colicins E7/E9 and from other nuclease-inhibitor complexes that have been studied. The structure of this complex provides valuable insights for the development of inhibitors for related non-specific nucleases that share the DRGH active site motif such as the Streptococcus pneumoniae nuclease EndA, which mediates infectivity of this pathogen, and mitochondrial EndoG, which is involved in recombination and apoptosis.
Keywords: Nuclease A, NuiA, nuclease-inhibitor complex, non-specific nuclease, molecular mimicry
Introduction
Non-specific nucleases are involved in a broad range of functions that includes extra- and intracellular digestion, programmed cell death, defense, replication, recombination, and repair (1–3). They also have proven useful for the determination of nucleic acid structures, mapping mutations, studying the interaction of DNA and RNA with various ligands (4), and for RNA sequencing (5). Most recently, an important role for these nucleases in microbial infectivity has been demonstrated, based on their ability to digest the DNA component of host neutrophil extracellular traps (NETs) (6–8). Consequently, these nucleases are now recognized as significant drug targets, and information related to the inhibition of these enzymes is of potential use for inhibitor development. As a result of their ability to degrade nucleic acids non-specifically, they also represent an endogenous toxic challenge. Therefore, regulation of their activity is critical for the cells that produce them.
The ββα Me superfamily of nucleases (9) comprises non-specific, structure-specific and sequence-specific enzymes that share a structurally-conserved active site scaffold and utilize a divalent metal ion. They can be grouped according to sequence motifs into three families: His-Cys box nucleases (e.g., I-PpoI, (10)), HNH nucleases (e.g., colicins E7 and E9 (11,12) and I-HmuI, (13)) and DRGH nucleases (e.g. the extracellular nuclease from Serratia marcescens (14), the DNA entry nuclease EndA from Streptococcus pneumoniae (15), Syncephalastrum racemosum nuclease(16), nuclease C1 from Cunninghamella echinulata (17), yeast Nuc1 (18), mitochondrial EndoG (19), and the Anabaena nuclease NucA (20)). Whereas the eukaryotic nucleases of the DRGH family represent the major mitochondrial nuclease activity, the prokaryotic members of this family are responsible for extracellular DNA degradation. Notably the DNA-entry nucleases EndA from Streptococcus pneumoniae and the related Streptodornase (Sda1) from Streptococcus sp. allow their host organisms to escape from neutrophil extracellular traps by digesting the DNA-scaffold of these structures thereby evading the first line of defense against microbial infection in mammals (6–8).
NucA, a member of the DRGH family, is one of the most potent nucleases known, and degrades both single and double-stranded DNA and RNA. Its activity is regulated by a potent and specific protein inhibitor, NuiA, that forms a tight 1:1 complex, Ki = 1 × 10−12 M(21). The structure of the active site is closely analogous to that of the Serratia nuclease (22,23), while the activity of the Serratia enzyme is dependent on the presence of cystine bonds and hence is determined by the redox level of the medium (24). A deletion analysis had shown that N- and C-terminal residues, directly or indirectly, are involved in the NucA-NuiA interaction (21). Nevertheless, the molecular basis for the strong inhibitory interaction has not yet been determined.
In comparison with the vast literature on proteinase inhibitors, nuclease inhibitors have received relatively little study. The most detailed investigations have focused on the Bacillus amyloliquefaciens RNase (barnase) inhibitor (barstar) (25), the RNaseA inhibitor (RI) (26), and the immunity proteins that protect E. coli from the colicin DNase activity (27), (28). Consideration of the structures of these nuclease-inhibitor complexes as well as the structure of the NucA-NuiA complex determined in the present study suggests few common modes of inhibition. As result of the recently recognized role of the non-specific nucleases in the infectivity process via their action on neutrophil extracellular traps, they have emerged as attractive targets for drug development (7,8).
Here we present the crystal structure of NucA (28 kDa) in complex with NuiA (15 kDa) at a resolution of 2.3 Å. Many of the features of the NucA-NuiA complex are unique. NuiA interacts directly with residues in the active enzyme site, displaying target site mimicry, and interacting directly with the active site Mg2+ ion through coordination with the C-terminal Thr135Nui residue. Binding of NuiA results in no significant change of the backbone atoms of NucA (22) but in several minor sidechain rearrangements. The structure of NucA-complexed NuiA shows some differences relative to the previously determined solution structure (29) of the uncomplexed inhibitor, primarily in the loop regions.
Methods
Protein Expression and Purification
The recombinant NucA construct, containing a D121A mutation to reduce activity and related cellular toxicity, lacking the N-terminal export signal peptide, and containing an N-terminal His-tag to facilitate purification, was produced as described previously (22). Recombinant NuiA, also containing an N-terminal His-tag, was similarly produced as described previously (29). E. coli cells containing the required plasmid were grown to mid-log phase (A600 ~ 0.6) at 37 °C in LB medium containing 30 μg/ml kanamycin. NuiA protein expression was induced with 0.4 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 37 °C for 6 hrs. Cells were harvested by centrifugation (at 7000 g), resuspended in 20 mM Tris/HCl, pH 8.0, 100 mM NaCl and lysed by sonication with a Branson Sonifier 200 using a microtip probe. The lysate was centrifuged at 30,000 g for 40 minutes. The supernatant was applied to a Ni2+-NTA resin (Qiagen) equilibrated with extraction buffer and eluted with 20 mM Tris/HCl, pH 8.0, 100 mM NaCl, 200 mM imidazole. The eluted fractions containing NuiA protein were then concentrated using a Millipore concentrator to a desired volume of 10 ml (~10 mg/ml concentration) and applied to the Superdex-75 gel filtration 2.6 × 60 cm column equilibrated with 20 mM Tris–HCl pH 8.0, 100 mM NaCl. The protein corresponding to the major absorbance peak at 280 nm was found to be pure NuiA, as judged by SDS/polyacrylamide gel electrophoresis.
The N-terminal His-tags on both proteins were cleaved by overnight incubation of the protein samples with thrombin (Novagen) at a concentration of 50 U/100 ml at 4°C. The preparations were then once again passed through Ni2+-NTA resin (Qiagen) equilibrated with 50 mM Tris/HCl, pH 8.0, 200 mM NaCl to remove any residual His-tagged protein as well as the cleaved N-terminal His-tag. The two proteins were then mixed together and applied to a Superdex-200 gel filtration 2.6 × 60 cm column previously equilibrated with 50 mM Tris-HCl pH 7.5, 200 mM NaCl and 2 mM DTT. The peak corresponding to the NucA-NuiA complex was identified by SDS/polyacrylamide gel electrophoresis.
