

Abstract
Tau protein, which binds to and stabilizes microtubules, is critical for neuronal survival and function. In the human brain, tau pre-mRNA splicing is regulated to maintain a delicate balance of exon 10-containing and exon 10-skipping isoforms. Splicing mutations affecting tau exon 10 alternative splicing lead to tauopathies, a group of neurodegenerative disorders including dementia. Molecular mechanisms regulating tau alternative splicing remain to be elucidated. In this study, we have developed an expression cloning strategy to identify splicing factors that stimulate tau exon 10 inclusion. Using this expression cloning approach, we have identified a previously unknown tau exon 10 splicing regulator, RBM4 (RNA binding motif protein 4). In cells transfected with a tau minigene, RBM4 overexpression leads to an increased inclusion of exon 10, whereas RBM4 down-regulation decreases exon 10 inclusion. The activity of RBM4 in stimulating tau exon 10 inclusion is abolished by mutations in its RNA-binding domain. A putative intronic splicing enhancer located in intron 10 of the tau gene is required for the splicing stimulatory activity of RBM4. Immunohistological analyses reveal that RBM4 is expressed in the human brain regions affected in tauopathy, including the hippocampus and frontal cortex. Our study demonstrates that RBM4 is involved in tau exon 10 alternative splicing. Our work also suggests that down-regulating tau exon 10 splicing activators, such as RBM4, may be of therapeutic potential in tauopathies involving excessive tau exon 10 inclusion.
Microtubule-associated protein tau regulates the organization and stability of microtubules (MTs)2 in the neurons. In humans, the Tau protein is encoded by a single gene on chromosome 17. The tau gene is expressed at a high level in neurons and at lower levels in glia and certain nonneuronal cells. Involved in maintaining cell morphology, axonal extension, and vesicle transport, Tau is critical for the formation and function of neurons (1–3) (for recent reviews, see Refs. 4–9). The expression of the tau gene is under complex regulation at multiple steps, including both post-transcriptional and post-translational levels. In the human brain, six tau isoforms are expressed as a result of alternative splicing of exons 2, 3, and 10 (10–12). Alternative splicing of exon 10 (Ex10), which encodes for one of the four MT-binding domains, gives rise to tau isoforms containing either four MT-binding repeats (Tau4R, Ex10+) or three MT- binding repeats (Tau3R, Ex10−). In the adult human brain, the regulated splicing of exon10 results in a ratio of Tau4R to Tau3R of approximately 1.
Genetic studies have revealed a number of mutations in the human tau gene in patients with tauopathy. More than 30 different mutations have been associated with frontotemporal dementia with parkinsonism linked to chromosome-17 (FTDP-17) (13–15). This is an autosomal dominant disorder with clinically heterogeneous manifestations that include behavioral, cognitive, and motor abnormalities. FTDP-17 mutations can be classified into two groups, missense mutations that affect Tau protein activity and splicing mutations that alter the ratio of distinct tau splicing isoforms (for recent reviews, see Refs. 4–9). Almost all splicing mutations characterized so far affect the regulation of tau exon 10 splicing. In vitro experiments suggest that Tau4R and Tau3R proteins bind and stabilize MTs in different manners (16–18). This delicate balance between exon 10+ to exon 10− tau isoforms is crucial for neuronal function in learning and memory. However, the underlying mechanism remains to be elucidated (13, 19, 20) (reviewed in Ref. 5).
A large number of genes in the human genome utilize alternative splicing to generate functionally distinct gene products. Understanding how these alternative splicing events are regulated is an important issue in functional genomics. In the past 2 decades, a number of alternative splicing regulators have been identified. Many of these trans-acting factors were initially identified using biochemical approaches (reviewed in Refs. 21–23). In this study, we have developed an expression cloning approach using a tau exon 10 splicing green fluorescent protein (GFP) reporter, Tau4R-GFP, in which GFP expression was dependent on the tau exon 10 inclusion. Using this system to screen a human brain cDNA library, we have identified a previously unknown tau exon 10 splicing activator, RBM4, a protein recently shown to play a role in alternative splicing of α-tropomyosin (24). Our experiments show that overexpression of RBM4 stimulates tau exon 10 inclusion, and RNA interference (RNAi)-mediated knock-down of RBM4 expression in transfected cells reduces tau exon inclusion. Immunohistochemical staining indicates that RBM4 is expressed in the brain regions involved in tauopathy. Our results support a role of RBM4 in regulating tau exon 10 splicing.
EXPERIMENTAL PROCEDURES
Expression Cloning Using the Tau Reporter Gene
We prepared a human adult brain cDNA library using the adult brain mRNA purchased from Clontech. The quality of this cDNA library was tested using reverse transcription-PCR and small scale screening before the library was subdivided into 400 cDNA pools. It was estimated that each pool contained ~5000 clones. Each of these 400 primary pools was amplified for 6–8 h at 37 °C after being carefully plated on 20 × 20-cm bacterial plates. cDNAs were prepared from each pool using an optimized medium scale plasmid preparation method combining polyethylene glycol precipitation with the endotoxin-free DNA preparation kit (Qiagen) to ensure the high quality of DNA preparations for transfection. The typical transfection efficiency is ~50–60%, as assessed by GFP expression in the control cells transfected with a GFP plasmid. For the first round of transfection, 40 combined pools (combined from 10 primary pools in a grid fashion to allow easy identification of individual positive primary pools) of cDNAs were transfected in cells stably expressing the Tau GFP reporter gene. Using this grid design, 400 cDNA pools were screened in 40 primary transfections in 15-cm tissue culture dishes and further screened in subsequent rounds of transfections.
Because HEK293 cells had a high efficiency of transfection and a low background GFP expression, we chose this cell line for preparing a stable cell line expressing the Tau reporter gene. Stable HEK293 cells expressing Tau4R-GFP reporter were selected with G418 and used for expression of cDNA clones following transfection with pooled cDNAs from the adult brain cDNA library. During the first two rounds of transfection, it appeared more efficient to visually examine individual transfected culture plates under an inverted fluorescent microscope than using a cell sorter. The primary positive pools that gave significant activation of GFP expression were identified, and cDNAs in these pools were further divided. For each primary positive pool, 20 subpools were prepared and individually transfected into stable cells expressing the Tau4R-GFP reporter gene in 10-cm dishes. The positive subpools were again identified using inverted fluorescent microscopy and subdivided for another round of transfection until individual single cDNA clones were isolated. After seven rounds of retransfection, we were able to isolate single cDNA clones that have significant effects on tau exon 10 splicing.
Plasmids and Antibodies
Mammalian expression plasmids for HA-tagged wild type or mutant RBM4 (25) and for raver were described previously (26). The plasmid expressing RNAi for RBM4 was described (24). Monoclonal anti-HA antibody was purchased from Covance. Affinity-purified polyclonal anti-RBM4 was prepared as described previously (25).
Transfections and RT-PCR
HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum. Transfection was carried out using a previously described calcium phosphate precipitation procedure with 1–3 µg of DNA (27). Cells were harvested 48 h after transfection, and the RNA was extracted using the RNAeasy kit (Qiagen). In RNAi experiments, Lipofectamine 2000 (Invitrogen) was used according to the manufacturer’s instructions. Splicing products of the expressed tau minigenes were detected using RT-PCR as previously described (27). RT-PCR was also performed to detect the endogenous Bcl-xL and γ-actin genes using the corresponding specific primers. The products were analyzed by electrophoresis using 6% polyacrylamide gels.
