

Abstract
Background
A subset of familial cases (FTDP-17) of frontotemporal dementia (FTD) are caused by mutations in the tau gene. The role of tau gene mutations and haplotypes in sporadic FTD and the functional consequences of tau polymorphisms are unknown.
Objectives
To investigate (1) the frequency of known FTDP-17 mutations in familial and sporadic FTD and compare these results with previous studies; (2) whether the tau H1 haplotype is associated with FTD; and (3) the functional effect of intronic tau sequence variations.
Patients and Methods
Patients with familial and sporadic FTD were screened for mutations in the microtubule-binding region of tau. The frequencies of tau haplotypes and genotypes were compared between patients with FTD and control subjects. We analyzed the splicing effect of novel intronic polymorphisms associated with FTD.
Results
The P301L mutation was detected in 11% of familial FTD cases. The H1 haplotype was not overrepresented in patients with FTD, but the P301L mutation appeared on the background of the H2 tau haplotype. We identified 4 novel single nucleotide polymorphisms in intron 9 and a 9–base pair deletion in intron 4A. A C-to-T transition 177 base pairs upstream from exon 10 was significantly increased in patients with FTD compared with controls. Direct analysis of brain tissue from a patient with this variant showed an increase in exon 10–containing tau transcripts.
Conclusions
Sequence variations in intronic or regulatory regions of tau may have previously unrecognized consequences leading to tau dysfunction and neurodegeneration.
The term frontotemporal dementia (FTD) refers to a group of primary degenerative dementias producing focal atrophy of the frontal and/or temporal lobes, which is frequently associated with parkinsonism and amyotrophic lateral sclerosis.1,2 Frontotemporal dementia is familial in up to 40% to 50% of the cases, and inheritance is most often consistent with an autosomal dominant pattern of inheritance.3 Two classes of mutations in the microtubule-associated protein tau have been found in 10% to 40% of subjects with familial FTD—those directly affecting the microtubule binding sites of tau, and those that alter tau splicing.4-6 The majority of the currently known tau mutations are located in exons 9 to 13, the region coding for the microtubule-binding domains of the protein. Tau mutations have not been identified in sporadic FTD cases in previous studies.7,8
Tau is also implicated in other related neurodegenerative conditions, eg, progressive supranuclear palsy and corticobasal degeneration (CBD).9,10 Several polymorphisms throughout the tau gene are in complete linkage disequilibrium and inherited as 2 distinct haplotypes, H1 and H2. The H1 haplotype is overrepresented in progressive supranuclear palsy and other tauopathies, although the biological basis for this association is currently unknown. The absence of detectable exonic mutations in many familial and in virtually all sporadic cases of FTD, together with the identification of tau polymorphisms of unknown significance, raises the question of whether these polymorphisms or other sequence variations may have functional consequences that are not yet understood.
The present study was undertaken to evaluate the prevalence of the major currently recognized tau mutations in patients with familial FTD and patients with sporadic FTD from a well-characterized clinical sample,3,11,12 to determine whether common tau polymorphisms confer an increased genetic risk of developing FTD, and to investigate the potential functional effects of rare sequence variations on tau splicing.
METHODS
SUBJECTS
The patients included in this study were recruited under a protocol approved by the University of California, Los Angeles, (UCLA) Institutional Review Board. The diagnosis of FTD was based on the extended Lund-Manchester criteria, including neuroimaging and neuropsychological testing.12 Autopsy results confirmed the diagnosis of FTD in 12 cases. De-identified, anonymous control samples of different ethnic origins were obtained from the Molecular Pathology core at UCLA for testing single nucleotide variants identified.
SCREENING FOR TAU MUTATIONS AND POLYMORPHISMS
Exons 9, 10, 12, and 13 and flanking intronic regions were amplified by polymerase chain reaction using the primer pairs shown in Table 1. Mutations in exons 9 (G272V) and 13 (R406W, G389R), and the −177C→T variant were screened by polymerase chain reaction amplification followed by restriction digestion. Direct sequence analysis was performed for exons 10 and 12 in all samples. In addition, exons 9 and 13 were sequenced in 3 cases in which either tau immunoblot or splicing analysis showed a pattern of imbalance in the ratio of tau isoforms (cases 9, 30, and 31). A mismatched primer pair (ModF+ModR) (Table 1) introducing a restriction site for TaqI was designed to screen for a G-to-A single nucleotide polymorphism (SNP) at position −58 upstream from exon 10. The −176G→A and the −47C→A polymorphisms were screened by direct sequencing or single-strand conformation polymorphism analysis of control samples.