Inhibition of NucA by NuiA-variants
The DNA-cleavage activity of NucA in the absence and presence of NuiA-variants was measured with a plasmid DNA cleavage assay. NucA at a final concentration of 5 nM was incubated alone or in the presence of a 5-fold molar excess of GST-tagged NuiA-variants with 24 ng/μl plasmid DNA (pBSK-VDEX, New England Biolabs) in a total volume of 80 μl at 37°C in a buffer consisting of 50 mM Tris-HCl, pH 8.2 and 5 mM MgCl2. After 0.5, 1, 2, 4, and 8 min, aliquots of 8 μl were taken from the reaction mixture and immediatedly mixed with gel loading buffer to quench the cleavage reaction. As a control, plasmid DNA was incubated under the same conditions for 8 min but without nuclease. DNA cleavage reactions were then analysed by electrophoresis on 0.8 % agaorse gels in Tris-borate-EDTA-buffer followed by ethidium bromide staining. In addition, we used GST-pull down assays to assess the structural integrity of the NuiA variants in order to easily distinguish between desired localized structural effects that should still allow complex formation and more global structural rearrangements introduced by the amino acid exchange that might prevent complex formation. To this end NucA and GST-NuiA were mixed at a final concentration of 6 μM in a total volume of 60 μl in a buffer consisting of 10 mM Tris-HCl, pH 8.2, 100 mM NaCl and 0.01 % Triton-X100.
Crystallization and data collection
The NucA-NuiA complex purified by gel filtration chromatography was concentrated to 9 mg/ml and exchanged into 25 mM Tris pH 7.5, 100 mM NaCl, 2 mM DTT buffer. Crystals of the protein complex were obtained using the hanging drop vapor diffusion technique at 4 °C by mixing 2 μl of the protein solution with 2 μl of the reservoir solution consisting of 100 mM MES pH 5.5 and (17–21) % PEG 6000. The crystals were transferred to 100 mM MES pH 5.5, 100 mM NaCl and 20 % PEG 6000 buffer and soaked in 100 mM MES pH 5.5, 100 mM NaCl, 20% PEG 6000 and 20% ethylene glycol as cryoprotectant.
For data collection, crystals were flash cooled by submersion in liquid nitrogen and placed on the goniometer in a stream of nitrogen gas cooled to −180 °C. A lower resolution data set was collected at 2.9 Å using a Rigaku 007HF X-ray generator equipped with a Saturn 92 CCD. A higher resolution data set was then collected at 2.3 Å at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Crystals of the NucA:NuiA complex belong to space group P 43212 and contain one molecule each of NucA and NuiA in the asymmetric unit.
Structure determination and refinement
The crystal structure of NucA (pdb id code: 1ZM8 (22) was used as the model for molecular replacement using the 2.9 Å resolution data set. The program Molrep (30) from CCP4 (31) was used to calculate phases by molecular replacement. The model for NuiA was built into the electron density following the trace of the NMR structure for NuiA (pdb id code 1J57 (29) that had been placed manually into the electron density. The model was then refined against the 2.3 Å data set by iterative cycles of model building using the program O(32) and refinement using the program CNS (33). The quality of the final structure was assessed using the programs Procheck (34) and Molprobity (35). The final model includes residues 34-274 of NucA and 5-135 of NuiA. The statistics for the data collection and results from refinement are reported in Table I. The structure of the NucA:NuiA enzyme complex has been submitted to the Protein Data Bank (accession number:ABCD).
Table I.
| Crystallographic data statistics | |
|---|---|
| data set | NucA-NuiA |
| unit cell | a= b= 87.23 c= 138.98 α= β= γ=90° |
| Space Group | P4(3)2(1)2 |
| Resolution (Å) | 50-2.3 |
| # of observations | 283,171 |
| unique reflections | 24,604 |
| Rsym(%)(last shell)1 | 10.0 (57.3) |
| I/σI (last shell) | 9.4 (3.0) |
| mosaicity | 0.23–0.77 |
| completeness(%) (last shell) | 99.7 (97.5) |
| Refinement statistics | |
| Rcryst(%)2 | 20.1 |
| Rfree(%)3 | 24.1 |
| # of waters | 229 |
| Mean B value (Å) | 46.9 |
| r.m.s. deviation from ideal values | |
| bond length (Å) | 0.008 |
| bond angle (°) | 1.3 |
| dihedral angle (°) | 24.0 |
| improper angle (°) | 0.82 |
| Ramachandran Statistics4 | |
| residues in: | |
| favored (98%) regions (%) | 95.8 |
| allowed (>99.8%) regions (%) | 100 |
Rsym = Σ (| Ii − < I>|)/Σ(Ii) where Ii is the intensity of the ith observation and <I> is the mean intensity of the reflection.
Rcryst = Σ|| Fo| − | Fc ||/Σ| Fo| calculated from working data set.
Rfree was calculated from 5% of data randomly chosen not to be included in refinement.
Ramachandran results were determined by MolProbity.
Results
Crystal structure of the NucA-NuiA complex
The crystal structure of the NucA-NuiA complex was determined at 2.3 Å resolution using recombinant NucA and NuiA expressed in E. coli (Table I). A ribbon model representing the secondary structure of the complex is shown in Figure 1A. As seen in the previously determined structure of NucA (22), the secondary structure is composed of 13 α helices and two β sheets. The root mean square deviation between the complexed and free NucA structure is 0.34 Å for all Cα atoms. A divalent metal ion is located in the active site of NucA and two additional divalent metal ions were observed in the secondary metal ion binding site previously identified in the NucA structure (22). The coordination geometry of the active site metal ion is consistent with Mg2+, the endogenous catalytic metal. Alternatively, the identities of the two metal ions at the secondary site were found to be most consistent with Ni2+ ions. These were presumably acquired or exchanged into the NucA molecule as a result of passage through the Ni2+-NTA column (36). Consistent with the previously determined solution structure (29), NuiA consists of four helices and a central six-stranded β-sheet, arranged as an αβα sandwich. Helices A, C, and D are positioned on one side of the central β sheet, and the distorted helix B is positioned on the other side. The three central strands of the β sheet are located at the C-terminus of the protein, and all six of the strands are antiparallel.
Figure 1. Overall fold of the NucA-NuiA complex.