RNA-Protein Interactions
The RNA probes were prepared by in vitro transcription in the presence of [32P]UTP using T7 RNA polymerase from linearized DNA templates as described (28). The RNA probes were gel-purified and used for the UV-cross-linking assay. The recombinant RBM4 protein was expressed as a His6-tagged protein and purified from Escherichia coli as described by Lai et al. (25). For UV cross-linking, ~20 fmol of RNA probe was incubated at 30 °C with 1 µg of purified RBM4 or 30 µl of cell lysate in a 40-µl reaction mixture under in vitro splicing conditions (28). The samples were irradiated on ice under 254-nm light for 10 min. The probes were digested in equal volumes of RNase A (5 mg/ml) (Sigma) at 30 °C for 20 min. For immunoprecipitation, following the RNase treatment, samples were incubated with HA.11 anti-HA monoclonal antibody (Covance) at 4 °C for 2 h. Protein A/G-agarose beads were then added with further incubation and gentle rocking. Following washing, the RNA-bound proteins immunoprecipitated were eluted and resolved on SDS-PAGE followed by autoradiography.
Immunohistochemical Staining
Human brain samples from autopsy material were fixed in formalin and paraffin-embedded. 6-µm-thick sections from the frontal and hippocampal regions were used for immunohistochemical staining. The Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) staining kit for rabbit polyclonal antibody was used to perform immunostaining with the affinity-purified rabbit anti-RBM4 antibody. Antigen retrieval was carried out in the presence of 1 mM EDTA, pH 9.0. Primary antibody was used at a 1:1000 dilution. Immunostaining was carried out following the instruction manual, color development was performed with 3,3′-diaminobenzidine (DAB) and counterstaining was with hematoxylene.
RESULTS
Establishing a GFP Reporter-based Strategy for Cloning Trans-acting Splicing Regulators That Stimulate Exon 10 Inclusion
In our previous studies, a human tau minigene has been established that recapitulates tau exon 10 alternative splicing in the human brain (28, 29). In this minigene system, alternative inclusion or exclusion of the exon 10 leads to the formation of Tau4R and Tau3R, respectively. To develop a system for identifying splicing activators that enhance tau exon 10 inclusion, we prepared a tau exon 10-splicing GFP reporter (Tau4R-GFP) by fusing the tau minigene with a cDNA fragment encoding GFP (Fig. 1). This Tau4R-GFP reporter gene was constructed by adding a translation start codon upstream of exon 9 so that the tau minigene construct produced a fusion protein containing tau peptide fragment fused to GFP (Fig. 1A). One base pair was inserted by site-directed mutagenesis in exon 10. This Tau4R-GFP reporter contained the GFP gene in frame with the 94-bp exon 10 so that the GFP gene was expressed as a functional protein product only when exon 10 was included (Fig. 2). The Tau4R-GFP reporter gene enabled us to use GFP expression as an indicator for exon 10 inclusion. When the Tau4R-GFP reporter gene was transfected into HeLa or HEK293 cells, the tau minigene underwent splicing in a similar pattern as the wild type tau gene, producing both Tau3R and Tau4R splicing isoforms. No aberrant splicing, such as activation of cryptic splice sites, was detected (data not shown). The Tau4R-GFP reporter showed a low background of GFP expression in HEK293 cells, facilitating the expression cloning screening as described below.
FIGURE 1.
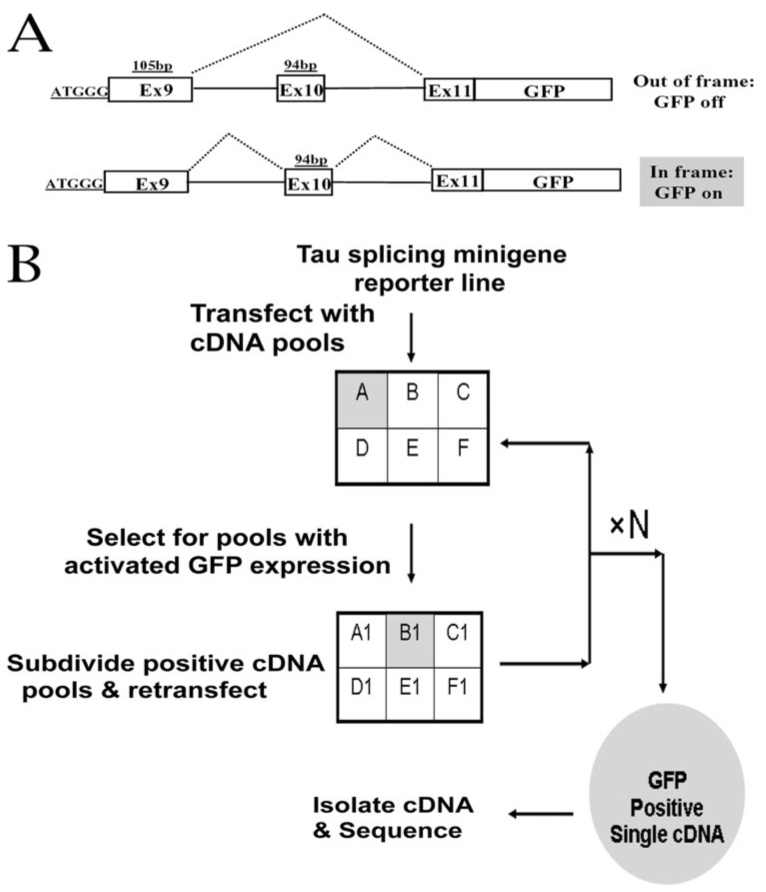
A, a schematic diagram of the Tau4R-GFP reporter constructs. The tau splicing reporter gene is designed in such a way that GFP is only expressed when the exon 10 is included (for a detailed description of the reporter gene construction, see "Experimental Procedures"). B, a diagrammatic representation of the cDNA library screening protocol based on GFP expression following progressive division and subdivision of cDNA pools and transfection of cDNA pools into the stable cell line expressing Tau4R-GFP reporter.
FIGURE 2. The expression of Tau-GFP reporter during the cDNA library screening.
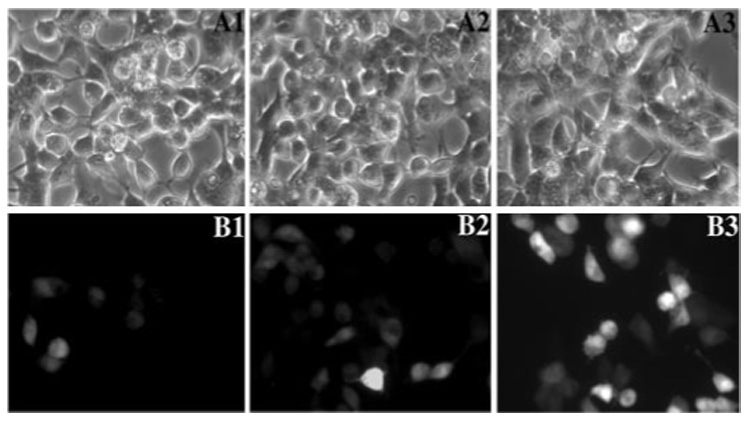
The stable HEK293 cell line expressing the Tau4R-GFP reporter gene was used for screening cDNA library. A progressive subdividing-retransfecting method was used to identify cDNAs encoding potential regulators of tau alternative splicing, as described under "Experimental Procedures." GFP fluorescence was monitored by fluorescent microscopy following the transfection of pools of cDNAs prepared from the cDNA library. A1–A3, phase-contrast images; B1–B3, corresponding fluorescent microscopic images. These experiments show progressive enrichment of GFP-expressing cells following the primary round (B1), the third round (B2), and the sixth round (B3) of subdivision and retransfection of the positive cDNA pools.
Identifying Trans-acting Factors Enhancing Tau Exon 10 Inclusion
We have been interested in identifying protein factors important for regulating tau exon 10 alternative splicing. By examining their biochemical function and molecular mechanisms, we wish to understand how these splicing regulators control the balance of different tau splicing isoforms. To search for splicing factors that stimulate tau exon 10 splicing, we made use of the Tau4R-GFP reporter gene cassette (Fig. 1A). We developed a progressive subdivision and screening protocol (Fig. 1B) to screen a human brain cDNA library (for details, see “Experimental Procedures”). Following seven rounds of resubdivision and retransfection, seven individual cDNA clones that influence tau exon 10 splicing were isolated. Five cDNA clones are still under characterization. Sequencing analyses of the remaining cDNAs and data bank searches showed that two cDNA clones encode an RNA-binding protein known as RNA-binding motif protein 4, RBM4 (25). RBM4 protein will be the focus of this report.
