

Abstract
Invasion of brain tumor cells has made primary malignant brain neoplasms among the most recalcitrant to therapeutic strategies. We tested whether the secreted protein Slit2, which guides the projection of axons and developing neurons, could modulate brain tumor cell invasion. Slit2 inhibited the invasion of medulloblastoma cells in a variety of in vitro models. The effect of Slit2 was inhibited by the Robo ectodomain. Time-lapse videomicroscopy indicated that Slit2 reduced medulloblastoma invasion rate without affecting cell direction or proliferation. Both medulloblastoma and glioma tumors express Robo1 and Slit2, but only medulloblastoma invasion is inhibited by recombinant Slit2 protein. Downregulation of activated Cdc42 may contribute to this differential response. Our findings reinforce the concept that neurodevelopmental cues such as Slit2 may provide insights into brain tumor invasion.
Keywords: Slit, medulloblastoma, glioma, invasion, inhibitor
Introduction
Medulloblastoma is the most common malignant brain tumor in children, and originates from granule precursor cells of the cerebellum (Raffel, 2004). Glioblastoma multiforme (GBM) is the most common adult malignant brain tumor and may arise from astrocytes or astrocytic precursors (Sanai et al., 2005). In these tumors, invading cells disrupt normal tissue architecture leading to early recurrence and poor patient prognosis (Sanai et al., 2005).
The Slit family of secreted proteins is known to be important for guiding the projection of normal axons and developing neurons (reviewed byWong et al., 2002). There are three Slit proteins in mammals, and all are expressed at the ventral midline of the neural tube.
The receptor for Slit is the transmembrane protein Robo (Roundabout), and four ROBO genes have been identified (Wong et al., 2002; Park et al., 2003). Recent studies have demonstrated a role for Slit-Robo signaling in leukocyte chemotaxis and tumor angiogenesis (Wu et al., 2001; Wang et al., 2003). Findings of SLIT gene inactivation suggest SLIT2 as a potential tumor-suppressor gene in gliomas, lung, breast and colorectal cancer, as well as in neuroblastoma (Dallol et al., 2002, 2003a, Dallol et al., b; Astuti et al., 2004).
Our study focuses on the expression and functional significance of Slit2 and Robo1 in medulloblastoma and glioblastoma cell invasion. Our results demonstrate a role for Slit2 as an inhibitor of medulloblastoma but not glioma cell invasion in vitro.
Results
Medulloblastoma spheroids respond to Slit2
To test the hypothesis that malignant brain tumor cells respond to Slit2, medulloblastoma and glioma spheroids were cocultured with control human embryonic kidney (HEK) or Slit2 aggregates in collagen gels for 48 and 72 h (Figure 1a and b). There was no significant difference in cell number in the proximal versus distal quadrant for UW3, Daoy (medulloblastoma) and C6 (glioma)-HEK cocultures (Figure 1c, e and g). There was a significant increase in the number of cells in the proximal quadrant for U251 glioma-HEK cocultures (t-test: P < 0.05; n = 5 cocultures) (Figure 1i). The presence of Slit2 aggregates resulted in a 56% inhibition of UW3 and a 31% inhibition of Daoy cells found in the proximal quadrant to the Slit2 source (t-test: P < 0.01; n = 5 UW3 cocultures; P < 0.05; n = 7 Daoy cocultures) (Figure 1d and f). There was no significant difference for C6 glioma-Slit2 cocultures. The number of cells in the proximal quadrant of U251-Slit2 cocultures was significantly higher, as seen in HEK control cocultures, consistent with the presence of a chemoattractant or motility inducer secreted by wild-type HEK cells (t-test: P < 0.05; n = 5 cocultures) (Figure 1h and j, respectively). These results suggest that medulloblastoma but not glioma cells respond to the guidance cue Slit2.
Figure 1.

Medulloblastoma spheroids respond to Slit2. (a) Model of the tumor spheroid cocultured with human embryonic kidney (HEK) or Slit2 aggregate in a collagen gel. Arrow denotes placement of HEK or Slit2 aggregate. (b) Still photograph of Cell Tracker labeled UW3 medulloblastoma spheroid implanted 550 μm from a Slit2 aggregate after 72 h. Arrow denotes placement of Slit2 aggregate. Scale bar = 250 μm. (c–j) Proximal and distal quadrant distribution of UW3 (c–d), Daoy (e–f) medulloblastoma cells and C6 (g–h), U251 (i–j) glioma cells cocultured with HEK control aggregates (c, e, g, i) or Slit2 aggregates (d, f, h, j) after 72 (UW3 and Daoy) and 48 h(C6 and U251). Error bars represent s.e.m. Asterisks denote significance at P < 0.05(*) and P < 0.01(**).
Slit2 inhibits medulloblastoma cell invasion rate
The Daoy cell line exhibits p53 and CDKN2 gene mutations which are commonly found in GBM (Raffel et al., 1993, 1995). These abnormalities are rarely encountered in medulloblastoma tumors (Raffel, 2004). Therefore, further experiments utilized the UW228-3 (UW3) cell line that displays mutations more closely associated with the classic medulloblastoma variant such as loss of heterozygosity at the distal end of chromosome 17p in a region distinct from that of the p53 gene (Keles et al., 1995). The UW3 cell line also expresses several neuronal antigenic markers also seen in medulloblastoma tumors (Keles et al., 1995).
To investigate whether Slit2 functions as a repellent or inhibitor of cell invasion, we performed time-lapse videomicroscopy of individual UW3 medulloblastoma cells detached from UW3 spheroids 24–44 h after implantation adjacent to either an HEK or Slit2 aggregate (Figure 2). We found a 54% inhibition of medulloblastoma cell invasive rate for Slit2 compared to HEK cocultures (t-test; P < 0.001) (Figure 2a–c). This kinetic effect is consistent with the 56% inhibition of total UW3 cell number seen for UW3-Slit2 cocultures in Figure 1. In addition to this kinetic effect, a subset of cells (seven of 32 or 22%) in Slit2 cocultures appeared to stall and remained stationary (Figure 2b). They maintained a ‘ruffied’ appearance and failed to further invade the gel during the observation period. When these stationary cells were excluded from the data, the UW3 cell invasion rate for Slit2 cocultures was still 38% less than HEK coculture controls (t-test; P < 0.001). There was no significant difference in cell direction or proliferation for Slit2 and HEK cocultures (Table 1).