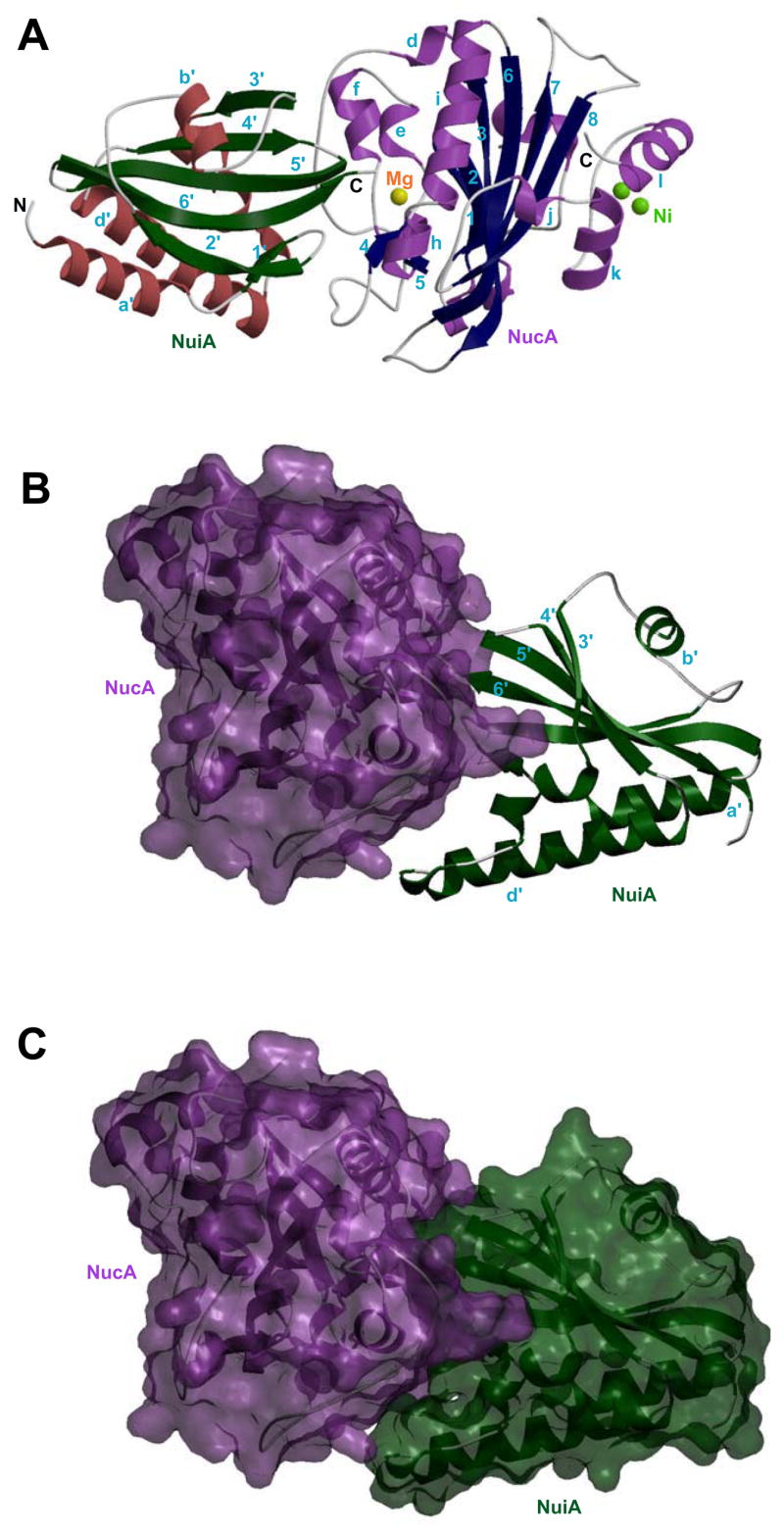
A) Ribbon diagram showing the crystal structure of NucA-NuiA complex. The two-stranded and six-stranded β-sheets (navy) and 13 α-helices (lavender) form the framework of NucA, and the 6 stranded β-sheet (dark green) and 4 helices (brick red) constitute the framework of the NuiA molecule. The active site metal in NucA is a magnesium ion (yellow) and the secondary divalent metal ion binding site contains two proposed nickel ions, shown in green. Structure of the NucA-NuiA complex, B) using a GRASP (semi transparent) surface for NucA (lavender), and ribbon diagram for NuiA (dark green) and C) semi transparent GRASP surfaces for both molecules depicting the “open jaw” structure of NuiA.
In the previously determined structure of NucA, the secondary metal ion binding site was found to constitute a lattice contact, with two carboxyl oxygen atoms of a Glu136Nuc residue on a symmetry-related NucA molecule making contact with both metal ions in the secondary metal ion binding site (22). In the structure of the NucA-NuiA complex, there is also a lattice contact involving the secondary metal ion binding site on NucA, and the Asp87Nui residue of a second, symmetry-related NuiA molecule.
The NucA-NuiA interface
The structure of NuiA can be described as an ‘open jaw’ biting into one side of the NucA molecule in the complex (Figure 1B & 1C). The Thr135 and Glu24 residues of the upper jaw enter the NucA active site, while Asp75 and Trp76 residues of the lower jaw engulf a protruding section of the nuclease that includes a long loop of NucA running from Arg93Nuc into the beginning of β-strand 4 (Arg122Nuc) (Figure 2A). Close contact is also made with several residues on coaxial helices H (Thr151Nuc-Asn155Nuc) and I (Thr158Nuc-Gln172Nuc) of NucA, including Asn155Nuc and Glu163Nuc (Figure 2B & 2C). The major hydrogen bonding and salt bridge interactions between NucA and NuiA are summarized in Table II and illustrated in Figure 2A. These include salt bridge interactions between Arg93Nuc and Glu24Nui, Arg93Nuc and the C-terminal Thr135Nui carboxyl oxygen, and Lys101Nuc and Asp75Nui, in addition to a network of direct and water-mediated hydrogen bonding interactions. The solvent accessible surface areas calculated individually are 10200 Å2 for NucA and 7488 Å2 for NuiA. After complex formation, the buried solvent accessible surface area at the interface is calculated to be 1391 Å2.
Figure 2. Interface and active site of the NucA-NuiA complex.
