

Abstract
Neurofibrillary tangles containing filaments of the microtubule-associated protein tau are found in a variety of neurodegenerative diseases. Mutations in the tau gene itself cause frontotemporal dementia with parkinsonism, demonstrating the critical role of tau in pathogenesis. Many of these mutations in tau are silent, are found at the 5′-splice site of exon 10, and lead to increased inclusion of exon 10. These silent mutations are predicted to destabilize a stem loop structure at the exon 10 5′-splice site; however, the existence of this stem loop under physiological conditions and its role in splice regulation are controversial. Here we show that base changes that stabilize this stem loop in vitro substantially decrease exon 10 inclusion in a wild type tau minigene and rescue the increase in exon 10 splicing caused by a dementia-causing point mutation. Moreover, we probed the intracellular structure of the tau stem loop with antisense RNA and demonstrate that the stability of the stem loop dictates antisense effectiveness. Together these results validate the stem loop as a bona fide structure regulating tau exon 10 splicing.
Tauopathies are neurodegenerative disorders characterized by neurofibrillary tangles, intraneuronal masses of paired helical filaments that are composed of highly phosphorylated forms of the microtubule-associated protein tau (1,2). Wild type tau containing neurofibrillary tangles are a common feature in a number of neurodegenerative diseases, including Alzheimer disease, Creutzfeldt-Jakob disease, Pick disease, progressive supranuclear palsy, frontal temporal dementia, and dementia pugilistica (2).
Tau is expressed predominantly in neurons where it promotes microtubule assembly, reduces microtubule instability, and plays a role in maintaining neuronal integrity and axonal transport (3). The human gene is found on chromosome 17q21 and consists of 16 exons that are alternatively spliced to generate six tau isoforms (4–6). The interaction between tau and microtubules is mediated by C-terminal microtubule binding domains encoded by exons 9–12 (7). Alternative splicing of exon 10 produces tau isoforms with either three (exon 10 missing) or four (exon 10 present) repeat domains, known as 3R and 4R tau, respectively. In the adult human brain, the overall ratio of 4R to 3R tau is generally ~1.
The critical role of tau in neurodegenerative diseases became clear with the discovery that mutations in tau could cause frontotemporal dementia and parkinsonism linked to chromosome 17 (FTD-17) (8) and 32 such mutations have been identified to date (9). Among these, three are silent mutations in exon 10 (synonymous changes in the nucleotide sequence that do not change the coding of the protein and therefore have no effect on protein function), and seven are found in the intron following exon 10. All these mutations alter the 4R-to-3R ratio, implicating impaired splicing as the mechanism of pathogenesis.
The exon-intron interface at the 3′ end of exon 10 displays a high degree of self-complementarity, suggesting the presence of a stem loop (8). Seven of the silent mutations are clustered in this stem loop region, and all result in the disruption of complementarity and predicted destabilization of the stem loop structure. It has been proposed that, as a consequence of destabilization, this region of the mRNA becomes more available for association with the U1snRNP, resulting in increased splicing of exon 10 (8). While these mutations have a profound effect on the ratio of 4R to 3R tau splice isoforms and are at the root of pathogenesis in FTD-17, the exact nature of the stem loop is still unclear. D’Souza and co-workers (10–12) have even questioned its existence, with data suggesting that the regulatory sequences in this region are not structured. We seek to demonstrate conclusively that these sequences form an RNA structure that is a key regulatory element in tau mRNA splicing.
Exon trapping studies have clearly shown that intronic mutations that increase the splicing of exon 10 (8, 13–16) also destabilize the stem loop in vitro. If the stem loop exists under physiological conditions in vivo as a regulatory element, then stabilizing the stem loop should lead to a decrease in exon 10 splicing. We have made several stem loop variants that increase the thermal stability of the stem loop in vitro and lead to a decrease in exon 10 splicing in vivo. More importantly, we probe the nature of these stem loop variants in cells with an antisense RNA directed against the junction of exon 10 to show that the effectiveness of antisense treatment depends on the stability of the tau stem loop.