Figure 2.
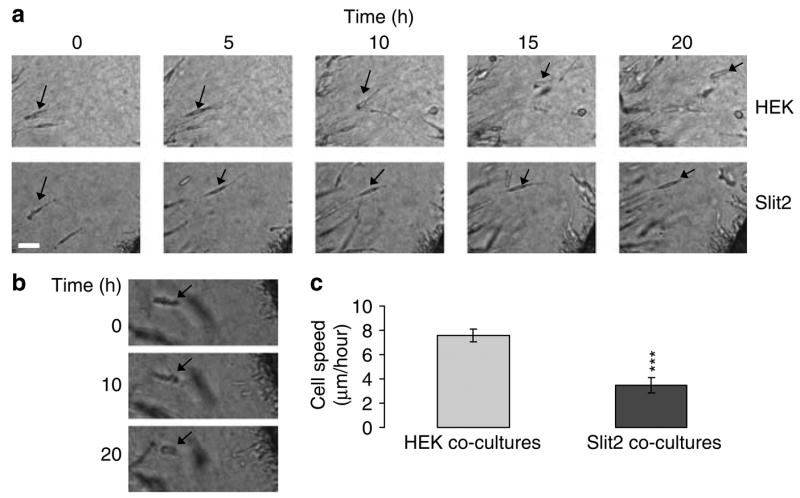
Slit2 inhibits individual medulloblastoma cell rate. (a) Still photographs depicting individual UW3 cell invasion from spheroids cocultured with human embryonic kidney (HEK) (upper panels) or Slit2 aggregates (lower panels) over 20 h. Arrows denote individual cell tracking. Scale bar = 50 μm (b) Image of ‘ruffling’ cell in UW3-Slit2 cocultures. Arrows denote individual cell tracking. Scale bar = 50 μm. (c) Mean invasion rate of individual UW3 cells cocultured with HEK (n = 35) or Slit2 aggregates (n = 32). Error bars represent s.e.m. Asterisks indicate a significant difference at P < 0.001 (***).
Table 1.
Analysis of cell proliferation and direction for UW3 spheroids cocultured with HEK or Slit2 aggregates in three-dimensional collagen type I gels
| Cell type | % of cells dividing in 20 h (%) | Mean deviation from expected angle (°) |
|---|---|---|
| HEK cocultures | 14 | −0.43 |
| Slit2 cocultures | 16 | −5.07 |
N = 35 and 32 cells for HEK and Slit2 cocultures, respectively. Abbreviation: HEK, human embryonic kidney.
A uniform concentration of Slit2 conditioned medium significantly inhibited UW3 medulloblastoma invasion into collagen type I gels, compared with HEK control conditioned medium and Dulbecco’s modified Eagle medium (DMEM) (Supplementary Figure 1). If Slit2 had only a chemorepellent role in medulloblastoma cell invasion, only a gradient, and not a uniform concentration, of Slit2 would have an effect. Our results suggest that Slit2 inhibits medulloblastoma cell invasion rate without affecting direction or proliferation.
Sustained Slit2 secreted from sodium alginate beads inhibits medulloblastoma cell invasion
We assessed the effect of prolonged, sustained Slit2 release from microencapsulated cells by placing four beads encapsulating Slit2 cells around individual spheroids, and monitoring invasion for 6 days (Figure 3a). Empty beads were used to assess the effect of their incorporation into the matrix on implanted spheroids, and wild-type HEK beads were used as negative controls. Western blot analysis confirmed the presence of Slit2 in the conditioned medium of encapsulated Slit2 cells (Figure 3b). A band corresponding to Slit2 protein (~200 kDa) was observed for the 6-day in vitro experiments. Sodium alginate beads, encapsulating Slit2 cells had a significant inhibitory effect on UW3 medulloblastoma cell invasion (40 and 25% compared with empty bead and HEK cocultures, respectively) (Figure 3c–f). Slit2 beads had no significant effect on C6 and U251 glioma cell invasion (Figure 3g–h). To confirm that the effects in our encapsulation studies are attributed to cell invasion and not proliferation or cytotoxicity, cultures of medulloblastoma and glioma cells were exposed to sodium alginate encapsulated HEK or Slit2 cells for 6 days (4 days for C6). Slit2 encapsulated sodium alginate beads did not significantly affect tumor cell proliferation or doubling time compared to controls (Table 2).
Figure 3.

Sustained Slit2 delivery using sodium alginate bead microencapsulation inhibits medulloblastoma cell invasion. (a) Schematic showing the position of sodium alginate beads relative to the tumor spheroid. Arrow represents placement of sodium alginate bead. (b) Western blot analysis of Slit2 expression (~200 kDa) in conditioned medium of encapsulated Slit2 cells over 6 days. Lanes 1 and 2 represent conditioned medium from human embryonic kidney (HEK) and Slit2 monolayer cultures as negative and positive controls, respectively. Lanes 3–5 represent conditioned medium from Slit2 beads on days 1, 3 and 6 in culture. Lanes 6–7 represent conditioned medium from HEK beads on days 3 and 6. (c–e) Photographs of UW3 spheroids surrounded by (c) four empty beads, (d) four beads encapsulating HEK cells and (e) four beads encapsulating Slit2 cells. Scale bar = 250 μm. (f–h) Quantification of total invasion by (f) UW3 spheroids, (g–h) C6 and U251 glioma spheroids implanted with empty, HEK or Slit2 beads. Error bars represent s.e.m. Asterisks indicate significance at P < 0.05, P < 0.01** and P < 0.001***. (i–k) Confocal images of UW3 spheroids (DiI labeled-red) confronted with human fetal astrocyte aggregates (DiO labeled-green) in the presence of (i) HEK sodium alginate beads or (j) Slit2 sodium alginate beads after 5 days. Scale bar 250 μm. Results are representative of three independent trials. (k) Quantification of the number of medulloblastoma cells infiltrating astrocyte aggregates after 5 days.