Confirming the Activity of RBM4 in Regulating Tau Exon 10 Splicing
To validate the screen and rule out potential indirect effects on the GFP reporter, we examined the effect of RBM4 on tau exon 10 splicing using the original tau exon 9-10-11 splicing substrate (29). Another RNA recognition motif (RRM)-containing splicing factor, Raver (40), was also tested. The tau minigene was co-transfected with either control vector or RBM4 or Raver expression plasmid (Fig. 3). Following transfection, tau exon 10 alternative splicing pattern was examined by RT-PCR using tau minigene-specific primers (Fig. 3A). RBM4 expression was measured both at the RNA level (by specific RT-PCR with actin as the internal control, Fig. 3, B and C) and at protein level (by Western blotting; Fig. 3D). Overexpression of RBM4 led to an increased inclusion of exon 10 (Fig. 3A, lane 2), whereas transfection of Raver (Fig. 3A, lane 3) or control vector transfection (Fig. 3A, lane 1) did not affect exon 10 splicing (Fig. 3A, compare lane 2 with lane 1 or lane 3). These results confirmed that RBM4 stimulates tau exon 10 inclusion, supporting that the effect of RBM4 detected in expression cloning is due to its activity in stimulating tau exon 10 splicing rather than other effects on GFP expression.
FIGURE 3. Overexpression of RBM4 protein increases the inclusion of tau exon 10 in transfected cells.
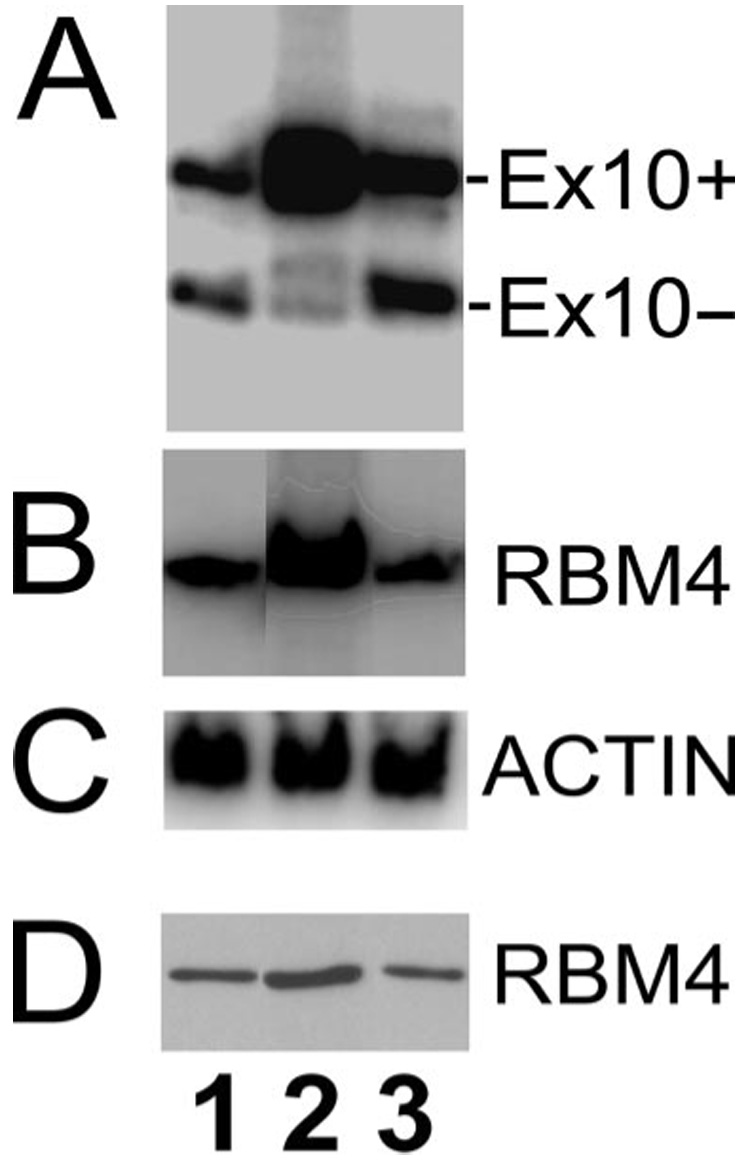
The corresponding cDNA plasmids expressing either the vector control (lane 1), RBM4 (lane 2), or Raver (lane 3) were co-transfected with Tau minigene tauEx9-10-11 into HEK293 cells. A, tau exon 10 alternative splicing was detected using RT-PCR. B, the expression level of RBM4 transcript in the corresponding groups using RT-PCR with RBM4 specific primers. C, the levels of actin transcript in the corresponding groups, showing comparable amounts of RNA used in each reaction. D, RBM4 protein levels in the corresponding groups as detected by Western blotting using the anti-RBM4 antibody.
The RNA Recognition Motif in RBM4 Is Essential for the Stimulation of Tau Exon 10 Inclusion
RBM4 is a protein containing an RNA recognition motif. We examined whether the interaction of RBM4 with RNA was required for its activity in stimulating tau exon 10 splicing. We made use of an RBM4 mutant containing mutations in the conserved aromatic residues of the RRM domain of RBM4, RBM4-mRRM. This mutant is deficient in RNA binding (25). Mammalian expression plasmids encoding wild type or mutant (RBM4-mRRM) proteins containing a hemagglutinin (HA) tag were co-transfected into HEK293 cells together with the tau minigene. Tau exon 10 alternative splicing was then measured using RT-PCR. Although the wild-type and mutant RBM4 proteins were expressed at a similar level (Fig. 4, B and D), the stimulation of tau exon 10 splicing was observed only in cells transfected with the wild-type and not mutant RBM4 (Fig. 4A). The quantification of tau splicing isoforms in these co-transfection-coupled RT-PCR experiments indicated that the ratio of exon 10-including to exon 10-skipping tau isoforms (Ex10+/Ex10−) was significantly increased in cells transfected with the wild-type RBM4 as compared with those of the vector control or cells expressing the RRM mutant RBM4 (p < 0.001). On the other hand, overexpression of the RRM mutant RBM4 protein did not affect tau exon 10 splicing, similar to the vector control (Fig. 4E). These experiments demonstrate that the RNA recognition domain of RBM4 is required for its stimulation of exon 10 inclusion, suggesting that RBM4 may regulate tau exon 10 splicing by interacting with tau pre-mRNA.
FIGURE 4. The RNA recognition domain of RBM4 is essential for its activation of tau exon 10 inclusion.