EXPERIMENTAL PROCEDURES
Cell Culture
HeLa cells (ATCC) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 2 mm glutamine. Minigenes harboring the tau stem loop variants were transiently transfected using Lipo-fectamine 2000. RNA was isolated 24 h after transfection.
Vectors
The coding region for the Photinus pyralis luciferase gene, minus the initiating ATG, with XhoI restriction sites on both the 5′ and 3′ ends, was amplified using the following primers: LucF, 5′-GCCCTCGAGGAAGACGCCAAAAA-CATAAAGAAA-3′ and LucR, 5′-CGACACTCGAGCACG-GCGATCTTTCC-3′. The PCR product was digested with XhoI and ligated in to the Tau-4R vector (17). The pTK-RL (Promega) vector expresses the Renilla luciferase gene from Renilla reniformis. All mutations in the tau stem loop were generated by site-directed mutagenesis using the following primers (nucleotide changes are italicized): DDPAC, 5′-GTGTGAGTACCTTCATACGTCCCATGCGCC-3′, WT-10C, 5′-GCAGTGTGAGTACCCTCACACGTCCCATGC-3′, DDPAC-10C, 5′-GCAGTGTGAGTACCCTCATACGTCCCATG-3′, WT-I-17T, 5′-TGAGTACCTTCACACTGTCCCATGCGCCG-3′, DDPAC-I17T, 5′-TGAGTACCTTCATACTGTCCCATGCGCCG-3′.
UV Melting
The six stem loop variant RNAs used in the UV melting experiments had the following sequences: wild type (WT),25′-GGCAGUGUGAGUACCUUCACACGUC-3′; DDPAC, 5′-GGCAGUGUGAGUACCUUCAUACGUC-3′; WT-10C, 5′-GGCAGUGUGAGUACCCUCACACGUC-3′; DD-10C, 5′-GGCAGUGUGAGUACCCUCAUACGUC-3′; WT-I-17T, 5′-GGCAGUGUGAGUACCUUCACACUGUC-3′; and DD-I17T, 5′-GGCAGUGUGAGUACCUUCAUACUGUC-3′. These were synthesized by Dharmacon. UV melting experiments were performed as described previously (18). Briefly, RNA oligonucleotides at a concentration of 1 µM in 150 NaCl, 0.1 mm EDTA in 10 mm NaPO4, pH 6.7, were heated at 0.5°C/min. The absorbance at 260 nm was collected using a Cary UV/VIS spectrophotometer with a temperature controlled heating block. Melting temperatures were calculated as described previously (19).
RT-PCR
Total RNA was isolated from transfected cells using RNAqueous (Ambion). cDNA synthesis and real time PCR with SYBR Green using the ABI 7700 from Applied Bio-systems was performed as described previously (20). Real-time PCR to quantify 4R tau mRNA was done using primers directed against exon 10, (5′-GTCCAAGTGTGGCTCAAAG-3′) and luciferase (5′-GCCTTATGCAGTTGCTCTCC-3′) in tau minigene-transfected cell lines. PCR reactions were performed at 60°C for a combined annealing and extension. Real-time PCR to quantify tau 3R expression was done using primers specific to the exon 9-exon 11 junction (5′-GAGGCGGGAAGGTGCAA-3′) and luciferase. PCR reactions were performed at 64°C for this set of primers to distinguish between the exon 9-exon 10 junction and the exon 9-exon 11 junction. Non-quantitative PCR was also done with primers specific to exon 9 (5′-GCTCCACTGAGAACCTGAA-3′) and the luciferase primer to visualize both 3R and 4R splice products by PAGE. Samples were run on a 10% TBE-acrylamide gel and visualized by post staining with 0.005% ethidium bromide in 1✕ TBE buffer.