Table 2.
Doubling time of medulloblastoma and glioma cells grown as monolayer cultures in the presence of human embryonic kidney or Slit2 beads after 4 or 6 days
| HEK beads (h) | Slit2 beads (h) | |
|---|---|---|
| UW3 | 28.7 | 30.6 |
| Daoy | 36.2 | 35.9 |
| U251 | 30.4 | 31.6 |
| C6 | 18.5 | 18.9 |
Furthermore, UW3 spheroids and human fetal astrocyte aggregates were cocultured in collagen gels with Slit2 or HEK control beads for 5 days (Figure 3i–k). Compared with HEK sodium alginate beads, Slit2 beads significantly inhibited the number of cells infiltrating the astrocyte aggregates (Figure 3i–k) demonstrating that Slit2 also inhibits UW3 invasion of a more biologically relevant substrate.
The effect of Slit2 is rescued by the Robo1 ectodomain
To examine the specificity of the effects seen here to Slit2-Robo1 signaling, we added R5, an IgG2b mono-clonal antibody to the first immunoglobulin domain of Robo1, to the medium to block the effect of secreted Slit2 from microencapsulated cells. At the concentration tested, R5 rescued the inhibitory effect of Slit2 on UW3 medulloblastoma invasion by 60% (data not shown). However, the addition of R5 just on days 0 and 3 may not be able to completely block the continuous sustained delivery of Slit2 into the collagen matrix over 5 days. To address this issue, mixed sodium alginate beads containing Slit2 and RoboN secreting cells were implanted with UW3 spheroids and compared with the effect of mixed Slit2-HEK beads or HEK beads alone. Western blot analysis confirmed the presence of Slit2 and RoboN in the conditioned medium of microencapsulated Slit2-RoboN cells (Figure 4a). After 5 days, mixed Slit2-HEK beads had a significant inhibitory effect on invasion; however, mixed Slit2-RoboN beads were not different from HEK controls (Figure 4b). The invasion inhibition for Slit2-HEK beads was 28% compared with Slit2-RoboN mixed beads and 26% compared with HEK beads. Slit2-RoboN beads demonstrated a 100% rescue of the inhibitory effect of sustained Slit2 secretion on UW3 invasion. These results show that Slit2 is responsible for the inhibitory effects seen in our invasion assays.
Figure 4.

RoboN rescues Slit2 inhibition of UW3 medulloblastoma cell invasion using sodium alginate bead microencapsulation. (a) Western blot analysis of Slit2 and RoboN expression (~200 kDa and ~100 kDa, respectively) in conditioned medium of encapsulated mixed Slit2-human embryonic kidney (HEK) and Slit2-RoboN cells at days 3 and 6. Conditioned medium from Slit2 (lane 1) and RoboN (lane 2) monolayer cultures were used as positive controls for Slit2 and RoboN, respectively. (b) Quantification of UW3 total invasion when cocultured with HEK, Slit2-HEK or Slit2-RoboN beads. Error bars represent s.e.m. Asterisks indicate significance at P < 0.05*, and P < 0.01**.
Brain tumor cell lines and human brain tumors express Robo1 and Slit2
We conducted reverse transcription-polymerase chain reaction (RT–PCR) analysis of Slit2 and Robo1 expression in our brain tumor cell lines and human tumor samples. All medulloblastoma and glioma cell lines tested expressed Robo1 and Slit2 mRNA (Figure 5a). Human fetal astrocytes and mouse cerebellum from P4 and P8 also expressed both Slit2 and Robo1 (Figure 5a). Slit1 was not detected in any cell lines tested (data not shown). Representative human brain tumor samples are shown in Figure 5b and c. In total, 57% (four of seven) and 86% (six of seven) of medulloblastomas express Robo1 and Slit2, respectively; 100% (two of two) of meningiomas and pilocytic astrocytomas express both Robo1 and Slit2; 100% (four of four) GBM’s express Robo1 and 75% (three of four) express Slit2, and the normal brain sample expresses both Robo1 and Slit2. These results show that Slit2 and Robo1 are expressed in a variety of tumor cell lines, brain tumors and both neonatal and embryonic tissues.
Figure 5.

Reverse transcription-polymerase chain reaction (RT–PCR) analysis of Robo1 and Slit2 expression. (a) Robo1, Slit2 and GAPDH mRNA in medulloblastoma and glioma cell lines. Human fetal astrocytes (HFA) and mouse neonatal cerebellum (C P4 and C P8) were screened as positive controls. (b) Robo1, Slit2 and GAPDH mRNA in medulloblastoma (MD), meningioma (MN) and pilocytic astrocytoma (PA) primary human tumors. (c) Robo1, Slit2 and β-actin mRNA in glioblastoma multiforme (GBM) tumors and normal brain (NB). (d) Slit2 decreases activated Cdc42-GTP in medulloblastoma cells. Cdc42, Rac1 and RhoA loading controls levels were also estimated by Western blot analysis. Results are representative of three independent experiments.
Slit2 and Robo1 are not inactivated by methylation silencing
Gene silencing by methylation could be one mechanism to explain the nonresponsiveness of the invasive glioma cell lines studied. We used the demethylating agent 5′ aza-2′-deoxycytidine to test for methylation silencing in our cell lines. Our results show that 5′ aza-2′-deoxycytidine treatment did not affect the mRNA expression levels of Slit2 and Robo1 in U251, U343, UW3 and Daoy cells (see Supplementary Figure 2a). These results suggest that other mechanisms account for the differences seen in our invasion assays, where medulloblastoma cells respond to exogenous Slit2 but glioma cells do not.