Table 1.
Primers and Restriction Enzymes Used
| Exon/Intron | Mutations/Polymorphisms | Enzyme | Primer Pair |
|---|---|---|---|
| Exon 4A | 735C→T | MspI | 4AF: 5′-AGCCGCCAGAGAAGCCACCAG-3′ |
| 4AR: 5′-GGAACGTCAGAAGCAGCAGGAGTC-3′ | |||
| Exon 9 | G272V | Hinf I | 9F: 5′-GCCCAGGGCCTTTTCTGACC-3′ |
| 9R: 5′-CACTCTCACTTCCCGCCTC-3′ | |||
| Intron 9 | −177C→T* | Hinf I | 10F: 5′-AGCCCTCTATCCCTTCAG-3′ |
| 10RM: 5′-GCATGGGACGTGTGAAGGTAC-3′ | |||
| −58G→A* | TaqI | ModF: 5′-TCATCGAAAGTGGAGGCGTCCTTTC-3′ | |
| ModR: 5′-GGCTACATTCACCCAGAGGTCGC-3′ | |||
| Intron 10 | 10F: 5′-AGCCCTCTATCCCTTCAG-3′ | ||
| 10R: 5′-GGCTACATTCACCCAGAGGT-3′ | |||
| Intron 12 | 12F: 5′-GCACAGAACCACAGAAGATGATGGCA-3′ | ||
| 12R: 5′-AGCATCCAACCCACCCTACCC-3′ | |||
| Intron 13 | G389R | NciI | 13F: 5′-GTCCCTCCCTTCCTCTTCTTG-3′ |
| R406W | NcoI | 13R: 5′-GAGTGACAAAAGCAGGTTAAGTGAT-3′ |
New sequence variations described in the present study (see “Results” and Table 3).
CONSTRUCTION OF TAU HAPLOTYPES
Two intragenic polymorphic markers in exon 4A and intron 9 were used to infer tau haplotypes.8,13 The frequency of the tau H1 and H2 haplotypes in patients with FTD was compared with that in 36 nondemented white volunteers with normal results on neuropsychological evaluation.
WESTERN BLOTTING
Frontal cortex (area 9) from 3 familial and 2 sporadic cases of FTD was used for biochemical analyses. Gray and white matter was dissected and processed separately. Soluble and sarcosyl-insoluble (PHF) tau was extracted from 1 g of tissue, and tau dephosphorylation and Western blotting were performed.14
TAU SPLICING ASSAY
A tau minigene construct15 including the wild-type tau genomic DNA fragments containing a segment of the genomic region between exons 9 and 11 and the corresponding fragments containing the −177T allele was used to evaluate the potential effect of sequence variations on the alternative splicing of exon 10. Total RNA was obtained from frozen brain tissue (A9) of the same cases used for the Western blot analysis, as well as from formalin-fixed, paraffin-embedded frontal cortex from one of the patients with the −177C→T variation. The RNA was prepared from the cell lines and FTD tissues by means of a reagent (Trizol; Life Technologies, Rockville, Md), and reverse transcriptase polymerase chain reaction was performed in the presence of γ-phosphorus 32–labeled (γ-32P) deoxycytidine-trinucleotide phosphate (Amersham Biosciences Corp, Piscataway, NJ). Blots were quantitated with a phosphor screen (PhosphorImager; Molecular Dynamics, Sunnyvale, Calif).
STATISTICAL ANALYSIS
The χ2 or Fisher exact tests were used to compare allele and haplotype frequencies between white patients and controls and to test for an association between novel sequence variants and tau haplotype. The statistical analysis was performed with the program SPSS 10.0 for Windows (SPSS Inc, Chicago, Ill), and significance level was set at P=.05.