Antisense RNA Treatment of Cells
Twenty-four hours after transfection, HeLa cells expressing the various tau minigene constructs were trypsinized and plated into either 96-well plates for luciferase measurements or 12-well plates for RNA isolation. These cells were then transfected with a 50 nm concentration of either a 2′-O-methyl antisense RNA with a phosphorothioate backbone (5′-UGAAGGUACUCACACUGCCGC-3′) or a randomized 2′-O-methyl RNA with a phosphorothioate backbone (Dharmacon) using Lipofectamine 2000 (Invitrogen).
Luciferase Measurements
Luciferase and Renilla activity levels were measured using the Dual Glo™ Luciferase Assay System from Promega.
RESULTS
Stabilizing Base Changes Decrease 4R Tau mRNA
In the stem loop-regulated model of tau mRNA splicing, destabilizing FTD-17 mutations lead to an increase in 4R mRNA. Likewise, stabilizing mutations should lead to an increase in 3R mRNA. We tested this supposition with exon trapping experiments based on minigenes previously described that express exons 9–11 and intronic sequences (21) with either a WT stem loop or one with the FTD-17 DDPAC mutation. This mutation is a C to T change in the +14 position relative to the exon 10 splice donor site (Fig. 1A) and results in nearly exclusive expression of 4R tau mRNA. Base changes that were predicted to increase the stability of the stem loop were made in minigenes containing both the WT and DDPAC mutated stem loops to address the effect of stabilizing the stem loop on tau splicing in vivo.
FIGURE 1. Stabilizing the tau stem loop in vitro.
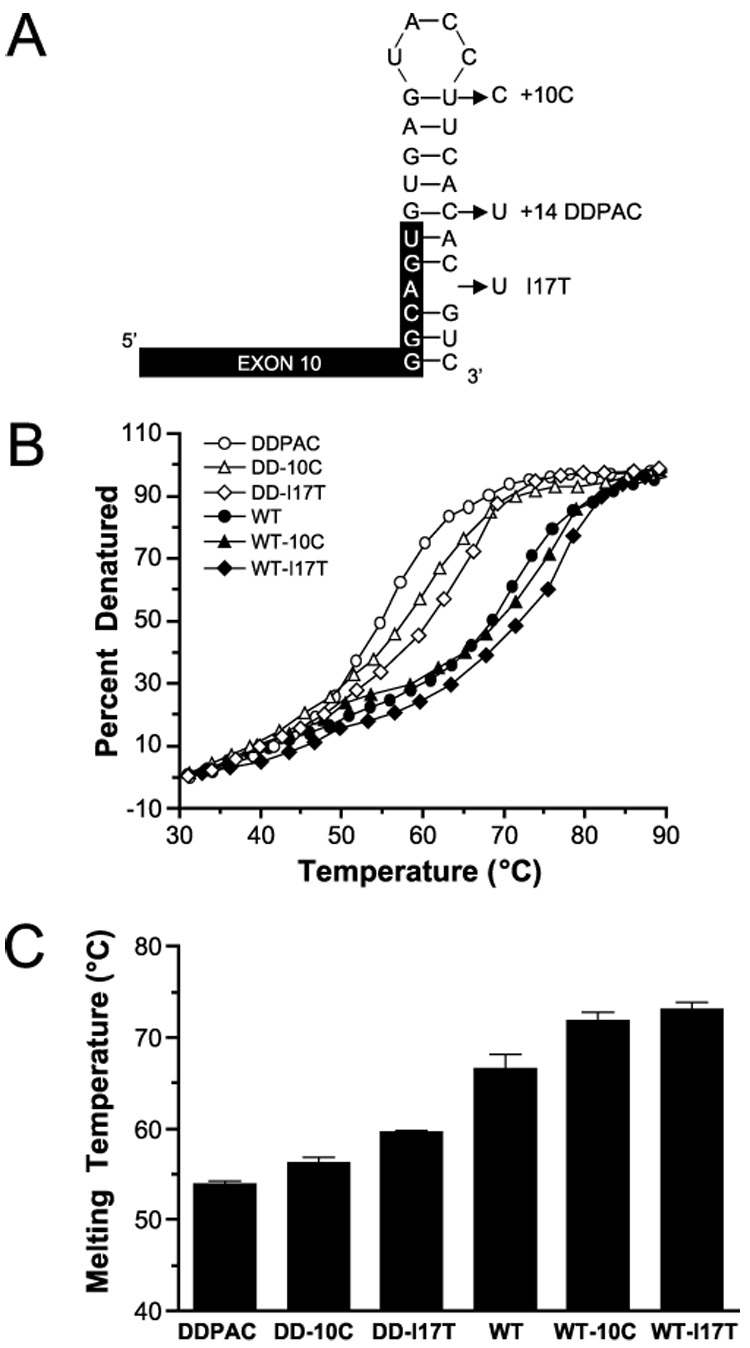
A, several base changes predicted to stabilize the tau mRNA stem loop in vitro were made in both the WT and DDPAC (+14) stem loops. B, UV melting curves were generated for all 6 stem loop variants: DDPAC (○), DD-10C (△), DD-I17T (◊) WT (●), WT-10C (▲), and WT-I17T (♦). C, melting temperatures (Table 1) derived from the melting curves are plotted here of increasing stability. These nucleotide changes enhanced the thermal stability of both the WT and +14 mutated stem loops.