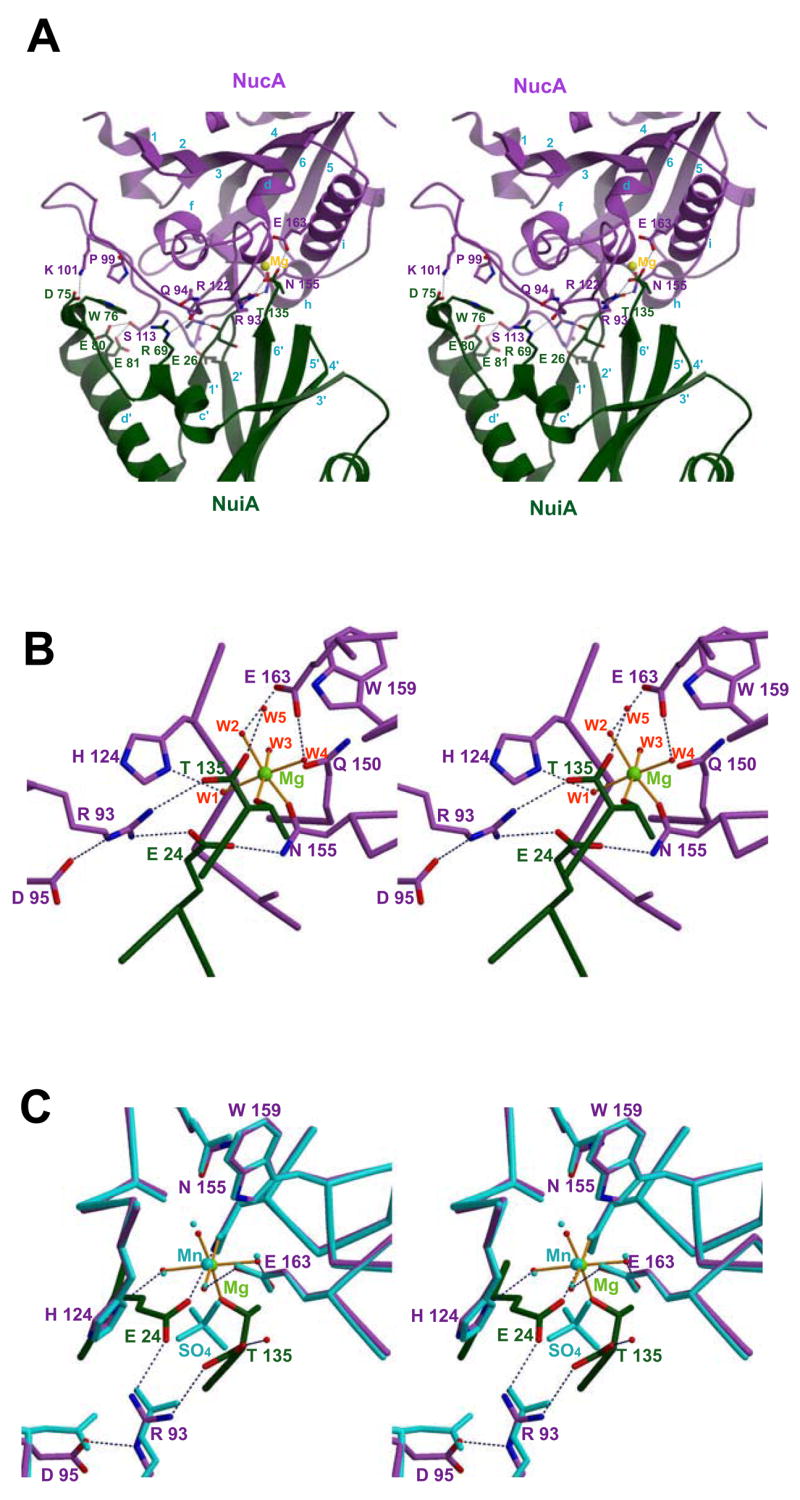
A) A stereoview showing the interface residues involved in salt bridge, hydrogen bonding or stacking interactions in the NucA (lavender) –NuiA (dark green) complex. The residues shown explicitly correspond to some of those identified as contributing to the interface as summarized in Table II. NucA residues are in lavender. B) A stereoview of the active site of NucA-NuiA complex showing divalent metal ion (in green) and the six coordinating ligands showing its octahedral coordination to N155Nuc (lavender), four water molecules (W1–W4), and to the T135Nui residue of NuiA (dark green), illustrated by orange lines. C) Stereo view of the active site residues of NucA-NuiA complex (lavender & dark green) and NucA (cyan), including their respective metal ions Mg2+ in complexed NucA (green) and Mn2+ in uncomplexed NucA (cyan). The OE1 and OE2 atoms of E24 and OG1 atom of T135Nui (darkgreen) occupy the same positions of the oxygen atoms of the sulfate ion of uncomplexed NucA (cyan). The dotted lines shown in navy represent the network of hydrogen bonds in all the panels.
The substantial electrostatic contribution to the NucA-NuiA interaction is illustrated by the GRASP-generated surfaces in Figure 3A & 3B. In order to reveal the electrostatic potential of the interface, NucA (NuiA) is represented by its electrostatic surface, while NuiA (NucA) is represented using a ribbon diagram. The electrostatic representations in Figure 3 are consistent with the entries in Table II, which include one acidic and five basic residues for NucA, and one basic and seven acidic residues for NuiA. This electrostatic pattern is consistent with the proposal that to a significant extent, NuiA binds to NucA as a substrate mimic. The principal hydrophobic contributions to the interface include: Phe97Nuc -Glu24Nui (methylenes), Pro99Nuc -Trp76Nui, and several other sidechain interactions, however, there is no concentration of hydrophobic residues in the interface. The structure of the complex also contains two bound MES molecules, one of which contacts both NucA and NuiA. Its primary contacts include Glu92Nuc and Arg106Nui.
Figure 3. Electrostatic surfaces of NucA and NuiA in the complex.

Electrostatic surface rendering of: (A) NucA, and (B) NuiA generated using GRASP. The protein:protein interface is revealed by using a coiled (yellow) representation of NuiA in panel (A) and NucA in panel (B), respectively.
Table II.