RESULTS
TAU MUTATION SCREENING
Forty-eight patients with FTD (23 women, 25 men) were screened for the common known FTDP-17 mutations. Demographic and clinical characteristics of the patients included in the present study, including ethnicity, as well as all haplotype and mutation data are summarized in a supplemental data table (available at: http://geschwindlab.medsch.ucla.edu). The mean age at onset was 54.2 years (range, 25-75 years). Eleven patients (23%) developed motor neuron signs in addition to dementia. The P301L C→T mutation was observed in 2 families of French Canadian origin. None of the previously reported tau mutations were detected in sporadic cases. One of the families has been described in detail in a previous report.16 It should be emphasized that the abnormality in this family shared significant features with CBD and Pick disease, a finding that highlights the overlap between FTD, Pick disease, and CBD. The second patient with the P301L mutation (case 16) presented at age 45 years with a progressive syndrome of aggressive behavior, paranoid delusions, apathy, and dementia. Several other family members had a similar condition. Three patients had 4R tau imbalance on biochemistry, but had no tau mutations in exons 9 to 13.
TAU HAPLOTYPES
The results of haplotype and genotype analysis are summarized in Table 2. A trend toward a more frequent presentation of the H2 haplotype was observed among patients with FTD with a positive family history, compared with both sporadic FTD cases and controls (P<.08) (Table 2). In both families, the P301L mutation was associated with the H2 haplotype (not shown).
Table 2.
Tau Haplotypes and Genotypes Among Patients With FTD and Control Subjects
| No. (%)
|
||||
|---|---|---|---|---|
| Controls | FTD
Global |
FTD
Sporadic |
FTD
Familial |
|
| Haplotypes | ||||
| No. of subjects | 72 | 90 | 54 | 36 |
| H1 | 54 (75) | 67 (74) | 43 (80) | 23 (64) |
| H2 | 18 (25) | 23 (26) | 11 (20) | 13 (36) |
| Genotypes | ||||
| No. of subjects | 36 | 45 | 27 | 18 |
| H1/H1 | 23 (64) | 26 (58) | 17 (63) | 8 (44) |
| H1/H2 | 8 (22) | 15 (33) | 9 (33) | 7 (39) |
| H2/H2 | 5 (14) | 4 (9) | 1 (4) | 3 (17) |
Abbreviation: FTD, frontotemporal dementia.
NOVEL TAU INTRONIC VARIATIONS
Four novel SNPs were detected in the intron upstream from exon 10 (see supplemental table and Table 3). A C-to-A change at position −47 in intron 9 was present in 3 patients with FTD and was also seen in 1 control chromosome, associated with the H1/H1 genotype. A common G→A SNP was identified −176 base pairs (bp) upstream from exon 10. The A allele in this position was in linkage disequilibrium with the H1 haplotype (Fisher exact test, P=.01). Another novel SNP was detected at position −177 (C→T) 5′ of exon 10 (Figure 1) in 3 sporadic cases. Since this variant was not detected in any of 229 white (American) control samples, 83 additional French Canadian controls were analyzed and the −177T allele was detected in 1 (0.6%) of 166. This change was not seen in any of 100 Native American, 100 Hispanic, 100 African American, and 100 Asian control chromosomes. The T allele at this position was transmitted with the H2 haplotype (Fisher exact test, P=.03).
Table 3.
Novel Sequence Variations in Tau*
| Position | Cases, No. (%) | Controls, No. (%) | P |
|---|---|---|---|
| −47 (C→A) | 3/90 (3.3) | 1/120 (0.8) | .42† |
| −58 (G→A) | 1/90 (1.1) | 0/112 | NA |
| −176 (G→A) | 33/90 (36.7) | 78/152 (51.3) | .04† |
| −177 (C→T) | 3/90 (3.3) | 1/624 (0.2) | .003† |
| Intron 4A deletion | 1/90 (1.1) | 6/170 (3.5) | .45† |
Abbreviation: NA, not applicable.
Frequencies refer to number of chromosomes.
χ2 with Yates correction.
Figure 1.