The predicted structure of the tau stem loop has a G:U base pair at the top of the stem (Fig. 1A). To enhance the stability of the loop, this U at position +10 in the intron was changed to a C to generate a G:C pair (WT-10C). This change results in a predicted −1.2 kcal/mol increase in stability. Additionally, the predicted structure of the stem loop has a bulging A in the −3 position in the stem. One nucleotide, T, was inserted into the intron at position +17 (WT-I17T) to create an additional A:U pair in the stem. This additional base pair is predicted to increase stability by −5.3 kcal/mol (Table 1).We also generated these base changes in combination with the DDPAC mutation (DD-10C and DD-I17T) to determine whether the increase in stability conferred by these changes could mitigate the destabilizing effects of the DDPAC +14 mutation and reduce the expression of 4R tau. In addition to the predicted increase in stability the changes at these positions confer, they were selected because they are not located in the splice donor site or in the U1 snRNP binding site and should not interfere with the splicing machinery.
TABLE 1.
Relative stabilities of stem loop variants
| Stem loop variant | Predicted ΔGa | Measured Tm |
|---|---|---|
| kcal/mol | °C | |
| DDPAC | −6.1 | 53.9 ± 0.6 |
| DD-10C | −7.3 | 56.2 ± 1.3 |
| DD-I17T | −11.4 | 59.6 ± 0.6 |
| WT | −8.2 | 68.0 ± 1.1 |
| WT-10C | −9.4 | 71.7 ± 1.5 |
| WT-I17T | −13.5 | 73.0 ± 2.2 |
Stem loop RNAs (Fig. 1A) with the sequence changes described above were synthesized, and their melting temperatures, Tm, were determined by UV melting curves (Fig. 1, B and C). The stabilizing base changes increased the melting temperature of the WT stem loop by 3.7 and 5.0°C for WT-10C and WT-I17T, respectively. Additionally, the DD-10C and DD-I17T sequence changes increased the stability of the DD-Tau stem loop by 2.3 and 5.7°C, respectively. However, neither of these variants was as stabilizing as predicted by the RNA folding algorithm MFOLD (22,23) (Table 1). For instance, the DD-I17T change is predicted to be more stabilizing than the WT or WT-10C variants, but this is clearly not the case.
To test the effects of these base changes on splicing in vivo, we generated tau minigene constructs with the various stem loops described above. These constructs are based on the previously described tau minigene, TauEx9—11wt (21) (Fig. 2A). The minigenes were transiently expressed in HeLa cells and total RNA was isolated from transfected cells. Relative 3R and 4R mRNA levels were measured using real-time PCR with primers specific to the 3R and 4R splice isoforms, and the ratio of 4R to 3R was calculated (Fig. 2B). The relative ratio of 4R to 3R tau mRNA can be seen in Fig. 2C where semi-quantitative RT-PCR products were separated on a 10% TBE-acrylamide gel.