Hydrogen bonding interaction* between NucA and NuiA
| NucA residues | Distance (Å) | Water | Distance (Å) | NuiA residues |
|---|---|---|---|---|
| Arg 93 NH2 | 3.0 | Glu24 OE2 | ||
| Arg93 NH1 | 3.1 | Thr135 O | ||
| Gln94 O | 3.3 | Arg69 NE | ||
| Lys101 NZ | 3.1 | Asp75 OD2 | ||
| Ser113 OG | 3.3 | Glu80 OE1 | ||
| Ser113 OG | 3.4 | Glu81 OE2 | ||
| Gly117 N | 3.2 | Glu26 OE1 | ||
| Arg122 NH1 | 3.0 | Ser23 O | ||
| Arg122 N | 2.8 | Glu24 O | ||
| Asn155 ND1 | 2.8 | Glu24 OE1 | ||
| Asn155 OD1 | 2.9 | Thr135 OG1 | ||
| Glu163 OE1 | 3.4 | Thr135 OXT | ||
| MG | 2.0 | Thr135 OG1 | ||
| Arg122 O | 3.0 | W1 | 3.0 | Thr135 OG1 |
| His124 ND1 | 2.8 | W1 | ||
| Asn155 OD1 | 3.0 | W1 | ||
| W1 | 3.0 | Glu24 OE1 | ||
| Ser128 N | 3.0 | W38 | 2.6 | Thr135 O |
| Glu163 OE1 | 2.6 | W86 | 2.9 | Thr135 OG1 |
| Asn155 O | 2.8 | W38 | ||
| Glu163 OE2 | 2.7 | W87 | 3.3 | Thr135 OG1 |
| MG# | 2.3 | W87 | ||
| Ala129 N | 2.9 | W165 | 2.7 | Thr135 OXT |
| 2.8 | Thr135 O | |||
| Arg167 NE2 | 3.0 | W166 | 2.4 | Thr135 OXT |
| Glu163 OE1 | 2.5 | W166 | ||
| Arg122 NH1 | 3.2 | W196 | 2.7 | Glu26 OE1 |
Metal ion at the active site
Acceptor-donor distances based on ≤ 3.4 Å
Effect of NuiA interface mutations
Previous deletion mutants of NuiA demonstrated the importance of residues Glu134 and Thr135 for tight binding to NucA(21). Based on NMR data indicating the involvement of residues in the loop immediately preceding helix D of NuiA in the NucA-NuiA interface(29), we investigated the contributions of this second interaction site by mutating residues Gln74Nui, Asp75Nui, and Trp76Nui (Figure 4). Replacement of Gln74Nui with alanine did not significantly impair the inhibition and the W76A mutation exerted a modest, although significant effect. A double mutant combining amino acid replacements Q74A and W76A was found to result in a more significant loss of inhibitory potency. However, replacement of Asp75Nui with asparagine or glutamic acid significantly reduced the level of inhibition. These results are consistent with a significant contribution of the Asp75Nui – Lys101Nuc salt bridge to the stability of the complex. Hydrophobic interactions between Trp76Nui and the Pro99Nuc and Thr111Nuc residues of NucA presumably form the basis for the contribution of Trp76Nui to NuiA inhibition. Although the Q74A mutation by itself did not appear to significantly interfere with NuiA inhibition, the greater effect of the Q74A/W76A double mutant could result from an additional structural destabilization of this region of NuiA.
Figure 4. Mutational data corresponding to the NucA-NuiA complex.
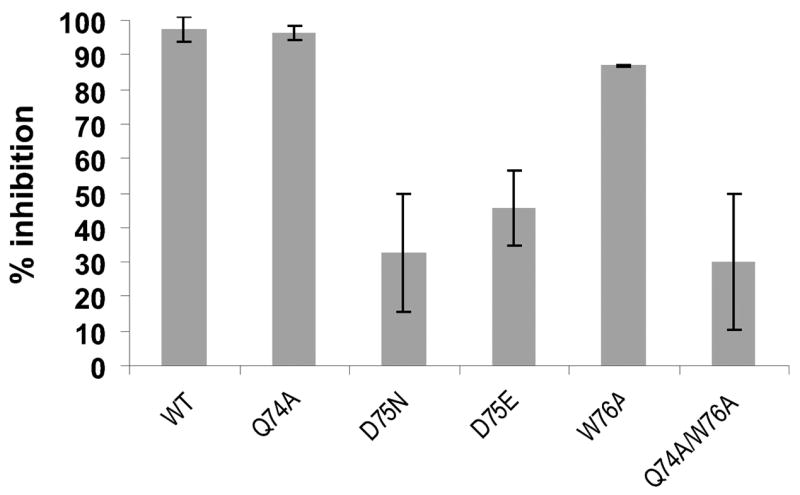
Inhibition of NucA wild type by NuiA-variants (Q74A, D75N, D75E, W76A and the double mutant Q74A/W74A) at a 5-fold molar excess over NucA. The data from three independently performed cleavage experiments are shown with relative inhibition (% inhibition) on the y-axis.
Active site of NucA in the complex
The structure of the NucA active site observed in the NucA-NuiA complex is essentially identical to that previously determined for isolated NucA (Figures 2B) (22). It is characterized by similar divalent metal ion coordination geometry and hydrogen bonding network. The only significant conformational change within the NucA molecule upon binding NuiA to the active site were rearrangements of the side chains of residues Arg93Nuc and Asp95Nuc. The catalytic divalent metal ion coordinates with both Asn155Nuc of NucA and with the OG1 atom of the C-terminal Thr135Nui (Figure 2B), as well as with 4 water molecules. The Thr135Nui OG1 ligand thus substitutes for the coordinated sulfate oxygen ligand that was present in the structure of uncomplexed NucA determined previously and has been suggested to be the 5′ phosphate binding site of the substrate (Figure 2C) (22). The catalytically important Arg93Nuc residue, which is positioned by an extensive secondary hydrogen bonding network, now forms salt bridges with the Thr135Nui terminal carboxylate and with Glu24Nui OE2. The other important active site residue of NucA, Asn155Nuc, which is the only NucA residue directly coordinating the active site Mg2+, also forms a hydrogen bond with the Glu24Nui OE1 in the NucA-NuiA complex. The W1 water molecule that is coordinated to the catalytic metal and has been previously suggested to function as the catalytic nucleophile required to break the phosphodiester bond, forms hydrogen bonds with Glu24Nui and Thr135Nui. In summary, Glu24Nui and Thr135Nui apparently play a significant role in forming the inhibitory complex by interacting with critical active site residues.
Other active site residues, particularly His124Nuc and Glu163Nuc, do not make direct contact with NuiA. Replacement of the mutated Ala121Nuc residue with a modeled Asp121 residue increases the computed NucA-NuiA interface area, indicating that this residue may also contribute to complex formation but is apparently not an absolute requirement. The side chain of Asp121 was modeled by adding the preferred rotamer. In this position there are no steric conflicts and the OD1 atom is 3.3 Å from OG1 of Ser25Nui, 3.7 Å from NE of Arg156Nuc, and 3.8 Å from ND2 of Asn155Nuc. Thus, an Asp121Nuc – Ser25Nui interaction may also contribute to the stability of the complex.