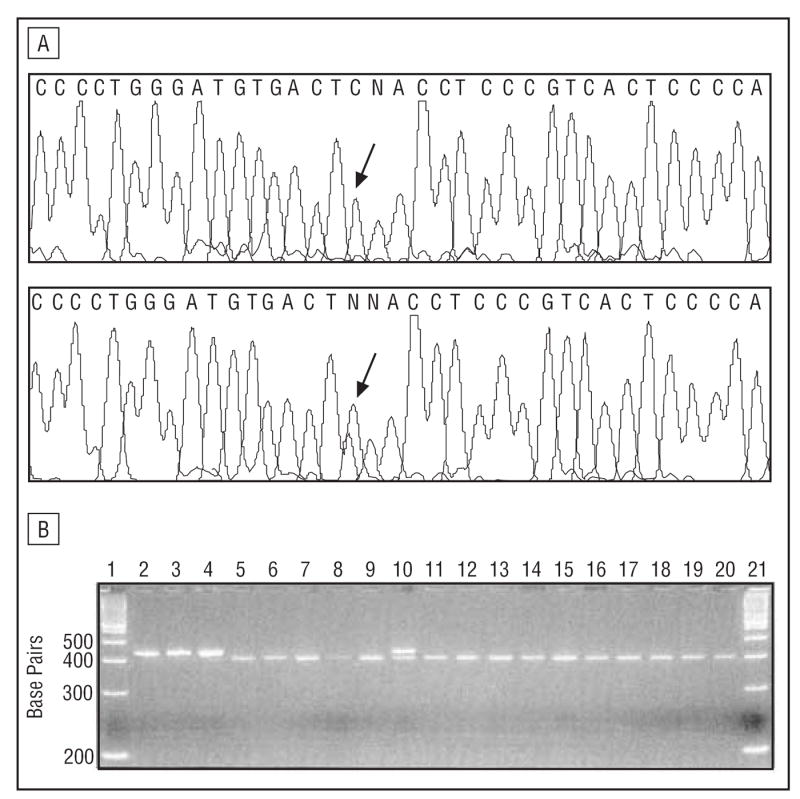
A, C-to-T substitution at position −177 of intron 9; case 30 (lower panel); control sample (upper panel). Both samples are also heterozygous for the −176G→A polymorphism. B, Exon 10 polymerase chain reaction products were digested with Hinf I and run on an agarose gel. Lanes 2 to 4 contain undigested polymerase chain reaction products. Case 30 is shown in lane 10.
A novel 9-bp deletion polymorphism 46 bp 3′ of exon 4A was also detected. This deletion was present in 1 patient with sporadic FTD and in 6 of 170 controls (Table 3).
TAU PROTEIN ISOFORMS
Frozen postmortem samples from 4 familial (cases 20, 31, 42, and 43) and 1 sporadic (case 9) cases of FTD were analyzed (Figure 2). The PHF tau isolated from the cortex of a patient with the P301L mutation (C43) showed an over-expression of 4R tau.14 Another case (case 9) without a known tau mutation showed a similar pattern of increased 4R tau in PHF preparations. Neuropathologically, this case was characterized by the presence of Pick bodies and ballooned neurons typical of Pick disease or CBD, similar to those observed in the P301L-positive case.16 This observation supports recent findings that tau isoform composition in Pick disease is not restricted to the 3R isoform only.17
Figure 2.

Biochemical profile of tau in brains of patients with familial frontotemporal dementia. Soluble and insoluble (PHF) tau were isolated from gray (G) and white (W) matter. An equal amount of total protein was loaded in each lane. Case 43 is a P301L-positive case. A mixture of 6 recombinant tau isoforms (Rt) was used as a standard. AD indicates Alzheimer disease.
TAU SPLICING
To assess the functional consequences of the intron 10 –177 C-to-T polymorphism that was significantly increased in patients with FTD relative to controls, an in vitro splicing assay was performed with the use of a tau minigene containing either the C or T variant. A 2- to 3-fold increase of tau transcripts lacking exon 10 was observed in the presence of the T variant compared with wild-type tau in all 3 cell lines analyzed (Figure 3A). To extend this observation in vivo, the pattern of tau exon 10 splicing in frozen and fixed brain specimens of patients with familial and sporadic FTD was assessed. Rather than a decrease, a relative increase of exon 10–containing tau was detected in 1 patient (case 30) with the −177C→T polymorphism and in 1 additional patient (case 31), both of whom had familial FTD (Figure 3B). In addition, case 31, without mutations in or around exon 10, was homozygous for several tau SNPs inherited as part of the H2 haplotype.
Figure 3.