Slit2 downregulates activated Cdc42 in medulloblastoma cells
To further understand the mechanism responsible for the differential response to Slit2, we biotinylated cell surface proteins to examine expression levels of Robo1 at the surface of U251 cells stably transfected with Robo1-HA. U251 cells overexpressing Robo1-HA did not respond to Slit2 using both coculture and sodium alginate bead assays (data not shown). Biotinylation showed that Robo1 protein was highly expressed at the cell surface of stably transfected U251 cells (see Supplementary Figure 2b). These results demonstrate that the lack of glioma responsiveness to Slit2 cannot be explained by aberrant receptor shuttling or expression and suggests an intracellular mechanism.
Cdc42 and Rac1 play roles in Slit-mediated repulsion (Wong et al., 2001; Fan et al., 2003; Lundstrom et al., 2004). To assess Rho-GTPase activation by Slit2, medulloblastoma and glioma cells were cultured in medium conditioned by HEK or Slit2 cells and pull-down assays of activated Cdc42, Rac1 and RhoA were conducted. Here, we show a decrease in Cdc42-GTP and a minor decrease in Rac1-GTP activities for UW3 cells cultured with Slit2 conditioned medium (Figure 5d). There was no change in RhoA-GTP activity for both glioma and medulloblastoma cells (Figure 5d). C6 cells and U251 glioma cells (data not shown) did not display any changes in Rho-GTPase activity (Figure 5d). These results suggest that Cdc42-GTP levels, and possibly Rac1-GTP levels, may play a role in the inhibitory effect of Slit2 on medulloblastoma cells. The inability of Slit2 to modulate activated Cdc42 and Rac1 in glioma cells may contribute to their resistance to Slit2 in our invasion assays.
Discussion
Treatment strategies for medulloblastoma include surgery, irradiation and/or chemotherapy (Raffel, 2004). These approaches do not always control invading tumor cells and local recurrence, cerebral spinal fluid and systemic metastasis result in a 60–70% 5 year survival rate (Louis et al., 2002).
We provide evidence for Slit2 inhibition of invading medulloblastoma cells. This was shown using medulloblastoma spheroid-Slit2 aggregate cocultures, sodium alginate bead bioreactors in collagen and also in confrontation cocultures with astrocyte aggregates. Slit proteins have been associated with chemorepellent roles in axon guidance; however, inhibitory effects have been described. Cells migrating from neurospheres generated from SVZ of P0-P3 Slit1 −/− mice were found to migrate further and in a more dispersed pattern than cells from Slit1 +/− neurospheres suggesting that Slit1 plays an inhibitory role in addition to its function as a repellent (Nguyen-Ba-Charvet et al., 2004). Other studies have suggested that Slit inhibits migration from subventricular zone explants; however, when combined with an astrocyte-derived migration inducing activity, Slit acts as a repellent (Mason et al., 2001). Slit2 has also been shown to inhibit leukocyte chemotaxis induced by chemokines (Wu et al., 2001).
Microencapsulation bioreactor technology has been used to assess the efficacy of antiangiogenic compounds on glioma growth (Joki et al., 2001; Read et al., 2001). Sodium alginate bioreactors provide an isolated environment for continuous protein delivery. We have shown sustained bioreactor Slit2 secretion for several weeks in our models (unpublished results). This model also demonstrates that the inhibition of medulloblastoma cell invasion by continuous Slit2 delivery can be regulated and controlled by mixed beads encapsulating both Slit2 and RoboN cells. Bioreactor technology can potentially be used for combination treatment with encapsulated cells overexpressing multiple anti-invasive, antiproliferative and proapoptotic molecules.
Marillat et al. (2004) have demonstrated a role for Rig1/Robo3 in controlling midline crossing of hindbrain precerebellar neurons and axons. Human patients with horizontal gaze palsy with progressive scoliosis (HGPPS) were reported to have mutations in Rig1/Robo3, and functional studies have shown defects in commissural hindbrain projections and pontine nuclei (Jen et al., 2004). Gilthorpe et al. (2002) have shown that early and late stage chick cerebellar rhombic lip fragments are repelled by Slit2. Medulloblastoma cells are thought to arise from external granular layer cerebellar precursors derived from the rhombic lip, and these results provide an obvious developmental parallel with our studies of Slit2 inhibition of medulloblastoma cell invasion. Slit also inhibits CXCR4-induced motility in breast cancer cells and CXCR4 antagonists have been shown to inhibit medulloblastoma tumor growth in vivo (Rubin et al., 2003; Prasad et al., 2004). However, in contrast to our studies, Slit treatment alone did not influence the motility of the breast cancer cells (Prasad et al., 2004).
The lack of responsiveness of highly invasive glioma cells to Slit2 may be attributed to a variety of mechanisms. Dallol et al. (2003a) have found by quantitative real-time RT–PCR that Slit2 expression was downregulated in some gliomas with methylated SLIT2 promoter compared to gliomas and normal brain samples showing no methylation of this promoter. Our results demonstrate that Slit2 and Robo1 are expressed by a variety of glioma and medulloblastoma cell lines and primary tumors. Marillat et al. (2002) have shown that most CNS neurons in the rat brain express at least one Robo and one Slit during their development, and that levels are maintained from the embryonic to the adult stage. These authors suggested that neurons expressing Robo mRNA could be unresponsive to Slit if molecules or mechanisms regulating Robo expression and function were present (Marillat et al., 2002). We utilized the demethylating agent 5′ aza-2′-deoxycytidine to test for methylation silencing as a potential mechanism for loss of Robo1 and Slit2 protein expression in our cell lines. This treatment had no effect on expression levels of both Slit2 and Robo1 in the cell lines tested.