Discussion
The structure of the NucA-NuiA complex presented here is the first example of an inhibitor complex for a DNA/RNA-non-specific nuclease and it reveals fundamental differences in the mechanism of inhibition when compared to the related DNase-colicin:immunoprotein complexes. In the DNase colicins the active site itself is not part of the enzyme:inhibitor interface and prevention of DNA cleavage is achieved by blocking the DNA-binding site adjacent to the catalytic center (Figure 5). In the present structure, the C-terminal threonine residue of NuiA inserts directly into the active site, binds to the catalytic metal ion and functionally replaces a coordinated sulfate molecule previously observed in the structure of uncomplexed NucA (22) which has been suggested to mimic the 5′-phosphate group of the cleaved product. The structure of the NucA-NuiA complex is, to the best of our knowledge, is the first structure in which the catalytic divalent metal ion interacts directly with both a nuclease and an inhibitor protein. Furthermore, one of the Glu24Nui OG oxygen atoms of NuiA occupies a position close to that of a second sulfate oxygen in the uncomplexed NucA structure (22). Thus, the Glu24Nui sidechain very likely mimics the charge of a DNA phosphate oxygen. The role of the C-terminal structural elements of NuiA for NucA inhibition as discovered in this structural analysis is consistent with mutagenesis studies indicating that the C-terminal deletion of Glu134Nui and Thr135Nui dramatically reduces the KI of NuiA and requires a 50- to 100-fold molar excess of NuiA over NucA at nanomolar concentrations to achieve efficient suppression of nuclease activity (21).
Figure 5. Structural comparison of nuclease inhibitor complexes.

Although both NucA and the E7 DNase domain are members of the ββα Me nuclease family, the inhibition modes differ dramatically. (A) NuiA (dark green) binds directly to NucA (lavender) in the active site containing the metal ion (magnesium shown in yellow). (B) Im7 (navy) binds to the E7 DNase domain (brick red) but neither interferes with the active site of the DNase nor directly blocks the catalytic metal ion (zinc shown in yellow) in the active site (dashed red circle).
Although nucleases have often been suggested as drug targets (8,37,38) there is considerably less information on nuclease-inhibitor complexes than is available for proteinase-inhibitor complexes. Comparisons among these diverse structures reveal limited homologies, with the present structure probably most similar to the barnase-barstar complex, although the latter nuclease lacks a catalytic metal ion. Interestingly, in the barnase-barstar complex an aspartic acid residue of barstar (Asp39) mimics the charge of the scissile phosphate group and substrate mimicry is further achieved by aromatic amino acid residues (Tyr29, Trp38) occupying the positions of the bases of an RNA-substrate. While the barnase and ribonuclease A inhibitor complexes involve direct insertion of the inhibitor protein into the active site, as stated above, inactivation of the colicin DNase domains by the immunity proteins is more indirect, blocking critical binding interactions without directly filling the active site (39).
In general, the active sites of the nucleases are matched to the hydrophilic properties of the nucleic acids substrates. As in the NucA-NuiA structure described here, the barstar-barnase interaction is also based primarily on hydrophilic hydrogen bonding and salt bridge interactions, and contains relatively few hydrophobic contacts (Table II). In contrast, Im9, which interacts near but not directly in the active site of the colicin E9 DNase, forms a more hydrophobic interface that includes Tyr54 (Im9) – Phe86 (E9), Tyr55 (Im9) – Pro88 (E9), and Im9 peptide backbone – Tyr83 (E9) interactions, in addition to several salt bridges. The ribonuclease A-inhibitor complex studied by Kobe and Deisenhofer (1996) (26) is characterized by a largely hydrophilic interface which relies primarily on electrostatic interactions. Although the inhibitor occupies most of the active site, it only partially mimics the RNase-RNA interaction and does not utilize the p1 phosphate-binding pocket of ribonuclease A, where a sulfate ion remains bound. In this example, the inhibitor forms a large, concave surface which surrounds the ribonuclease so that a large contact area compensates for more modest shape complementarity (26). Based on this fairly limited set of examples, inhibitors which block the active site directly appear to rely more heavily on hydrophilic/electrostatic interactions and probably function more as substrate analogs. An excellent example for molecular mimicry of an inhibitor interacting with an enzyme acting on DNA is provided by the uracil DNA glycosylase: inhibitor complex, UDG-Ugi (40). The interacting surfaces display near perfect electrostatic and shape complementarity. A negatively charged ridge of the Ugi β-sheet binds to the positively charged DNA binding site of UDG, thereby preventing access of the DNA substrate to the enzyme. Mimicry of the phosphate backbone of DNA has been discovered in a range of other proteins interacting with DNA (41).
The insertion of the C-terminal Thr135 into the active site and its direct interaction with the active site metal ion is a particularly interesting feature of the complex described here. One of the more widely used nuclease inhibitors, aurintricarboxylic acid (ATA) can form bidentate chelate structures involving carboxyl and hydroxyl groups, and is believed to act via chelation of the active site metal ion (42). This molecule has been shown to inhibit EndoG, a structural analog of NucA (43). Though Thr135 does not form a bidentate interaction with the active site Mg2+ ion, as would be expected for Mg2+-ATA binding, complexation of the catalytic metal ion apparently is a useful structural approach for effecting nuclease inhibition.
As noted previously, NucA contains a second metal ion binding site remote from the active site, which is capable of binding two divalent metal ions (22). Based on the structural parameters observed for this site, it appears to have picked up two Ni2+ ions from the nickel column used to purify the enzyme. In the previous study of NucA, this site contained either two Zn2+ ions, when the enzyme was crystallized in the presence of 10 mM ZnCl2, or two Mn2+ ions, after incubation of the enzyme with Mn2+. Interestingly, in both the previous structure of NucA as well as the present NucA-NuiA complex, the second metal binding site is positioned at a lattice contact – although with a different molecule in each case. In both cases, a carboxylate ligand from a symmetry related molecule interacts such that each carboxylate oxygen atom binds to a different metal ion. Although the role of the second divalent ion binding site is unknown, its ability to support intermolecular complex formation suggests that metals may play a role in regulating NucA interactions with other proteins, possibly by anchoring NucA to the periplasm. Since, as noted previously, this second metal site does not appear to be shared by other nucleases in the ββα Me nuclease family, it may represent a unique mechanism for cellular localization of NucA.