FIGURE 2. Stabilization of the stem loop reduces the 4R to 3R ratio.
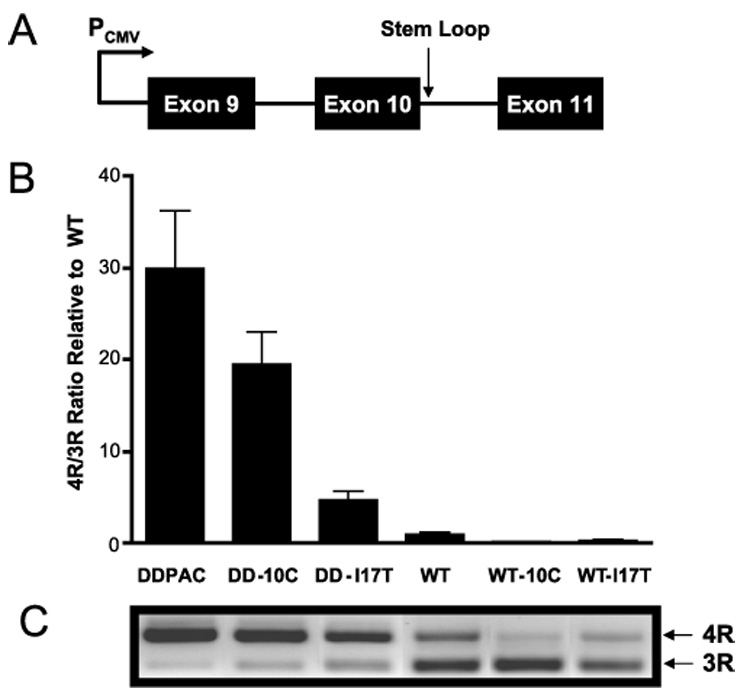
HeLa cells were transfected with minigenes (A) that expressed each of six tau stem loops. Quantitative RT-PCR (B) revealed that the ratio of 4R to 3R tau was reduced to the extent that the mutations stabilized the stem loop. C, relative amounts of 4R and 3R PCR products reflect the ratio of tau 4R and 3R isoforms quantitated by RT-PCR.
The linear model of tau splicing regulation proposes that the +14 DDPAC mutation leads to an increase in 4R due to disruption of a binding site for an intronic splicing silencer (10–12). Here the effect of the +14 mutation on splicing is suppressed in the same rank order as the Tm of the stem loop variants. Additionally, the ratio of 4R to 3R directly correlates (r² = 0.75, p < 0.05) with the predicted stabilities of the stem loop variants, with the 10C and I17T mutations having the strongest suppressive effects on exon 10 splicing.
Antisense Inhibition of Splicing Depends on the Stability of the Stem Loop
It has been previously reported that an antisense RNA directed against the splice donor site of exon 10 inhibits exon 10 splicing and leads to an increase in 3R tau mRNA (24). We used this antisense RNA to probe the stability of the stem loop in vivo. The antisense RNA binding site is from −8 to +13 relative to the splice site. The stem loop sequences as defined here span from positions −6 to +19. If the stem loop folds in vivo then the antisense RNA will need to compete with the RNA structure for binding (25,26). The more stable the structure, the less accessible the splice site is to binding by the antisense RNA, resulting in reduced antisense effectiveness in preventing splicing.
To quantitate the effects of antisense treatment on tau mRNA splicing, we constructed a splicing reporter based on the minigenes previously described above. In our modified construct, the P. pyralis luciferase cDNA is ligated to the 3′ end of exon 11. This construct has been designed to act as a reporter for 4R tau mRNA; the luciferase is only in frame when exon 10 is present. Additionally, the luciferase cDNA cloned into the minigene lacks it own translation initiating ATG and depends on an initiating ATG in exon 9 for expression. Therefore, luciferase acts as a reporter only when splicing has occurred. When transiently transfected into HeLa cells, this construct produces 4R and 3R in a ratio similar to what is observed in the adult brain (data not shown).