Wong et al. (2001) have demonstrated a role for the srGAPs (Slit-Robo GAPs) in Slit-Robo signaling. Interaction of srGAP’s with the Rho-GTPase, Cdc42 is essential for the repulsive activity of this guidance cue (Wong et al., 2001). Other studies support a role for Phosphatidyl inositol 3 kinase, Dock and Pak, and the Abelson (Abl) kinase and Enabled (Ena) proteins further implicating actin and microtubule reorganization in Slit-Robo signaling (Bashaw et al., 2000; Fan et al., 2003; Wang et al., 2003). Our time-lapse studies showed a change in cell morphology, as the presence of Slit2 aggregates increased the number of noninvading ruffling cells. We have shown that Slit2 does not influence cell proliferation suggesting that these stationary cells are not undergoing cell death. The presence of noninvading ruffling cells has also been demonstrated in glioblastoma spheroid cocultures in response to a secreted chemorepellent (Werbowetski et al., 2004). This prolonged cell ruffling may be associated with a failure of the cell to organize its actin-myosin cytoskeleton (Del Maestro et al., 2001).
We have assessed Robo1 surface expression levels by biotinylation of cell surface proteins and our results confirm that differential responses to Slit2 are attributed to mechanisms downstream of the cell membrane. We have demonstrated decreased Cdc42 activation for medulloblastoma cells in response to Slit2. This agrees with previous results obtained by Wong et al. (2001). Therefore, there may be a conserved mechanism regulating both the repellent and inhibitory response to Slit2. Cdc42 is known to regulate actin-rich filopodia formation and nuclear repositioning during the initiation of cell migration (reviewed by Luo, 2000; Gomes et al., 2005). The inhibitory effect of Slit2 on medulloblastoma cell invasion may be mediated by Cdc42-dependent effects on the actin cytoskeleton and/or cell polarity (reviewed by Luo, 2000; Etienne-Manneville and Hall, 2001, 2003; Palazzo et al., 2001; Gomes et al., 2005). Slit2-Robo1 interaction may be insufficient to inhibit the extensive invasion seen in glioblastoma cells lines such as U251 and C6 but effective in modulating the cell movement in the less invasive medulloblastoma cell lines.
Slit2 and Robo1 expression were not correlated with the degree of malignancy of the tumor samples or cell lines tested in this study. Rossi et al. (2005) obtained similar results using RT–PCR analysis of Slit2 expression in the cerebral tumors and glioma cell lines. Although Slit2-Robo1 mRNA can be detected in many human tumors, this does not necessarily correlate with Slit2 protein content or activity. Our results would suggest that only cells from specific tumors may respond to this signaling by modulating their invasive paradigm. Analysis of the regional expression of Slit2 and Robo1 in highly invasive tumor samples may shed light on this issue. In the absence of specific Slit2 and Robo1 antibodies, a comparison of Slit2 and Robo1 expression at the leading edge versus the tumor core by in situ hybridization would help to reconcile the RT–PCR and methylation data, and further delineate the role of Slit2 signaling in invading medulloblastoma cells.
We have demonstrated a slight decrease in Rac1 activity in response to Slit2. Slit stimulation leads to recruitment of a Dock and Pak complex to the Robo receptor and increases Rac activation (Fan et al., 2003). In contrast, Vilse, a conserved family of RhoGAPs, regulate the repellent response to Slit by binding to the intracellular domain of Robo receptors and promoting Rac inactivation (Lundstrom et al., 2004). Our results are indicative of an inhibitory, and not a directional, effect of Slit2 on medulloblastoma cells. A number of different signaling pathways may be involved in Slit2 inhibition of medulloblastoma invasion.
A new role for Slit2 as an inhibitor of medulloblastoma invasion has been identified. Our results reinforce the concept that selective neurodevelopmental cues may provide significant insights into tumor invasion and outline the need for detailed assessments of how to implement this strategy for other tumor types.
Methods
Cell culture
C6 (murine astrocytoma) cells were cultured and seeded into spinner culture flasks as previously described (Werbowetski et al., 2004; Werbowetski-Ogilvie et al., 2006). All culture reagents were obtained from Gibco BRL (Invitrogen, Burlington, ON, Canada) unless otherwise stated.
Confluent cultures of U251 (human glioblastoma) UW228-3 (UW3) (human medulloblastoma cell line derived from one of three cell aliquots taken from the same tumor resected from a 9-year-old girl) (Keles et al., 1995), and Daoy (human medulloblastoma derived from tumor biopsy from a 4-yearold boy) (Jacobson et al., 1985), HEK 293 and HEK 293 cells stably overexpressing full-length human Slit2 with a c-Myc tag at the carboxy terminus were trypsinized (0.05% trypsin/0.53mM ethylenediamine-N, N, N′, N′-tetraacetic acid) and hanging drops were prepared (Corcoran and Del Maestro, 2003; Del Duca et al., 2004). Aggregates consisting of 40 000 cells/drop and 25 000 cells/drop (U251) were utilized. All cell lines were purchased from the American Type Culture Collection (ATCC) (Rockville, MD, USA) except the UW3 and UW1 cell lines, which were a gift from Dr John R Silber (University of Washington, Seattle, WA, USA).
Spheroids and hanging drop aggregates were placed in a 1ml 25μM Cell Tracker Green solution prepared as described (Werbowetski et al., 2004). Labeled spheroids were cocultured with HEK or Slit2 aggregates, implanted into collagen type I gels (Vitrogen 100) (COHESION, Palo Alto, CA, USA) and photographed after 48 and 72 h using the time-lapse imaging system and Northern Eclipse 6.0 software (Werbowetski et al., 2004). Spheroids were divided into four quadrants with respect to the position of the adjacent spheroid. The total cell number in the proximal and distal quadrants was calculated.
Time-lapse videomicroscopy
UW3 medulloblastoma spheroids and HEK or Slit2 hanging drop aggregates were cocultured for 24 h and then imaged for an additional 20 h using the imaging system described (Werbowetski et al., 2004). Invasion rate, deviation from the expected invasive path (angle change from perpendicular) and number of divisions were calculated for individual detached UW3 cells in regions proximal to the Slit2 or HEK aggregate.
Conditioned medium assay
Conditioned medium was collected from HEK and Slit2 confluent monolayer cultures to assess the effect of a uniform concentration of Slit2 on UW3 spheroids implanted in collagen gels. A measure of 500 μl of DMEM + 10% FBS, HEK or Slit2 conditioned medium was placed on top of the collagen gel, and invasion was measured over 5 days with a medium change on day 3.