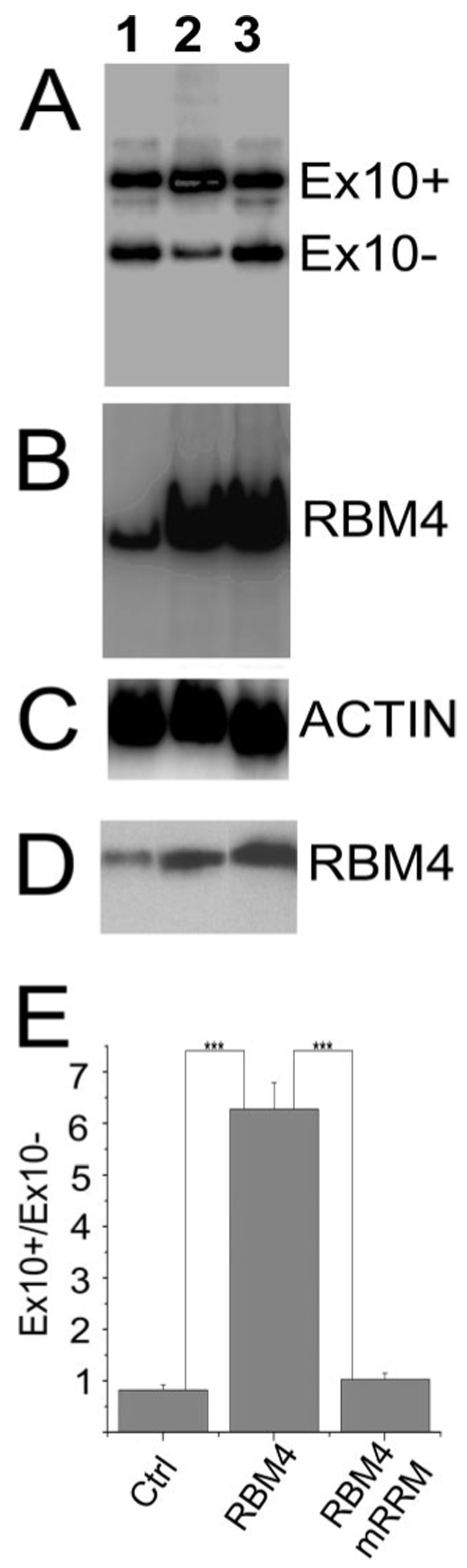
The tau minigene was co-transfected with either the vector control (lane 1) or cDNA expression plasmid encoding the wild-type RBM4 (lane 2) or the mutant RBM4 (RBM4mRRM; lane 3). A, the tau exon 10 splicing pattern as detected by RT-PCR. B, the level of RBM4 transcripts as detected by RT-PCR for each group. C, the actin transcript levels as detected by RT-PCR for internal controls. D, the level of RBM4 protein in each group as detected by Western blotting using a specific anti-RBM4 antibody. E, quantification of tau exon 10 splicing data derived from six independent experiments. The graph shows the average ratio Ex10+/Ex10− transcripts ± S.E. (***, p < 0.001).
RBM4 Interacts with an Intronic Element in Tau Pre-mRNA
The observation that the RNA recognition motif of RBM4 is necessary for its splicing stimulatory activity in tau exon 10 splicing prompted us to test whether RBM4 interacts with tau pre-mRNA. A UV-cross-linking-coupled immunoprecipitation assay was performed using 32P-labeled tau pre-mRNA and protein extracts prepared from HEK293 cells transfected with HA-tagged RBM4 (HA-RBM4) or the vector control. Following UV cross-linking, immunoprecipitation was carried out using a monoclonal anti-HA antibody, and immunoprecipitation products were analyzed by SDS-PAGE followed by autoradiography. A 40-kDa band was detected in the reactions containing HA-RBM4 (Fig. 5A, lane 2) but not in the vector control (Fig. 5A, lane 1), indicating that RBM4 interacts with tau pre-mRNA. To map the site of RBM4 interaction, different regions of tau pre-mRNA were tested. UV-cross-linking experiments using RNA fragments corresponding to different regions of tau pre-mRNA suggest that RBM4 interacts with an intronic region downstream of exon 10 in the tau pre-mRNA (Fig. 5, B and C). In the presence of control RNA, no cross-linking signal was detected with RBM4 (Fig. 5B, lane 1). Specific UV-cross-linking signal was detected when HA-RBM4 protein extract was incubated with tau pre-mRNA transcript containing the exon 10 with the 5′ end of intron 10, and the removal of exon 11 together with the 3′ end of intron 10 did not affect RBM4 binding (Fig. 5B, lane 2). The removal of exon 10 did not affect the UV cross-linking, suggesting that the intronic sequence immediately downstream of the 5′ splice site of exon 10 contained an RBM4 binding site (Fig. 5C). The UV-cross-linking signal was detected when a 260-nucleotide intronic fragment located 100 nucleotides downstream of the 5′ splice site of exon 10 was incubated with the wild-type HA-RBM4 protein extract but not the vector control (Fig. 5C, lane 1 and 2, respectively). Therefore, the RNA transcript containing the 260-nucleotide intronic sequence alone was sufficient to interact with RBM4 (Fig. 5C). This interaction was significantly reduced when the RRM mutant RBM4 was used (Fig. 5C, lane 3). These results suggest that RBM4 interacts with tau pre-mRNA either directly or indirectly through association with other protein(s) present in the cell extracts.
FIGURE 5. RBM4 interacts with an intronic element downstream of the 5′ splice site of exon 10 in tau pre-mRNA.
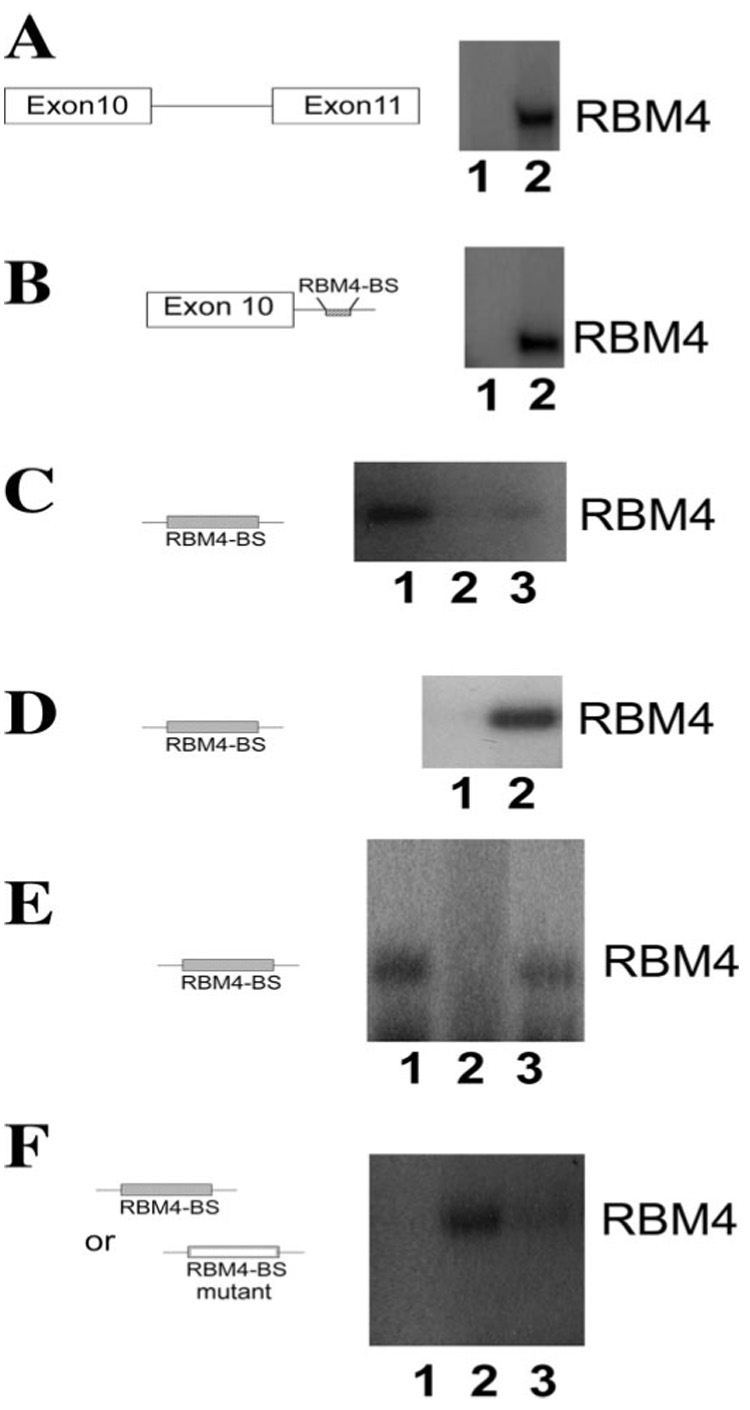
UV-cross-linking experiments were performed using either protein extracts prepared from cells transfected with HA-RBM4 expression plasmid (A–C) or purified recombinant RBM4 protein (D–F). Tau pre-mRNA or its fragments, as depicted, were radiolabeled and used in the cross-linking assay. Following UV cross-linking and RNase treatment, the reactions were analyzed using autoradiography following SDS-PAGE. A and B, different tau pre-mRNA transcripts (lane 2) or control RNA transcripts (lane 1) were incubated with cell lysates containing HA-RBM4. Following UV cross-linking and RNase digestion, RBM4 was immunoprecipitated using a monoclonal anti-HA antibody. Immunoprecipitates were analyzed on SDS-PAGE followed by autoradiography. C, radiolabeled RNA fragment containing the putative RBM4 binding site was incubated with protein extracts containing HA-tagged wild-type RBM4 (lane 1), vector control (lane 2), or HA-tagged RBM4-mRRM mutant (lane 3). D, recombinant RBM4 protein was incubated with the radiolabeled control RNA (lane 1) or the tau intronic RNA fragment containing the putative RBM4-binding element (lane 2) under splicing conditions followed by UV cross-linking. E, the UV-cross-linking experiment was performed with recombinant RBM4 protein and 32P-labeled RNA transcript containing the tau intronic RBM4-binding element under splicing conditions in the absence of competitor RNA (lane 1) or in the presence of a 100-fold molar excess of unlabeled tau RNA containing the RBM4-binding element (lane 2) or in the presence of a 100-fold molar excess of unlabeled control RNA (lane 3). F, the UV-cross-linking experiment was carried out using recombinant RBM4 protein with radiolabeled control transcript (lane 1) or with a RNA transcript containing either the wild-type (lane 2) or mutant (lane 3) RBM4-binding element under the splicing conditions. Autoradiographs of gels following SDS-PAGE of the UV-cross-linking reaction products are shown in all panels.