Effect of the −177T polymorphism on exon 10 splicing. A, Alternative splicing of exon 10 from 3 cell lines transfected with tau minigenes containing wild-type tau (WT) and the −177T intronic polymorphism. The intensity of tau transcript with (exon 10+) and without (exon 10−) exon 10 was measured, and the ratios are indicated below each lane. B, Autoradiograph of tau exon 10 reverse transcriptase polymerase chain reaction products from brain specimens with frontotemporal dementia. The percentage of exon 10 inclusion is expressed as mean (±SD) below each lane. Cases 20, 42, and 9 were not significantly different from normal tau.
COMMENT
TAU MUTATIONS UNDERLIE FEW CASES OF FAMILIAL FTD
In the current study, we report the analysis of known FTDP-17 causal tau mutations in a series of familial and sporadic FTD. The frequency of tau mutations among familial cases in our series was 11%, similar to the figures obtained by others.7,8 The absence of tau mutations in patients with sporadic FTD in our series is in agreement with previous reports.7,8 Therefore, mechanisms other than the currently recognized tau mutations must explain most familial cases, as well as sporadic FTD.
THE TAU H1 HAPLOTYPE IS NOT ASSOCIATED WITH FTD
Since an association between the tau H1 haplotype and other neurodegenerative disorders involving tau has been consistently reported,9,10,18,19 we investigated whether either the H1 or the H2 haplotype was also overrepresented in FTD. Unlike progressive supranuclear palsy and other sporadic tauopathies, no significant association was detected between either the H1 haplotype or the H1/H1 genotype and FTD in our series. A trend toward association of the H2 haplotype and H2/H2 genotype was observed in patients with familial FTD compared with both sporadic FTD and controls. Such an association has been recently reported in Pick disease.20 We also observed the P301L mutation coinherited with the H2 haplotype, as was seen in 4 of 5 other families.7 Larger samples are needed to assess a potential founder effect in this mutation.
TAU ISOFORMS AND SPLICING
The result of biochemical analysis of tau in pathological specimens was consistent with the genetic data. The PHF tau from the brain with the P301L mutation showed a biochemical profile typical of this mutation,14,16 whereas no tau mutations were identified in 3 patients with a biochemical profile similar to that typically observed in Alzheimer disease. Both patients with FTD with 4R tau deposition had Pick bodies and ballooned neurons that are observed in Pick disease and CBD, emphasizing the clinical and pathological overlap of FTD with these conditions.
Previous data have highlighted the involvement of abnormal splicing in the pathogenesis of FTD.5,6,14,21 Few studies have looked at regions more than 100 bp from the splice junctions. In the present series, we identified 4 novel SNPs within 200 bp in the intron upstream from exon 10, one of which (−177C→T) was significantly increased in patients with FTD relative to a large number of control chromosomes. In vitro experiments demonstrated that the −177C→T change causes an imbalance in the splicing of tau toward isoforms in which exon 10 has been omitted. There are no identifiable splicing regulatory elements in this region. However, the −177T variant alone cannot completely account for the final pathological alteration, since analysis of postmortem tissue from the same case demonstrated an increase in 4R to 3R tau transcript. Thus, other factors that may be in linkage disequilibrium with the −177T allele, or other splicing regulatory elements, must be involved. The common −176 G→A polymorphism was seen more frequently in controls and was in linkage disequilibrium with the H1 haplotype.
These results highlight the importance of confirming any in vitro splicing changes, especially those obtained with simple exon trapping vectors, in vivo. While the splicing alterations observed in vitro may reflect the effect of that specific sequence variant, the final balance of tau isoforms in vivo is likely to be the result of more complex interactions. Moreover, the mechanisms by which common tau polymorphisms confer increased risk for progressive supranuclear palsy,9,18 CBD,10 and primary progressive aphasia19 are unknown. Whether these extended tau haplotypes affect disease risk by altering splicing or other aspects of tau regulation or, alternatively, whether they implicate additional genes nearby, remains to be determined.
Acknowledgments
The John Douglas French Alzheimer’s Foundation (Los Angeles) partially supported this work. Dr Sobrido was supported by a fellowship from the Fundación Pedro Barrié de la Maza (La Coruña, Spain). This study was also supported by the UCLA Alzheimer’s Disease Research Center (National Institute on Aging [Bethesda, Md] grant AG16570 [Drs Cummings and Geschwind]); an Alzheimer’s Disease Research Center of California (California Department of Health Services [Sacramento]) grant (Dr Cummings); the Sidell-Kagan Foundation (Los Angeles) (Dr Cummings); and National Institute on Aging grant AG17518 (Dr Wu). Dr Wu is a Leukemia and Lymphoma Society (New York, NY) Scholar.