Patient samples and primary cultures
Fifteen human brain tumor samples were obtained from the Department of Molecular Pathology and Neurology, University of Lodz tissue bank, Simmelweiss University tissue bank in Hungary, Centre de Neuro-pathologie, Clermont-Ferrand tissue bank in France and the Brain Tumor Tissue Bank London Health Sciences Centre (London, ON, Canada). Seven samples were classified as medulloblastoma, two were low-grade pilocytic astrocytoma, four were childhood GBM, two were meningioma and one was normal brain. All relevant clinical information for each sample was available. Human fetal central nervous system (CNS) tissue obtained from 14- to 23-week-old embryos was provided by the Human Fetal Tissue Repository (Albert Einstein College of Medicine, Bronx, NY, USA). Human fetal astrocytes were prepared from the fetal CNS tissue following Canadian Institutes of Health Research (CIHR) guidelines as previously described (Ruffini et al., 2004). Populations of proliferating fetal astrocytes were obtained after three passages. Samples of P4 and P8 normal mouse cerebellar tissue were also analysed.
Reverse transcription-polymerase chain reaction
Total RNA was extracted from cell lines, human fetal astrocyte cultures, P4 and P8 mouse cerebellum using the RNeasy kit (Qiagen, Mississauga, ON, Canada). First strand cDNA was synthesized using the First-Strand cDNA Synthesis Kit (Amersham Biosciences, Baie D’Urfe, QC, Canada) and used directly for RT–PCR. See Supplementary Table 1 (1–4) for primer details. Total RNA was extracted from frozen medulloblastoma, pilocytic astrocytoma and meningioma tumors samples using the TRIzol Reagent (Gibco BRL) and used directly for RT–PCR. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. See Supplementary Table 1 (5–7) for primer details. Glioblastoma multiforme and normal brain samples were obtained after preparation for microarray analysis and processed separately. The RiboAmp™ RNA Amplification Kit (Arcturus, MountainView, CA, USA) was used for linear amplification of mRNA. The aRNA was used for RT–PCR analysis as described above. β-Actin was used as an internal control. See Supplementary Table 1 (4, 6, 7) for primer details. For all RT–PCR reactions, PCR in the absence of templates and/or PCR using templates generated without reverse transcriptase were used as negative controls. The following PCR conditions were used: 95°C for 2 min, 80°C for 3 min 30 s (during which Taq is added), 40 cycles at 95°C for 60 s, 55°C (60°Cfor β-actin) for 30 s, and 72°C for 60 s. Polymerase chain reaction products were separated by agarose gel electrophoresis.
Demethylation treatment
Treatment of brain tumor cell lines with the demethylating agent 5′ aza-2′-deoxycytidine (5-aza-dc) was carried out as previously described (Dallol et al., 2003a). Briefly, cells were cultured as described above and treated with 10 μM 5-aza-dc after 24 h. Medium was changed 24 h after treatment and then on day 3. RNA was prepared for RT–PCR on day 5.
Sodium alginate encapsulation
Sodium alginate bead cell encapsulation for treatment of brain tumors has been described (Read et al., 2001). Briefly, a stock solution of 0.137M NaCl, 5.5mM D-glucose, 5.45mM 2-[4-(2-hydroxyethyl)-1-piperazinyl]ethanesulfonic acid (HEPES) was prepared at a pH of 7.3. HEK and Slit2 cells were trypsinized and resuspended in alginate solution at a cell density of 1.6 × 106 cells/ml. From these solutions, droplets of cells dispersed in 1.5% sodium alginate were released into a 0.1M CaCl2 solution prepared from the stock. After polymerization, the alginate beads were washed two times in phosphate-buffered saline and once in DMEM + 10% FBS.
Four empty beads, HEK beads, or beads encapsulating Slit2 secreting cells were implanted approximately 1mm around spheroids in collagen gels. Invasion was measured for 6 days. RoboN (an extracellular fragment of Robo1 that inhibits Slit2-Robo1 interactions) (Wu et al., 2001; Wang et al., 2003) was used to block the effect of Slit2. RoboN-293 cells with an HA tag at the carboxy terminus were cultured and Slit2/RoboN or Slit2/HEK cells were encapsulated in sodium alginate at a cell density of 1.2 × 106 cells/ml. Mixed beads and HEK beads were implanted along with UW3 spheroids into collagen gels for 5 days.
Confrontation cocultures and confocal microscopy
Human fetal astrocytes were obtained as described above, and hanging drop aggregates (45 000 cells/drop) were prepared as described (Ruffini et al., 2004; Werbowetski et al., 2004). UW3 and astrocyte aggregates were labeled with 0.075 mg/ml DiI and DiO solutions in DMEM, respectively (Molecular Probes, Eugene OR, USA). Cocultures were implanted in collagen gels and surrounded by either four HEK or Slit2 alginate beads. After 5 days, cultures were analysed using a LSM 510 confocal scanning laser microscope (Carl Zeiss, Toronto, ON, Canada) as previously described (Werbowetski et al., 2004).
Western blot analysis
Conditioned medium from encapsulated HEK, Slit2-c-myc cells, encapsulated mixed Slit2 and HEK cells or Slit2 and RoboN-HA cells as well as Slit2 and RoboN monolayer cultures was collected, concentrated 10 × and 20 μg protein was subjected to 7.5% sodium dodecylsulfate-polyacrylamide electrophoresis (SDS–PAGE). Myc expression was detected using an anti-Myc antibody (1:1000) that recognizes a C-terminal myc epitope tag on Slit2 and HA expression was detected using an anti-HA antibody (1:500) (COVANCE, Berkeley, CA, USA). The horseradish peroxidase-conjugated goat anti-mouse and anti-rabbit IgG secondary antibodies (1:5000) were used and visualized by enhanced chemiluminescence (Perkin-Elmer Life Sciences Inc., Markham, ON, Canada).