Comparison with chemical shift mapping results
The identification of the NuiA interface on the basis of the crystal structure of the NucA-NuiA complex is in reasonable agreement with the previous identification using chemical shift mapping (29). In that study, the NuiA residues Leu20Nui, Met22Nui, Ser68Nui, Gln74Nui, Trp76Nui, Leu107Nui, Gly108Nui, Glu109Nui, Val133Nui, and Glu134Nui had been identified as being near the interface on the basis of the amide shift differences observed between uncomplexed and NucA-complexed NuiA, although the NuiA resonances corresponding to the complex were not fully assigned. Examination of the structure of the NucA-NuiA complex indicates that most of these residues are positioned in or near the interface. In several instances, residues adjacent to interacting residues are identified, probably due to the use of the amide shift as the reporter group rather than the resonances of the interacting sidechain. For example, the Gln74Nui and Trp76Nui amides were identified, while the Asp75Nui side chain carboxyl forms a salt bridge with NucA. Similarly, the amides of Val133Nui and Glu134Nui, were identified as part of the interface, although Thr135Nui interacts most directly with the active site. Despite these limitations, the chemical shift mapping approach appears to have done a reasonable job of identifying the NuiA interface.
Comparison of DNA and NuiA binding by NucA
We have previously obtained a useful model for the complex formed with cleaved or uncleaved DNA by superposition of our NucA structure (22) with the reported structures for DNA-complexed Vvn nuclease (1OUP) (44). This superposition places the DNA chain in a reasonable position relative to the catalytic groups of NucA. The 5′-terminal phosphate group of a hydrolyzed DNA substrate was found to superpose closely with the sulfate anion observed in the NucA structure (22). The superposition shown in Figure 6 compares the NucA-DNA complex modeled as described previously with the NucA-NuiA complex obtained in the present study. Based on this comparison, oxygen atoms of Glu24Nui and Thr135Nui in NuiA occupy the position of the scissile phosphate of the Vvn DNA product complex. Note in particular the similar relative positions of the 5′-phosphate from the DNA in Figure 6 and the sulfate from the superposition shown in Figure 2C. Additionally, the Thr135Nui carboxyl group appears to be positioned near a bridging phosphate from the 3′-terminus of the cleaved DNA. The carbonyl oxygen of Glu24Nui is also positioned fairly close to an oxygen atom in a bridging phosphate group at the 5′-side of the cleaved DNA. Thus, there is a more general mimicry of cleaved DNA by the NuiA inhibitor.
Figure 6. Modeled DNA interaction positioned in the NuiA-NucA complex.
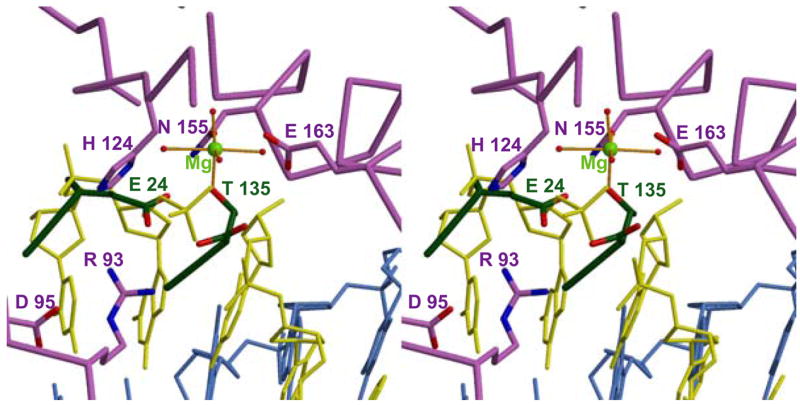
The extent to which NuiA mimics the substrate is illustrated by this stereoview of a docked model of a cleaved DNA octamer in the active site of NucA. Selected residues from NucA and NuiA are indicated in lavender and green, respectively. The model is based on the alignment of active site residues in the NucA structure (Arg122-Ile125 and Met147-Arg156) with active site residues in the structure of Vvn in complex with DNA (Trp78-Val81 and Leu119-Gly128; PDB ID 1OUP). The coordination of the active site Mg2+ (green) to four water molecules as well as to Asn155 (lavender) and to a phosphate oxygen of the cleaved DNA strand (yellow) are indicated. The active site residues are shown in grey and the complementary strand of the DNA is shown in blue.
The structure of the NucA-NuiA complex presented here provides unique insights into the basis for inhibition of sugar non-specific nucleases of the ββα Me nuclease superfamily. Despite the significant cellular toxicity that expression of this class of enzymes can pose, little is known about how their activity is controlled, and structure of the complex differs dramatically from that of the complexes formed between the colicins and their immunity proteins (39). Based on the recently discovered role of similar nucleases as mediators of infectivity resulting from their ability to destroy the DNA scaffold of neutrophil extracellular traps (NETs), they have emerged as potential drug targets (6–8). Hence, an understanding of the structural basis for their inhibition will be of increasing importance for the development of strategies to deal with infective microorganisms.
Acknowledgments
We gratefully acknowledge the assistance of Dr. Tom Kirby in the preparation of the NucA mutant, Dr. Robert Petrovich of the protein expression core facility for protein expression, Dr. Joseph Krahn for contributions to the structural refinement, and Dr. Zhongmin Jin for data collection using the mail-in crystallography at SER-CAT. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. This research was supported in part by the Intramural Research program of the NIH, and NIEHS. Work in the Giessen Lab is supported by the Deutsche Forschungsgemeinschaft (Pi 122/20-1).