HeLa cells were transiently transfected with each of the six stem loop variants for 24 h. Transfected cells were then trypsinized, replated, and transfected with 50 nm of a 2′-O-methyl antisense RNA with a phosphorothioate backbone, which had been previously shown to inhibit the inclusion of exon 10 in tau mRNA (24). Transfection of the antisense RNA resulted in a decrease in luciferase expression relative to cells transfected with a 50 nm concentration of a randomized control oligoribonucleotide (Fig. 3A). All cells were replated to the same density and transfected from the same batch of antisense RNA-Lipofectamine complex to ensure that all cell lines were treated the same. The degree to which luciferase levels were decreased correlated (r² = 0.78, p < 0.05) with increasing stability of the tau stem loop. The more stable the stem loop is, the less effective is the antisense treatment.
FIGURE 3. Antisense RNA inhibition of exon 10 splicing correlates with stem loop stability.
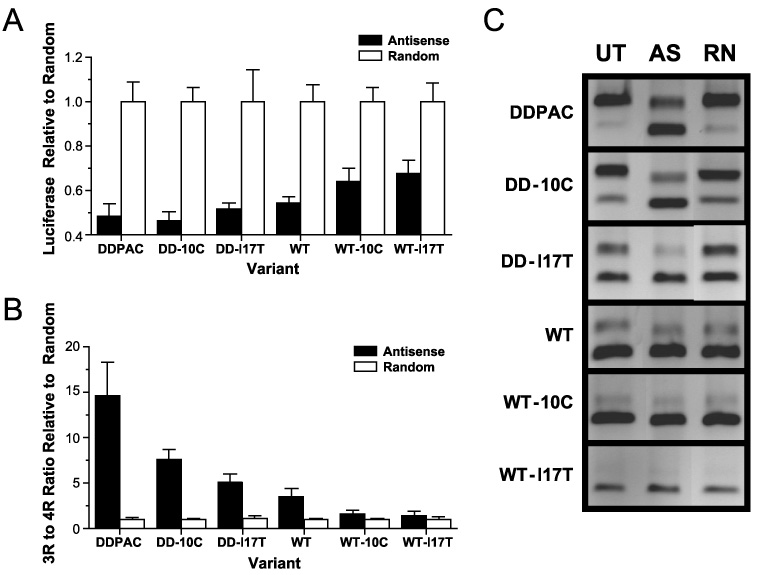
Antisense RNA directed against the intron-exon junction of exon 10 inhibits the inclusion of exon 10 in the mRNA. A, HeLa cells were transiently transfected with luciferase reporters for 4R tau expression for each of the 6 variants and then treated with antisense RNA. Luciferase levels decrease in cells treated with antisense RNA (filled columns) relative to cells treated with a randomized RNA (open columns). The degree to which luciferase levels decrease correlates with stem loop stability. B, this result is more evident when the ratio of 3R to 4R mRNA is measured using quantitative PCR on RNA collected from antisense treated cells (filled columns) relative to cells treated with a randomized RNA (open columns). C, PCR products of untreated (UT), antisense (AS), and random RNA (RN) treated cells are shown.
To confirm that the resulting increase in luciferase was in fact due to the conversion of 4R mRNA to 3R mRNA, real-time RT-PCR was performed on RNA extracted from the transfected cells. The resulting PCR products confirmed that the increase in luciferase is due to a dramatic increase in the 3R to 4R ratio of splice isoforms relative to cells transfected with the randomized oligoribonucleotide (Fig. 3, B and C). These results revealed a strong correlation between the antisense-induced changes in the 3R to 4R ratio and stem loop stability (r² = 0.85, p < 0.01). Antisense inhibition of exon 10 inclusion is most effective against the least stable stem loop (the DDPAC minigene).