We thank Amir Abu-Khalil for his technical assistance.
References
- 1.Lund and Manchester Groups. Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatry. 1994;57:416–418. doi: 10.1136/jnnp.57.4.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 3.Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia. Arch Neurol. 1999;56:817–822. doi: 10.1001/archneur.56.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. published correction appears in Ann Neurol. 1998;44:428. [DOI] [PubMed] [Google Scholar]
- 5.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 6.Clark LN, Poorkaj P, Wszolek Z, et al. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci U S A. 1998;95:13103–13107. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houlden H, Baker M, Adamson J, et al. Frequency of tau mutations in three series of non-Alzheimer’s degenerative dementia. Ann Neurol. 1999;46:243–248. doi: 10.1002/1531-8249(199908)46:2<243::aid-ana14>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 8.Poorkaj P, Grossman M, Steinbart E, et al. Frequency of tau gene mutations in familial and sporadic cases of non-Alzheimer dementia. Arch Neurol. 2001;58:383–387. doi: 10.1001/archneur.58.3.383. [DOI] [PubMed] [Google Scholar]
- 9.Conrad C, Andreadis A, Trojanowski JQ, et al. Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann Neurol. 1997;41:277–281. doi: 10.1002/ana.410410222. [DOI] [PubMed] [Google Scholar]
- 10.Di Maria E, Tabaton M, Vigo T, et al. Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann Neurol. 2000;47:374–377. doi: 10.1002/1531-8249(200003)47:3<374::aid-ana15>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 11.Geschwind D, Karrim J, Nelson SF, Miller B. The apolipoprotein E ∈4 allele is not a significant risk factor for frontotemporal dementia. Ann Neurol. 1998;44:134–138. doi: 10.1002/ana.410440122. [DOI] [PubMed] [Google Scholar]
- 12.Miller BL, Ikonte C, Ponton M, et al. A study of the Lund-Manchester research criteria for frontotemporal dementia. Neurology. 1997;48:937–942. doi: 10.1212/wnl.48.4.937. [DOI] [PubMed] [Google Scholar]
- 13.Lilius L, Froelich Fabre S, Basun H, et al. Tau gene polymorphisms and apolipoprotein E 4 may interact to increase risk for Alzheimer’s disease. Neurosci Lett. 1999;277:29–32. doi: 10.1016/s0304-3940(99)00833-2. [DOI] [PubMed] [Google Scholar]
- 14.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 15.Jiang Z, Cote J, Kwon JM, Goate AM, Wu JY. Aberrant splicing of tau pre-mRNA caused by intronic mutations associated with the inherited dementia frontotemporal dementia with parkinsonism linked to chromosome 17. Mol Cell Biol. 2000;20:4036–4048. doi: 10.1128/mcb.20.11.4036-4048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nasreddine ZS, Loginov M, Clark LN, et al. From genotype to phenotype. Ann Neurol. 1999;45:704–715. doi: 10.1002/1531-8249(199906)45:6<704::aid-ana4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 17.Zhukareva V, Mann D, Pickering-Brown S, et al. Sporadic Pick’s disease. Ann Neurol. 2002;51:730–739. doi: 10.1002/ana.10222. [DOI] [PubMed] [Google Scholar]
- 18.Baker M, Litvan I, Houlden H, et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:711–715. doi: 10.1093/hmg/8.4.711. [DOI] [PubMed] [Google Scholar]
- 19.Sobrido MJ, Abu-Khalil A, Weintraub S, et al. Possible association of the tau H1/H1 genotype with primary progressive aphasia. Neurology. 2003;60:862–864. doi: 10.1212/01.wnl.0000049473.36612.f2. [DOI] [PubMed] [Google Scholar]
- 20.Russ C, Lovestone S, Baker M, et al. The extended haplotype of the microtubule associated protein tau gene is not associated with Pick’s disease. Neurosci Lett. 2001;299:156–158. doi: 10.1016/s0304-3940(00)01785-7. [DOI] [PubMed] [Google Scholar]
- 21.Grover A, Houlden H, Baker M, et al. 5′ Splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J Biol Chem. 1999;274:15134–15143. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]