To investigate whether RBM4 directly interacts with tau pre-mRNA, we tested purified recombinant RBM4 protein using the UV-cross-linking assay. The purified RBM4 protein interacted with the tau pre-mRNA transcript but not the nonspecific control RNA (Fig. 5D, lane 2 and 1, respectively), indicating that RBM4 interacts with tau pre-mRNA directly rather than indirectly via other protein(s). To test the specificity of the interaction between RBM4 and the tau intronic region, the UV-cross-linking assay was carried out in the presence of excess amount of unlabeled RNA, either the tau pre-mRNA or nonspecific control RNA as competitors. The RBM4 binding signal was significantly reduced in the presence of the unlabeled tau RNA containing the 260-nucleotide intronic region (Fig. 5E, lane 2), as compared with the reaction in the absence of competitive RNA (Fig. 5E, lane 1) or with that in the presence of unlabeled nonspecific control RNA (Fig. 5E, lane 3). These experiments demonstrate that RBM4 specifically interacts with tau pre-mRNA in the intronic region downstream of the regulated exon 10.
Mutation of the Intronic RBM4 Binding Site Reduces the Activity of RBM4 in Stimulating Tau Exon 10 Inclusion
The UV-cross-linking experiments described above show that RBM4 interacts with tau pre-mRNA in the intronic region downstream of the 5′ splice site of exon 10. Sequence analysis indicates that a pyrimidine-rich element (UCCUUCUUG) is located 100 nucleotides downstream of the 5′ splice site in this intronic region. This element shares sequence similarity to the RBM4 binding site identified in α-tropomyosin pre-mRNA (24). To examine the potential role of this putative RBM4 binding site, we performed site-specific mutagenesis and prepared a mutant tau minigene by changing the pyrimidine nucleotides in the UCCUUCUUG element to purine nucleotides. The mutant tau pre-mRNA (tau-mRBMBS) was then tested for its interaction with the purified RBM4 protein. As compared with the wild-type tau pre-mRNA, the mutant tau transcript showed significantly reduced binding to RBM4 protein (Fig. 5F, compare lane 3 with lane 2).
To test the functional significance of this intronic RBM4 binding site in tau exon 10 splicing, we compared tau exon 10 splicing of the wild type and the mutant tau pre-mRNA in the cells co-transfected with the RBM4 expression plasmid. RBM4 overexpression was confirmed both at the RNA and at the protein levels (Fig. 6, B and D, with actin level as the internal control shown in Fig. 6C). The mutant tau pre-mRNA showed a significantly reduced response to RBM4 compared with the wild-type tau splicing substrate (Fig. 6A, compare lanes 1 and 2). Although the splicing stimulatory activity of RBM4 was not completely eliminated, the effect of the mutation in the RBM4 binding site on RBM4-mediated exon 10 inclusion was consistently observed. Fig. 6E shows the quantification of ratios of exon 10+/exon 10− tau transcripts from six independent experiments. When the intronic UCCUUCUUG was mutated, the stimulatory activity of exon 10 inclusion by RBM4 overexpression was reduced, changing the ratio of exon 10+/exon 10− from ~6 with the wild-type tau splicing substrate to 2 with the mutant tau pre-mRNA (Fig. 6E). The residual stimulatory activity by RBM4 on the mutant tau pre-mRNA is consistent with the observation that RBM4 binding was not completely abolished in the mutant tau pre-mRNA (Fig. 5E, lane 3).
FIGURE 6. Mutation in the RBM4 putative binding site in tau pre-mRNA affects the exon 10 splicing stimulatory activity of RBM4.
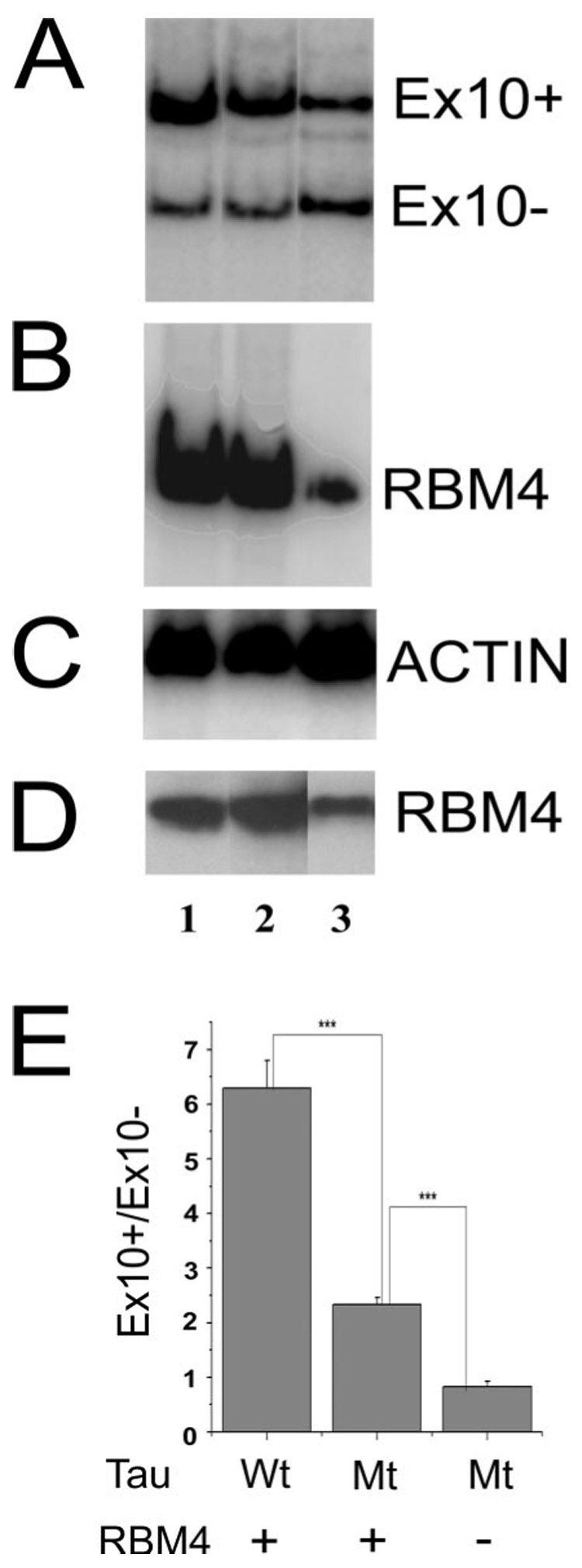
The expression plasmid for RBM4 (lanes 1 and 2) or the vector control (lane 3) was co-transfected into HEK293 cells with either the wild-type tau minigene (lane 1) or the mutant tau minigene (tauEx9-10-11mRBM4BS) in which the putative RBM4-binding site had been mutated (lanes 2 and 3). A, tau exon 10 alternative splicing was assayed using RT-PCR. B, the level of RBM4 transcript was detected by RT-PCR with RBM4-specific primers. C shows levels of actin used as loading controls in RT-PCR; D, the level of RBM4 protein was detected by Western blotting using a specific RBM4 antibody. E, quantification of results derived from six independent experiments. The graph shows the average ratio Ex10+/Ex10− transcripts ± S.E. (***, p < 0.001).