DISCUSSION
Synonymous and intronic mutations found in and around exon 10 that cause FTD-17 clearly increase the inclusion of exon 10 in tau mRNA in vivo. However, uncertainty remains regarding the stem loop regulation model of tau mRNA alternative splicing. We provide here strong evidence that exon 10 splicing is substantially regulated by a stem loop structure that forms from sequences at the end of exon 10 and the beginning of the following intron. The ratio of 4R to 3R mRNA levels in cells strongly correlates with the measured melting temperatures of these stem loop variants in vitro. If this region of the pre-mRNA were strictly an unstructured protein binding site, this correlation would not be expected. In fact, the putative stabilizing mutations would be expected to have the same effect as putative destabilizing mutations, altering the sequence of the binding element. In particular, the I17T base insertion is within the defined intronic splicing silencer where most of the FTD stem loop mutations are found (10–12). These results, however, do not exclude the possibility that the stem loop structure itself is a protein binding site.
Interestingly, previous attempts to correlate stability with 3R expression have not been conclusive (13). In this report, the stabilities of the stem loops were not measured experimentally. We show here that measuring the actual Tm values of the stem loop variants is critical to evaluating the relative effectiveness of the stabilizing changes. In designing the stem loop variants studied here, we used the program MFOLD (22,23) to predict the free energy of each of the structures. We found that the predicted free energies of the structures differed substantially from the relative experimentally measured melting temperatures of the stem loops. The predicted free energies of both the DD-10C and the DD-I17T structures suggested they would be nearly as stable and substantially more stable, respectively, than the WT stem loop. However, measured melting temperatures for these structures revealed that they were both less stable than the WT RNA. The in vivo ratio of 4R to 3R reflected these measured Tm values rather than the predicted free energies of these structures.
We provide further evidence that the stem loop structure exists in vivo by demonstrating that the effectiveness of antisense RNA inhibition of exon 10 splicing is dependent on the stability of the stem loop structure. It has long been recognized that the structural nature of the target mRNA can have a dramatic effect of the efficacy of antisense RNA, and this effect is predictable (27,28). We used antisense RNA to probe the RNA structure at the exon 10 donor site as it exists in vivo. Because the binding site for the antisense RNA is contained within the stem loop structure, antisense RNAs have to compete with the structure for binding; the more stable the structure is the less effective is the antisense. We show here that as the stability of the stem loop element increases, the effectiveness of the antisense RNA decreases.
These data taken together provide convincing evidence that the RNA stem loop element is involved in regulating the relative expression levels of tau 4R and 3R mRNA levels. Additionally, these results bolster the previously proposed hypothesis concerning the pathogenicity of FTD-17 mutations located at the exon 10 exon-intron interface (8). These mutations disrupt the structural integrity of a RNA stem loop in the tau pre-mRNA, leading to increased expression of 4R tau mRNA. The existence of this stem loop suggests that it may be a drug target for FTD-17 therapeutics: small molecules that bind to this structure and stabilize it may thereby regulate tau mRNA splicing.
Acknowledgment
We thank Dr. Eriks Rozners of Northeastern University for the use of his Cary UV/VIS spectrophotometer and helpful discussions.
Footnotes
The abbreviations used are: WT, wild type; RT, reverse transcription; TBE, Tris borate-EDTA.