Proliferation assay
UW3, Daoy, U251 and C6 cells were plated on six-well culture dishes (40 000 cells/well for UW3, Daoy and U251; 20 000 cells/well for C6, two replicates for each time point). Cells were allowed to adhere before being exposed to either HEK or Slit2 encapsulated beads. UW3 and Daoy medulloblastoma and U251 glioma cells were counted on days 2, 4 and 6 using a Coulter Z™ Series counter (Beckman-Coulter Inc., Mississauga, ON, Canada). C6 were counted on days 2 and 4 only because of the high proliferation rate of this cell line.
Transfection and biotinylation
A pCEP4 (Invitrogen) plasmid expressing full-length rat Robo1 tagged with HA was constructed as described (Li et al., 1999). U251 glioblastoma cells were transfected with the Robo1-HA vector and antibiotics were added after 48 h. Selection was carried out for 5 weeks with a medium change every 3 days. Hygromycin B (Sigma-Aldrich Co., Canada Ltd., Oakville, ON, Canada) was used for stable selection of U251-Robo1-HA cells at a concentration of 500 μg/ml. Stable cell lines were obtained after isolating clones and tested for protein expression by Western blot analysis using an anti-HA antibody. The surface proteins of U251 wild-type and U251-HA Robo1 cell lines were biotinylated as described (Zhao et al., 2004). Input total cell lysates and enriched cell surface proteins were resolved and subjected to Western blot analysis. Anti-HA antibody was used to detect recombinant Robo1 and anti p42/44 was used as a cell surface biotinylation control.
Rho-GTPase activity assays
Rac1, Cdc42 and Rho activity assays were performed using nonradioactive Activity Assay kits (Upstate Biotechnology, Lake Placid, NY, USA). Briefly, UW3 and C6 monolayer cultures were treated with concentrated Slit2 or HEK conditioned medium diluted in serum-free DMEM for 24 h, and lysed. Cell lysates were affinity precipitated with a GST fusion-protein corresponding to the p21-binding domain of human PAK1 bound to glutathione–agarose (Rac and Cdc42) or the Rho binding domain of Rhotekin bound to agarose beads and run on 15% SDS–PAGE. Western blot analysis was used to probe blots for activated Rho-GTPase proteins. Cdc42, Rac1 and Rho levels were determined by Western blot analysis and used as loading controls. Cdc42, Rac1 and Rho antibodies were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA), BD Transduction Laboratories (BD Biosciences, Pharmingen, Mississauga, ON, Canada) and Upstate Biotechnology, respectively.
Statistical analysis
All tests were performed using SPSS Graduate Pack 9.0 statistical software (SPSS Inc., Chicago IL, USA). Descriptive statistics including mean and standard error of the mean, one-way ANOVA’s, independent sample two-tailed t-tests, paired t-tests and Tukey’s test for multiple comparisons were utilized. P-values less than 0.05 were considered significant.
Acknowledgments
We thank Carmen Sabau, Roberta Waldkircher, and Samer Hussein for technical assistance (Montreal Neurological Institute, Montreal, Quebec). We also wish to thank Vinit Srivastava for providing the cerebellar tissue. TEWO is the recipient of a Canadian Institutes of Health Research (CIHR) Doctoral Research Award, MSS is the recipient of a Jeanne Timmins Costello Fellowship, NJ is a recipient of a Chercheur Boursier Salary award from Fonds de la Recherche en Sante du Quebec (FRSQ) and AAL is a recipient of the Terry Fox Studentship from the National Cancer Institute of Canada. RFDM holds the William Feindel Chair of Neurooncology and is a Killam Scholar at the Montreal Neurological Institute. This work was supported by the Goals for Lily and the Alex Pavanel Family, the Franco Di Giovanni, the Barbara Jacobson and Carol Cuthbertson, the Raymonde and Tony Boeckh Brain Tumor Research Funds, The Brain Tumour Foundation of Canada and the Maggie De Fontes Foundation.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- Astuti D, Da Silva NF, Dallol A, Gentle D, Martinsson T, Kogner P, et al. Br J Cancer. 2004;90:515–521. doi: 10.1038/sj.bjc.6601447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashaw GJ, Kidd T, Murray D, Pawson T, Goodman CS. Cell. 2000;101:703–715. doi: 10.1016/s0092-8674(00)80883-1. [DOI] [PubMed] [Google Scholar]
- Corcoran A, Del Maestro RF. Neurosurgery. 2003;53:174–184. doi: 10.1227/01.neu.0000072442.26349.14. [DOI] [PubMed] [Google Scholar]
- Dallol A, Da Silva NF, Viacava P, Minna JD, Bieche I, Maher ER, et al. Cancer Res. 2002;62:5874–5880. [PubMed] [Google Scholar]
- Dallol A, Krex D, Hesson L, Eng C, Maher ER, Latif F. Oncogene. 2003a;22:4611–4616. doi: 10.1038/sj.onc.1206687. [DOI] [PubMed] [Google Scholar]
- Dallol A, Morton D, Maher ER, Latif F. Cancer Res. 2003b;63:1054–1058. [PubMed] [Google Scholar]
- Del Duca D, Werbowetski T, Del Maestro RF. J Neurooncol. 2004;67:295–303. doi: 10.1023/b:neon.0000024220.07063.70. [DOI] [PubMed] [Google Scholar]
- Del Maestro RF, Shivers R, Mc Donald W, Del Maestro AGR. J Neurooncol. 2001;53:87–98. [Google Scholar]