Knockdown of RBM4 by RNAi Decreases Tau Exon 10 Inclusion, Suggesting a Role of Endogenous RBM4 in Stimulating Exon 10 Inclusion
To test the potential role of the endogenous RBM4 in regulating exon 10 splicing, we used RNAi. RBM4-specific RNAi or control RNAi was co-transfected using Lipofectamine along with the tau minigene into HEK293 cells. For reasons not clear yet, cells transfected under these conditions showed an increase in Tau4R even in mock or control transfections (data not shown; see Fig. 7B, lane 1). The effect of the RNAi was evaluated at both RNA and protein levels. RT-PCR with RBM4-specific primers and Western blotting with anti-RBM4 antibody showed that the RBM4-specific RNAi transfection led to a ~70–80% reduction in RBM4 expression level (RNA in Fig. 7C and protein in Fig. 7E, respectively; compare lanes 1 and 2). We examined tau exon 10 splicing by measuring the ratio of exon 10 splicing isoforms (Ex10+/Ex10−) following the quantitative RT-PCR as described before. RBM4 down-regulation by RNAi led to a decrease in the inclusion of exon 10 (Fig. 7A, compare lane 1 and 2). The results from six independent experiments were quantified. RNAi knockdown of RBM4 led to an ~7-fold decrease in exon 10 inclusion as compared with the control RNAi (Fig. 7F). This effect was specific because alternative splicing of other genes examined was not affected, including Bcl-x (Fig. 7B). This observation suggests relative specificity of RBM4 in regulation of tau exon 10 alternative splicing. These results indicate that the endogenous RBM4 protein plays a role in activating tau exon 10 inclusion.
FIGURE 7. RNAi-mediated knockdown of RBM4 decreases tau exon 10 inclusion.
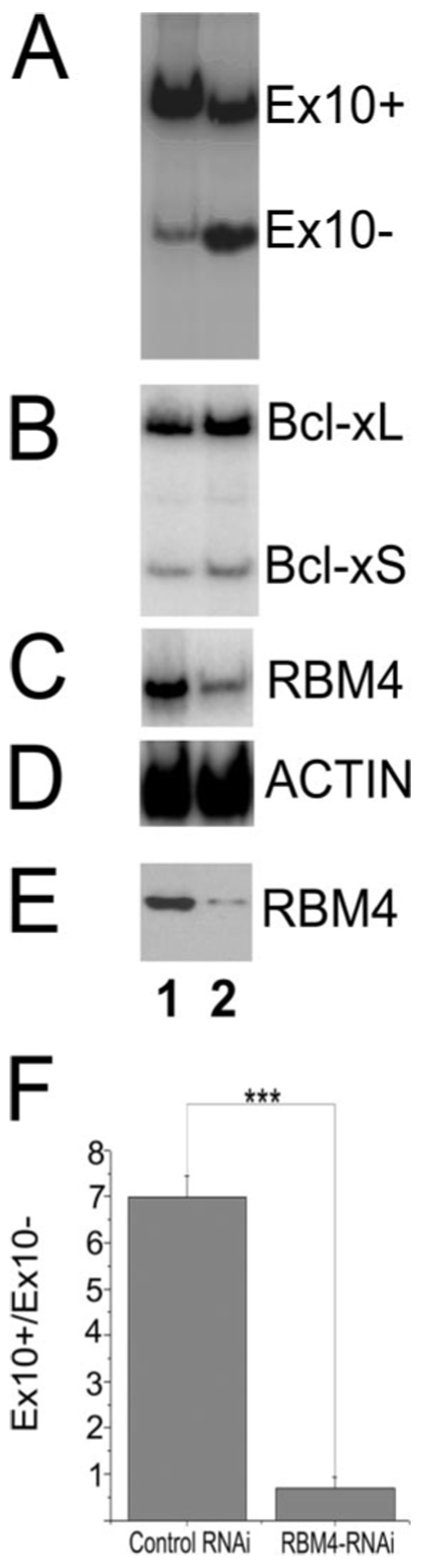
RNAi specific for human RBM4 (lane 2) or control RNAi (lane 1) was co-transfected with Tau minigene TauEx9-10-11 (wild type) into HEK293 cells as described under "Experimental Procedures." RNA preparations or protein lysates were made 48 h post-transfection and analyzed by RT-PCR or Western blotting. A, tau exon 10 alternative splicing isoforms as detected by RT-PCR. B, the alternative splicing pattern of Bcl-x detected by RT-PCR using Bcl-x-specific primers. C, the level of RBM4 RNA detected by RT-PCR.D, the level of actin transcript as detected by RT-PCR as the internal control. E, the levels of RBM4 protein expression detected by Western blotting using specific anti-RBM4 antibody. F, the quantification of tau Ex10 splicing isoforms as shown in A. The results are derived from six independent experiments. The graph shows the average ratio Ex10+/Ex10− transcripts ± S.E. (***, p < 0.001).
RBM4 Expression in the Human Neurons in the Frontal Cortex and Hippocampus
To begin to explore the potential involvement of RBM4 in tauopathy, we examined RBM4 expression in the human brain, particularly in the regions affected in tauopathy patients. Immunohistochemical staining of postmortem adult human brain samples was carried out using an affinity-purified anti-RBM4 antibody. As shown in Fig. 8, the frontal cortex (A and B) and hippocampal (C and D) sections showed strong signals, whereas such staining signal was not detectable in the negative controls either in the absence of the anti-RBM4 antibody (E) or in the presence of the antibody preparation after absorption with RBM4 protein (not shown). RBM4 immunostaining signals were detectable in both neurons and glial cells, including layers II neurons in the frontal cortex (marked by arrows in Fig. 8, A and B) and CA1 pyramidal neurons in the hippocampus (as indicated by arrowheads in Fig. 8, C and D). The signal was stronger in the nucleus than in the cytoplasm. Together with the activity of RBM4 in stimulating tau exon 10 inclusion, RBM4 expression in neurons suggests a possible role of RBM4 as a regulator of tau exon 10 alternative splicing in the human brain.
FIGURE 8. RBM4 expression in human frontal cortex and hippocampus was detected by immunohistochemical staining.