REFERENCES
- 1.Kosik KS, Shimura H. Biochim. Biophys. Acta. 2005;1739:298–310. doi: 10.1016/j.bbadis.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 2.Lee VM, Goedert M, Trojanowski JQ. Annu. Rev. Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 3.Hirokawa N. Curr. Opin. Cell Biol. 1994;6:74–81. doi: 10.1016/0955-0674(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 4.Neve R, Harris P, Kosik K, Kurnit D, Donlon T. Mol. Brain Res. 1986;1:271–280. doi: 10.1016/0169-328x(86)90033-1. [DOI] [PubMed] [Google Scholar]
- 5.Kosik KS, Orecchio LD, Bakalis S, Neve RL. Neuron. 1989;2:1389–1397. doi: 10.1016/0896-6273(89)90077-9. [DOI] [PubMed] [Google Scholar]
- 6.Andreadis A, Brown WM, Kosik KS. Biochemistry. 1992;31:10626–10633. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 7.Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Biochemistry. 1994;33:9511–9522. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- 8.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Moriis JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenbert T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 9.D’Souza I, Schellenberg GD. Biochim. Biophys. Acta. 2005;1739:104–115. doi: 10.1016/j.bbadis.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 10.D’Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, Schellenberg GD. Proc. Natl. Acad. Sci. U. S. A. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Souza I, Schellenberg GD. J. Biol. Chem. 2000;275:17700–17709. doi: 10.1074/jbc.M909470199. [DOI] [PubMed] [Google Scholar]
- 12.D’Souza I, Schellenberg GD. J. Biol. Chem. 2002;277:26587–26599. doi: 10.1074/jbc.M203794200. [DOI] [PubMed] [Google Scholar]
- 13.Grover A, Houlden H, Baker M, Adamson J, Lewis J, Prihar G, Pickering-Brown S, Duff K, Hutton M. J. Biol. Chem. 1999;274:15134–15143. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]
- 14.Miyamoto K, Kowalska A, Hasegawa M, Tabira T, Takahashi K, Araki W, Akiguchi I, Ikemoto A. Ann. Neurol. 2001;50:117–120. doi: 10.1002/ana.1083. [DOI] [PubMed] [Google Scholar]
- 15.Varani L, Hasegawa M, Spillantini MG, Smith MJ, Murrell JR, Ghetti B, Klug A, Goedert M, Varani G. Proc. Natl. Acad. Sci. U. S. A. 1999;96:8229–8234. doi: 10.1073/pnas.96.14.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yasuda M, Takamatsu J, D’Souza I, Crowther RA, Kawamata T, Hasegawa M, Hasegawa H, Spillantini MG, Tanimukai S, Poorkaj P, Varani L, Varani G, Iwatsubo T, Goedert M, Schellenberg DG, Tanaka C. Ann. Neurol. 2000;47:422–429. [PubMed] [Google Scholar]
- 17.Jiang Z, Tang H, Havlioglu N, Zhang X, Stamm S, Yan R, Wu JY. J. Biol. Chem. 2003;278:18997–19007. doi: 10.1074/jbc.M301800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varani L, Spillantini MG, Goedert M, Varani G. Nucleic Acids Res. 2000;28:710–719. doi: 10.1093/nar/28.3.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puglisi JD, Tinoco I., Jr Methods Enzymol. 1989;180:304–325. doi: 10.1016/0076-6879(89)80108-9. [DOI] [PubMed] [Google Scholar]
- 20.Donahue CP, Jensen RV, Ochiishi T, Eisenstein I, Zhao M, Shors T, Kosik K. Hippocampus. 2002:821–833. doi: 10.1002/hipo.10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang Z, Cote J, Kwon JM, Goate AM, Wu JY. Mol. Cell. Biol. 2000;20:4036–4048. doi: 10.1128/mcb.20.11.4036-4048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuker M. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathews DH, Sabina J, Zuker M, Turner DH. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 24.Kalbfuss B, Mabon SA, Misteli T. J. Biol. Chem. 2001;276:42986–42993. doi: 10.1074/jbc.M105113200. [DOI] [PubMed] [Google Scholar]
- 25.Toulme JJ, Tinevez RL, Brossalina E. Biochimie. (Paris) 1996;78:663–673. doi: 10.1016/s0300-9084(96)80012-5. [DOI] [PubMed] [Google Scholar]
- 26.Toulme JJ, Di Primo C, Moreau S, Sczakiel G, Tinevez RL, Brossalina E. Prog. Nucleic Acids Res. Mol. Biol. 2001;69:1–46. doi: 10.1016/s0079-6603(01)69043-3. [DOI] [PubMed] [Google Scholar]
- 27.Sczakiel G. Antisense Nucleic Acid Drug Dev. 1997;7:439–444. doi: 10.1089/oli.1.1997.7.439. [DOI] [PubMed] [Google Scholar]
- 28.Vickers TA, Wyatt JR, Freier SM. Nucleic Acids Res. 2000;28:1340–1347. doi: 10.1093/nar/28.6.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]