- Etienne-Manneville S, Hall A. Cell. 2001;106:489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Nature. 2003;421:753–756. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- Fan X, Labrado JP, Hing H, Bashaw GJ. Neuron. 2003;40:113–127. doi: 10.1016/s0896-6273(03)00591-9. [DOI] [PubMed] [Google Scholar]
- Gilthorpe JD, Papantoniou E, Chedotal A, Lumsden A, Wingate RJT. Development. 2002;129:4719–4728. doi: 10.1242/dev.129.20.4719. [DOI] [PubMed] [Google Scholar]
- Gomes ER, Shantanu J, Gundersen GG. Cell. 2005;121:451–463. doi: 10.1016/j.cell.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Jacobson PF, Jenkyn DJ, Papadimitriou JM. J Neuropathol Exp Neurol. 1985;44:472–485. doi: 10.1097/00005072-198509000-00003. [DOI] [PubMed] [Google Scholar]
- Jen JC, Chan WM, Bosley TM, Wan J, Carr JR, Rub U, et al. Science. 2004;304:1509–1513. doi: 10.1126/science.1096437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joki T, Machluf M, Atala A, Zhu J, Seyfried NT, Dunn IF, et al. Nat Biotechnol. 2001;19:35–39. doi: 10.1038/83481. [DOI] [PubMed] [Google Scholar]
- Keles GE, Berger MS, Srinivasan J, Kolstoe DD, Bobola MS, Silber JR. Oncol Res. 1995;7:493–503. [PubMed] [Google Scholar]
- Li HS, Chen JH, Wu W, Fagaly T, Zhou L, Yuan W, et al. Cell. 1999;96:807–818. doi: 10.1016/s0092-8674(00)80591-7. [DOI] [PubMed] [Google Scholar]
- Louis DN, Pomeroy SL, Cairncross JG. Cancer Cell. 2002;1:125–128. doi: 10.1016/s1535-6108(02)00040-5. [DOI] [PubMed] [Google Scholar]
- Lundstrom A, Gallio M, Englund C, Steneberg P, Hemphala J, Aspenstrom P, et al. Genes Dev. 2004;18:2161–2171. doi: 10.1101/gad.310204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Marillat V, Cases O, Nguyen-Ba-Charvet KT, Tessier-Lavigne M, Sotelo C, Chedotal A. J Comp Neurol. 2002;442:130–155. doi: 10.1002/cne.10068. [DOI] [PubMed] [Google Scholar]
- Marillat V, Sabatier C, Failli V, Matsunaga E, Sotelo C, Tessier-Lavigne M, et al. Neuron. 2004;43:69–79. doi: 10.1016/j.neuron.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Mason HA, Susumu I, Corfas G. J Neurosci. 2001;21:7654–7663. doi: 10.1523/JNEUROSCI.21-19-07654.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Ba-Charvet KT, Picard-Riera N, Tessier-Lavigne M, Baron-Van Evercooren A, Sotelo C, Chedotal A. J Neurosci. 2004;24:1497–1506. doi: 10.1523/JNEUROSCI.4729-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo AF, Joseph HL, Chen Y-J, Dujardin DL, Alberts AS, Pfister KK, et al. Curr Biol. 2001;11:1536–1541. doi: 10.1016/s0960-9822(01)00475-4. [DOI] [PubMed] [Google Scholar]
- Park KW, Morrison CM, Sorensen LK, Jones CA, Rao Y, Chien CB, et al. Dev Biol. 2003;261:251–267. doi: 10.1016/s0012-1606(03)00258-6. [DOI] [PubMed] [Google Scholar]
- Prasad A, Fernandis AZ, Rao Y, Ganju RK. J Biol Chem. 2004;279:9115–9124. doi: 10.1074/jbc.M308083200. [DOI] [PubMed] [Google Scholar]
- Raffel C. Neoplasia. 2004;6:310–322. doi: 10.1593/neo.03454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffel C, Thomas GA, Tishler DM, Lassoff S, Allen JC. Neurosurgery. 1993;33:301–305. doi: 10.1227/00006123-199308000-00018. [DOI] [PubMed] [Google Scholar]
- Raffel C, Ueki K, Harsh GR, IV, Louis DN. Neurosurgery. 1995;36:971–974. doi: 10.1227/00006123-199505000-00013. [DOI] [PubMed] [Google Scholar]
- Read TA, Sorensen DR, Mahesparan R, Enger PO, Timpl R, Olsen BR, et al. Nat Biotechnol. 2001;19:29–34. doi: 10.1038/83471. [DOI] [PubMed] [Google Scholar]
- Rossi MR, Huntoon K, Cowell JK. Gene. 2005;356:85–90. doi: 10.1016/j.gene.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K, et al. Proc Natl Acad Sci USA. 2003;100:13513–13518. doi: 10.1073/pnas.2235846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffini F, Arbour N, Blain M, Olivier A, Antel JP. Am J Pathol. 2004;165:2167–2175. doi: 10.1016/S0002-9440(10)63266-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanai N, Alvarez-Buylla A, Berger MS. N Engl J Med. 2005;353:811–822. doi: 10.1056/NEJMra043666. [DOI] [PubMed] [Google Scholar]
- Wang B, Xiao Y, Ding BB, Zhang N, Yuan X, Gui L, et al. Cancer Cell. 2003;4:19–29. doi: 10.1016/s1535-6108(03)00164-8. [DOI] [PubMed] [Google Scholar]
- Werbowetski T, Bjerkvig R, Del Maestro RF. J Neurobiol. 2004;60:71–88. doi: 10.1002/neu.10335. [DOI] [PubMed] [Google Scholar]
- Werbowetski-Ogilvie TE, Agar NYA, Waldkircher de Oliveira RM, Faury D, Antel JP, Jabado N, et al. Cancer Res. 2006;66:1464–1472. doi: 10.1158/0008-5472.CAN-05-1913. [DOI] [PubMed] [Google Scholar]
- Wong K, Park HT, Wu JY, Rao Y. Curr Opin Genet Dev. 2002;12:583–591. doi: 10.1016/s0959-437x(02)00343-x. [DOI] [PubMed] [Google Scholar]
- Wong K, Ren XR, Huang YZ, Xie Y, Liu G, Saito H, et al. Cell. 2001;107:209–221. doi: 10.1016/s0092-8674(01)00530-x. [DOI] [PubMed] [Google Scholar]
- Wu JY, Feng L, Park HT, Havlioglu N, Wen L, Tang H, et al. Nature. 2001;410:948–952. doi: 10.1038/35073616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zhang W, Kho Y, Zhao Y. Anal Chem. 2004;76:1817–1823. doi: 10.1021/ac0354037. [DOI] [PubMed] [Google Scholar]