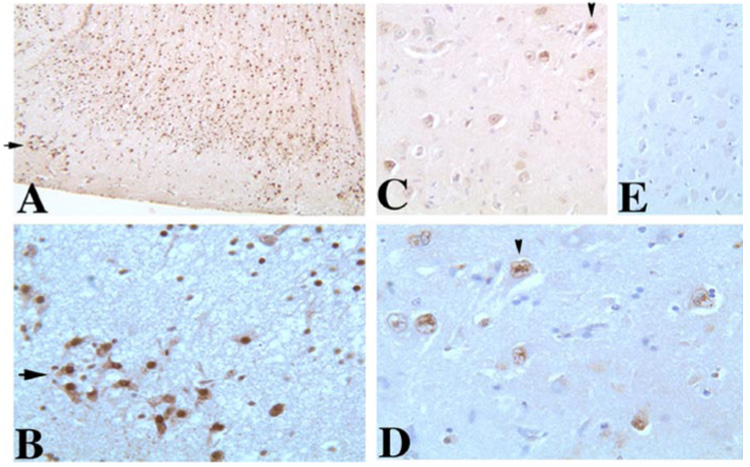
Adult human brain sections of frontal cortex (A and B, × 150 and × 600 magnification, respectively) or hippocampus (C (×300), D (×600), E (×300)) are shown. E, a negative control, staining in the absence of the primary antibody. The frontal cortical sections and hippocampal sections were used for immunohistochemical staining with the affinity-purified anti-RBM4 antibody. RBM4 shows strong signals in the frontal cortex neurons, including layer II neurons (marked by arrows) and scattered signals in CA1 pyramidal neurons in the hippocampus (as indicated by arrowheads) in the brain. The signal is stronger in the nucleus than in the cytoplasm.
DISCUSSION
Pre-mRNA splicing is the most upstream event in the post-transcriptional RNA processing. It is estimated that over 65% of human genes undergo alternative splicing to form functionally distinct gene products (30). Alternative splicing is an extremely powerful mechanism for generating proteomic diversity. Accumulating evidence supports an important role of aberrant splicing in the pathogenesis of human diseases. Cis-acting splicing mutations that affect either splice sites or splicing regulatory elements lead to the formation of defective or toxic gene products. Such splicing mutations can cause human diseases by four types of mechanisms: exon skipping, intron retention, cryptic splice site activation, or imbalance of different splicing isoforms (for recent reviews, see Refs. 21–23). The importance of maintaining the delicate balance of different splicing isoforms has only begun to be appreciated. A number of model systems have been established to investigate the cis-regulatory elements and trans-acting factors controlling alternative splicing events. However, relatively little is known about mechanisms regulating alternative splicing of genes associated with human pathogenesis.
Most splicing regulators have been identified using biochemical approaches. So far, there has been no report on using an expression cloning approach to systematically screen for alternative splicing regulators. Our study demonstrates that such an expression cloning approach based on alternative splicing-dependent GFP reporter can be successfully used to identify alternative splicing regulator(s). Further improvement of this expression cloning strategy may lead to development of high throughput systems for identifying genes important for splicing regulation. For example, a luciferase reporter constructed from our Tau-GFP reporter is being used to develop a high throughput system for tau exon 10 splicing regulation.3
RBM4 was initially identified in a yeast two-hybrid screen for proteins interacting with transportin-SR2 (25). Mammalian RBM4 contains, at its amino terminus, two RRMs and a CCHC-type zinc finger. At the C-terminal region of RBM4 are three alaninerich stretches. RBM4 has been shown to promote the inclusion of skeletal muscle exon of α-tropomyosin by binding to a CU-rich intronic element. Domain dissection experiments show that the RNA recognition motif of RBM4 is required for splice site modulation, whereas the CAD domain was required for trafficking to the nucleus (24, 25). In α-tropomyosin alternative splicing, RBM4 competes with PTB for common or overlapping intronic elements and activates the inclusion of skeletal muscle-specific exon (24).
In our study, RBM4 was identified by its functional activity in stimulating exon 10 inclusion during the expression cloning using the tau exon 10-alternative splicing-dependent GFP reporter. Both overexpression (Fig. 3 and Fig. 4) and RNAi-mediated down-regulation (Fig. 7) in transfected cells using the tau minigene support a role of RBM4 in enhancing tau exon 10 inclusion. The RNA-binding domain of RBM4 is required for its stimulatory activity on tau exon 10 inclusion, because mutating the conserved aromatic amino acid residues in the RNA recognition motif eliminated such splicing enhancement (Fig. 4). This is consistent with the previous observation that the same RRM mutant was inactive in stimulating alternative splicing in vitro (25).
Our UV-cross-linking experiments suggest that RBM4 interacts with an intronic region downstream of the 5′ splice site of exon 10. Further mutagenesis experiment indicates a CU-rich element in this region as a RBM4 binding site that is functionally important for the splicing-stimulatory activity of RBM4. Mutating the pyrimidine into purine residues in this RBM4 binding site significantly reduced the interaction between RBM4 and tau pre-mRNA (Fig. 5). The same mutation also remarkably decreased the stimulation of exon 10 inclusion by RBM4 (Fig. 6). Therefore, our study not only identified a new intronic splicing enhancer element for tau exon 10 splicing but also uncovered an important splicing regulator, RBM4, that interacts with this intronic splicing enhancer element.
Our immunocytochemical staining experiments indicate that human RBM4 is expressed in the neurons, including the hippocampus and the frontal cortex, regions affected in tauopathy. Interestingly, a previous study reported an association of RBM4 with Down syndrome in the fetal brain using a proteomics approach, suggesting that RBM4 is important for neuronal function (31). It has also been reported that RBM4 is phosphorylated by cyclin A1-Cdk2 complex (32). It is conceivable that cellular stresses could affect the activity of RBM4 and influence its regulation of tau exon 10 splicing, possibly contributing to the pathogenesis of a subset of tauopathy.
Tauopathy is one of most common forms of neurodegeneration. This is a group of genetically and phenotypically heterogeneous diseases, including Alzheimer disease, Down syndrome, several variants of prion diseases, progressive supranuclear palsy, amyotrophic lateral sclerosis, Pick disease, corticobasal degeneration, frontotemporal dementia. The formation of filamentous Tau protein-containing inclusions in the affected brain regions is the common neuropathological feature of these diseases. The underlying molecular defects for the majority of these diseases remain to be elucidated. Mutations in tau gene have been associated with only a subset of tauopathy, namely FTDP-17. Splicing mutations in the human tau gene, including all intronic mutations and several exonic mutations, are associated with changes in Tau4R/3R ratio. Studies of these splicing mutations in the tau gene in FTDP-17 patients demonstrate the importance of controlling the balance of different Tau isoforms by alternative splicing for normal brain function (for recent reviews, see Refs. 6 and 9). Identification of critical alternative splicing regulators for tau gene expression will not only help in our understanding of regulatory mechanisms of tau alternative splicing but also provide important information on how to correct the tau splicing defects in cells carrying FTDP-17 intronic mutations. For example, if certain proteins are identified that can reverse the Tau4R/Tau3R ratio to the normal 1:1 ratio in FTDP-17 mutant cells, it will provide potentially useful information for future designing of treatment strategy for the FTDP-17 patients carrying tau splicing mutations.
Several cis-acting regulatory elements have been identified to influence tau exon 10 alternative splicing, including a purine-rich element inside exon 10, a stem-loop at the 5′ splice site of exon 10, and intronic splicing silencer and enhancer elements inside or near exon 10 (29, 33–37) (reviewed in Refs. 7–9). Our study indicates that an intronic pyrimidine-rich element located 100 nucleotides downstream of the 5′ splice site of exon 10 also plays a role in tau exon 10 splicing.