References
- 1.Linn S, Lloyd RS, Roberts RJ. Nucleases. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1993. [Google Scholar]
- 2.D’Alessio GRJF, editor. Ribonucleases: Structures and Functions. Academic Press; San Diego, New York, Boston, London, Sydney, Tokyo, Toronto: 1997. [Google Scholar]
- 3.Rangarajan ES, Shankar V. FEMS Microbiol Rev. 2001;25(5):583–613. doi: 10.1111/j.1574-6976.2001.tb00593.x. [DOI] [PubMed] [Google Scholar]
- 4.Schein C. Nuclease Methods and Protocols. Humana Press Inc; 2001. [Google Scholar]
- 5.Krupp G, Gross HJ. Nucleic Acids Res. 1979;6(11):3481–3490. doi: 10.1093/nar/6.11.3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Science. 2004;303(5663):1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 7.Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. Curr Biol. 2006;16(4):401–407. doi: 10.1016/j.cub.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 8.Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, Feramisco J, Nizet V. Curr Biol. 2006;16(4):396–400. doi: 10.1016/j.cub.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 9.Kuhlmann UC, Moore GR, James R, Kleanthous C, Hemmings AM. FEBS Lett. 1999;463(1–2):1–2. doi: 10.1016/s0014-5793(99)01499-4. [DOI] [PubMed] [Google Scholar]
- 10.Flick KE, Jurica MS, Monnat RJ, Jr, Stoddard BL. Nature. 1998;394(6688):96–101. doi: 10.1038/27952. [DOI] [PubMed] [Google Scholar]
- 11.Hsia KC, Chak KF, Liang PH, Cheng YS, Ku WY, Yuan HS. Structure (Camb) 2004;12(2):205–214. doi: 10.1016/j.str.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Mate MJ, Kleanthous C. J Biol Chem. 2004;279(33):34763–34769. doi: 10.1074/jbc.M403719200. [DOI] [PubMed] [Google Scholar]
- 13.Shen BW, Landthaler M, Shub DA, Stoddard BL. J Mol Biol. 2004;342(1):43–56. doi: 10.1016/j.jmb.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 14.Nestle M, Roberts WK. J Biol Chem. 1969;244(19):5213–5218. [PubMed] [Google Scholar]
- 15.Puyet A, Greenberg B, Lacks SA. J Mol Biol. 1990;213(4):727–738. doi: 10.1016/S0022-2836(05)80259-1. [DOI] [PubMed] [Google Scholar]
- 16.Ho HC, Liao TH. Biochem J. 1999;339(Pt 2):261–267. [PMC free article] [PubMed] [Google Scholar]
- 17.Ho HC, Shiau PF, Liu FC, Chung JG, Chen LY. Eur J Biochem. 1998;256(1):112–118. doi: 10.1046/j.1432-1327.1998.2560112.x. [DOI] [PubMed] [Google Scholar]
- 18.Vincent RD, Hofmann TJ, Zassenhaus HP. Nucleic Acids Res. 1988;16(8):3297–3312. doi: 10.1093/nar/16.8.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cote J, Renaud J, Ruiz-Carrillo A. J Biol Chem. 1989;264(6):3301–3310. [PubMed] [Google Scholar]
- 20.Muro-Pastor AM, Flores E, Herrero A, Wolk CP. Mol Microbiol. 1992;6(20):3021–3030. doi: 10.1111/j.1365-2958.1992.tb01760.x. [DOI] [PubMed] [Google Scholar]
- 21.Meiss G, Gimadutdinow O, Haberland B, Pingoud A. J Mol Biol. 2000;297(2):521–534. doi: 10.1006/jmbi.2000.3586. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh M, Meiss G, Pingoud A, London RE, Pedersen LC. J Biol Chem. 2005;280(30):27990–27997. doi: 10.1074/jbc.M501798200. [DOI] [PubMed] [Google Scholar]
- 23.Miller MD, Cai J, Krause KL. J Mol Biol. 1999;288(5):975–987. doi: 10.1006/jmbi.1999.2729. [DOI] [PubMed] [Google Scholar]
- 24.Ball TK, Suh Y, Benedik MJ. Nucleic Acids Res. 1992;20(19):4971–4974. doi: 10.1093/nar/20.19.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guillet V, Lapthorn A, Hartley RW, Mauguen Y. Structure. 1993;1(3):165–176. doi: 10.1016/0969-2126(93)90018-c. [DOI] [PubMed] [Google Scholar]
- 26.Kobe B, Deisenhofer J. J Mol Biol. 1996;264(5):1028–1043. doi: 10.1006/jmbi.1996.0694. [DOI] [PubMed] [Google Scholar]
- 27.Ko TP, Liao CC, Ku WY, Chak KF, Yuan HS. Structure. 1999;7(1):91–102. doi: 10.1016/s0969-2126(99)80012-4. [DOI] [PubMed] [Google Scholar]
- 28.Kleanthous C, James R, Hemmings AM, Moore GR. Biochem Soc Trans. 1999;27(2):63–67. doi: 10.1042/bst0270063. [DOI] [PubMed] [Google Scholar]
- 29.Kirby TW, Mueller GA, DeRose EF, Lebetkin MS, Meiss G, Pingoud A, London RE. J Mol Biol. 2002;320(4):771–782. doi: 10.1016/s0022-2836(02)00460-6. [DOI] [PubMed] [Google Scholar]
- 30.Vagin A, Teplyakov A. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 31.Bailey S. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 32.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallographica, A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 33.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D. 1998;54(5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 34.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 35.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Proteins. 2003;50(3):437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 36.Hannan JP, Whittaker SB, Davy SL, Kuhlmann UC, Pommer AJ, Hemmings AM, James R, Kleanthous C, Moore GR. Protein Sci. 1999;8(8):1711–1713. doi: 10.1110/ps.8.8.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loverix S, Steyaert J. Curr Med Chem. 2003;10(9):779–785. doi: 10.2174/0929867033457845. [DOI] [PubMed] [Google Scholar]
- 38.Klarmann GJ, Hawkins ME, Le Grice SF. AIDS Rev. 2002;4(4):183–194. [PubMed] [Google Scholar]
- 39.Kleanthous C, Walker D. Trends Biochem Sci. 2001;26(10):624–631. doi: 10.1016/s0968-0004(01)01941-7. [DOI] [PubMed] [Google Scholar]
- 40.Mol CD, Kuo CF, Thayer MM, Cunningham RP, Tainer JA. Nature. 1995;374(6520):381–386. doi: 10.1038/374381a0. [DOI] [PubMed] [Google Scholar]
- 41.Dryden DT, Tock MR. Biochem Soc Trans. 2006;34(Pt 2):317–319. doi: 10.1042/BST20060317. [DOI] [PubMed] [Google Scholar]
- 42.Sharma RK, Garg BS, Kurosaki H, Goto M, Otsuka M, Yamamoto T, Inoue J. Bioorg Med Chem. 2000;8(7):1819–1823. doi: 10.1016/s0968-0896(00)00109-7. [DOI] [PubMed] [Google Scholar]
- 43.Ishihara Y, Shimamoto N. J Biol Chem. 2006;281(10):6726–6733. doi: 10.1074/jbc.M510382200. [DOI] [PubMed] [Google Scholar]
- 44.Li CL, Hor LI, Chang ZF, Tsai LC, Yang WZ, Yuan HS. Embo J. 2003;22(15):4014–4025. doi: 10.1093/emboj/cdg377. [DOI] [PMC free article] [PubMed] [Google Scholar]