It remains to be elucidated what transacting splicing factors in the brain regulate tau exon 10 alternative splicing. A number of known splicing regulators have been tested in a chimeric tau minigene. In this chimeric tau system, SRp30c, SRp55, SRp75, 9G8, U2AF, PTB, and hnRNPG repress tau exon 10 inclusion, whereas CELF3 and CELF4 activate exon 10 splicing (38, 39). Their effects on tau pre-mRNA splicing in the context of native tau exon 9 and exon 11 splice sites remain to be examined. We have established a tau minigene system using human tau gene that contains the native tau gene splice sites (29). Using the minigene and biochemical approaches, we identified Tra2beta as an SR-rich protein that binds to the purine-rich exonic splicing enhancer element and stimulates tau exon 10 splicing (28). A recent study showed that both Tra2beta and ASF/SF2 interact with this exon 10 purine-rich enhancer and play a role in exon 10 splicing (37). Here we report the role of RBM4, a previously unknown tau splicing regulator, in stimulating tau exon 10 inclusion. We further identified the intronic pyrimidine-rich intronic splicing enhancer element downstream of exon 10 and showed that this intronic element is required for the stimulatory activity of RBM4 in tau exon 10 inclusion. It will be interesting to test whether RBM4 is associated with the development of tauopathy, in particular among patients without mutations in the tau gene.
Acknowledgments
We thank Dr. Chris Smith for generously providing raver cDNA plasmid. We are grateful to members of the Wu laboratory for helpful discussions and/or critical reading of the manuscript.
Footnotes
This work was supported by National Institutes of Health Grants AG17518, EY014576, and GM070967 (to J. Y. W.), the Society for Progressive Supranuclear Palsy, Muscular Dystrophy Association (to J. Y. W.), and by a scholar award from the Leukemia Society of America (to J. Y. W.).
The abbreviations used are: MT, microtubule; Ex10, exon 10; FTDP-17, frontotemporal dementia with parkinsonism linked to chromosome-17; RT, reverse transcription; GFP, green fluorescent protein; RNAi, RNA interference; HA, hemagglutinin; RRM, RNA recognition motif.
C. Donahue, K. Kosik, and J. Y. Wu, unpublished data.
REFERENCES
- 1.Couchie D, Faivre-Bauman A, Puymirat J, Guilleminot J, Tixier-Vidal A, Nunez J. J. Neurochem. 1986;47:1255–1261. doi: 10.1111/j.1471-4159.1986.tb00748.x. [DOI] [PubMed] [Google Scholar]
- 2.Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. J. Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Neurobiol. Aging. 2003;24:1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Spillantini MG, Goedert M. Adv. Exp. Med. Biol. 2001;487:21–37. doi: 10.1007/978-1-4615-1249-3_3. [DOI] [PubMed] [Google Scholar]
- 5.Lee VM, Goedert M, Trojanowski JQ. Annu. Rev. Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 6.Cairns NJ, Lee VM, Trojanowski JQ. J. Pathol. 2004;204:438–449. doi: 10.1002/path.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andreadis A. Biochim. Biophys. Acta. 2005;1739:91–103. doi: 10.1016/j.bbadis.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 8.D’Souza I, Schellenberg GD. Biochim. Biophys. Acta. 2005;1739:104–115. doi: 10.1016/j.bbadis.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 9.Kar A, Kuo D, He R, Zhou J, Wu JY. Alzheimer Dis. Assoc. Disord. 2005;19 Suppl. 1:29–36. doi: 10.1097/01.wad.0000183082.76820.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Proc. Natl. Acad. Sci. U. S. A. 1988;85:4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. EMBO J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andreadis A, Brown WM, Kosik KS. Biochemistry. 1992;31:10626–10633. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 13.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 14.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Proc. Natl. Acad. Sci. U. S. A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bird TD, Sumi SM, Nemens EJ, Nochlin D, Schellenberg G, Lampe TH, Sadovnick A, Chui H, Miner GW, Tinklenberg J. Ann. Neurol. 1989;25:12–25. doi: 10.1002/ana.410250104. [DOI] [PubMed] [Google Scholar]
- 16.Makrides V, Shen TE, Bhatia R, Smith BL, Thimm J, Lal R, Feinstein SC. J. Biol. Chem. 2003;278:33298–33304. doi: 10.1074/jbc.M305207200. [DOI] [PubMed] [Google Scholar]
- 17.Panda D, Samuel JC, Massie M, Feinstein SC, Wilson L. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9548–9553. doi: 10.1073/pnas.1633508100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bunker JM, Wilson L, Jordan MA, Feinstein SC. Mol. Biol. Cell. 2004;15:2720–2728. doi: 10.1091/mbc.E04-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goedert M, Jakes R, Crowther RA, Hasegawa M, Smith MJ, Spillantini MG. Biochem. Soc. Trans. 1998;26:463–471. doi: 10.1042/bst0260463. [DOI] [PubMed] [Google Scholar]
- 20.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 21.Black DL. Annu. Rev. Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 22.Faustino NA, Cooper TA. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 23.Wu JY, Y L, Havlioglu N. Alternatively Spliced Genes. 2nd Ed. New York: Wiley-VCH; 2004. pp. 125–177. [Google Scholar]
- 24.Lin JC, Tarn WY. Mol. Cell. Biol. 2005;25:10111–10121. doi: 10.1128/MCB.25.22.10111-10121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai MC, Kuo HW, Chang WC, Tarn WY. EMBO J. 2003;22:1359–1369. doi: 10.1093/emboj/cdg126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gromak N, Rideau A, Southby J, Scadden AD, Gooding C, Huttelmaier S, Singer RH, Smith CW. EMBO J. 2003;22:6356–6364. doi: 10.1093/emboj/cdg609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang ZH, Zhang WJ, Rao Y, Wu JY. Proc. Natl. Acad. Sci. U. S. A. 1998;95:9155–9160. doi: 10.1073/pnas.95.16.9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang Z, Tang H, Havlioglu N, Zhang X, Stamm S, Yan R, Wu JY. J. Biol. Chem. 2003;278:18997–19007. doi: 10.1074/jbc.M301800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Z, Cote J, Kwon JM, Goate AM, Wu JY. Mol. Cell. Biol. 2000;20:4036–4048. doi: 10.1128/mcb.20.11.4036-4048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 31.Bernert G, Fountoulakis M, Lubec G. Proteomics. 2002;2:1752–1757. doi: 10.1002/1615-9861(200212)2:12<1752::AID-PROT1752>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 32.Diederichs S, Baumer N, Ji P, Metzelder SK, Idos GE, Cauvet T, Wang W, Moller M, Pierschalski S, Gromoll J, Schrader MG, Koeffler HP, Berdel WE, Serve H, Muller-Tidow C. J. Biol. Chem. 2004;279:33727–33741. doi: 10.1074/jbc.M401708200. [DOI] [PubMed] [Google Scholar]
- 33.Grover A, Houlden H, Baker M, Adamson J, Lewis J, Prihar G, Pickering-Brown S, Duff K, Hutton M. J. Biol. Chem. 1999;274:15134–15143. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]
- 34.Varani L, Hasegawa M, Spillantini MG, Smith MJ, Murrell JR, Ghetti B, Klug A, Goedert M, Varani G. Proc. Natl. Acad. Sci. U. S. A. 1999;96:8229–8234. doi: 10.1073/pnas.96.14.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Varani L, Spillantini MG, Goedert M, Varani G. Nucleic Acids Res. 2000;28:710–719. doi: 10.1093/nar/28.3.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Souza I, Schellenberg GD. J. Biol. Chem. 2000;275:17700–17709. doi: 10.1074/jbc.M909470199. [DOI] [PubMed] [Google Scholar]
- 37.D’Souza I, Schellenberg GD. J. Biol. Chem. 2006;281:2460–2469. doi: 10.1074/jbc.M505809200. [DOI] [PubMed] [Google Scholar]
- 38.Gao QS, Memmott J, Lafyatis R, Stamm S, Screaton G, Andreadis A. J. Neurochem. 2000;74:490–500. doi: 10.1046/j.1471-4159.2000.740490.x. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Gao QS, Wang Y, Lafyatis R, Stamm S, Andreadis A. J. Neurochem. 2004;88:1078–1090. doi: 10.1046/j.1471-4159.2003.02232.x. [DOI] [PubMed] [Google Scholar]