

Abstract
Systemic vasculitis, an inflammatory necrotizing disease of the blood vessel walls, can occur secondary to autoimmune diseases, including connective tissue diseases. Various pathogenic mechanisms have been implicated in the induction of vasculitis, including cell-mediated inflammation, immune complex-mediated inflammation and autoantibody-mediated inflammation. This inflammatory activity is believed to contribute to accelerated atherosclerosis, and also leads to increased risk for cardiovascular events in patients with rheumatoid arthritis and systemic lupus erythematosus. Endothelial cell activation is a common pathogenic pathway in the systemic vasculitis associated with rheumatoid arthritis and systemic lupus erythematosus, with elevated levels of endothelin-1 potentially inducing vascular dysregulation.
Introduction
The term 'systemic vasculitis' describes a heterogeneous group of rare diseases, the systemic vasculitides, characterized by inflammation and fibrinoid necrosis of blood vessel walls. Vasculitis may be primary in origin (with no identifiable cause) or it may be secondary to infection, malignancy, or autoimmune disease. Although rare, there is evidence to suggest that vasculitis accelerating atherosclerosis is a complicating feature of most, possibly all, autoimmune diseases. This includes connective tissue diseases (CTDs) such as rheumatoid arthritis (RA), scleroderma, sarcoidosis and systemic lupus erythematosus (SLE).
In this review, which focuses on vasculitis associated with CTDs, we look at the progress that has been made in classifying the systemic vasculitides and discuss the pathogenesis of systemic vasculitides in CTDs and their adverse clinical sequelae, giving particular attention to RA and SLE. The standardized treatment for vasculitis is effective in the majority of patients, but some relapse and need other therapeutic approaches. In evaluating new treatment strategies for the management of systemic vasculitis, we explore the role of endothelin (ET)-1 in systemic vasculitides and discuss the therapeutic potential of endothelin receptor blockade in these entities.
Classification of the systemic vasculitides
Early attempts to classify systemic vasculitis into discrete categories were based primarily on blood vessel size, and indeed that approach still underpins more recent classification schemes. These schemes include those of the American College of Rheumatology [1] and of the international consensus conference held in Chapel Hill, North Carolina, USA that gave rise to the Chapel Hill nomenclature [2]. Today, most physicians use the Chapel Hill nomenclature, which is based on clinical and histopathological features of vasculitis (Figure 1).
Figure 1.
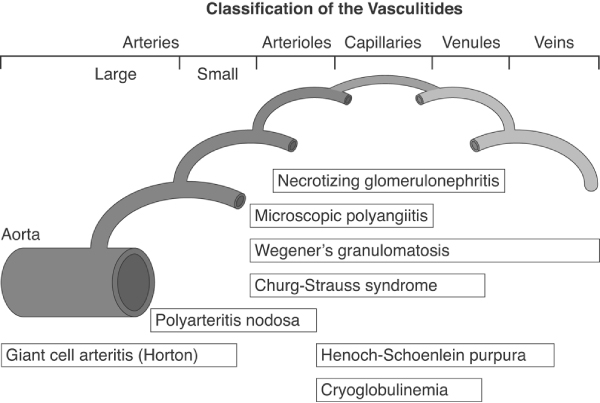
Classification of the systemic vasculitis. Reproduced with permission from Jennette et al. Arthritis Rheum 1994 © John Wiley & Sons/American College of Rheumatology [2].
In essence, classification schemes, including the Chapel Hill nomenclature, recognize two major groups of systemic vasculitides [2]: large vessel vasculitis, which consists of giant cell arteritis (GCA) and Takayasu arteritis, both of which involve the aorta and its major branches; and necrotizing vasculitis, which encompasses the rest of the vasculitides. The necrotizing systemic vasculitides include polyarteritis nodosa and Kawaski disease, which affect medium-sized arteries, and a large group of disorders in which the vasculitis affects arterioles, capillaries and venules (Figure 1). Within this latter group there are four disorders, namely Wegener's granulomatosis, Churg-Strauss syndrome, microscopic polyangiitis and necrotizing glomerulonephritis, which are characterized by the presence of anti-neutrophil cytoplasmic antibodies (ANCAs). Other types of small vessel vasculitis include Henoch-Schönlein purpura, which is characterized by IgA-dominant immune deposits in the small blood vessels, and essential cryoglobulinaemic vasculitis, in which cryoglobulin immune deposits are responsible for small blood vessel vasculitis (Figure 1).
The vasculitides that occur in autoimmune diseases usually affect small-sized vessels, as is the case in SLE, systemic sclerosis and Sjögren's syndrome. Where vasculitis occurs during the course of RA, the small arteries are involved in most patients but medium-sized arteries can also be affected, mimicking polyarteritis nodosa. A comprehensive overview of the classification of systemic vasculitides was recently reported by Sunderkötter and Sindrilaru [3].
Pathogenesis of vasculitis
Progress in the classification of the systemic vasculitides has facilitated better understanding of the pathogenesis underlying these inflammatory conditions, which can involve cell-mediated inflammation, immune complex (IC)-mediated inflammation, and ANCA-mediated inflammation. Of central importance is that many data have accumulated demonstrating that various inflammatory pathways lead to endothelial cell activation, which may induce complications such as vessel occlusion and tissue destruction in a predisposed host, and longstanding disease. Investigations into ANCA vasculitides have pioneered the field and expanded our understanding of the pathogenesis of vessel inflammation. Comprehensive overviews of the pathogenic mechanisms that underlie the vasculitides and the distinction between primary and secondary vasculitis were published elsewhere [3,4].
In GCA, for example, vasculitis is essentially a T-cell driven process that is triggered by exposure to antigens, most probably infectious agents. Dendritic cells present in the adventitia and media of the blood vessel wall are potent antigen-presenting cells that can prime naïve T cells. In patients with GCA they are responsible for activating CD4+ T cells, which orchestrate vascular injury by recruiting macrophages and monocytes to the vessel walls [5]. These cells induce systemic inflammation via the release of cytokines, such as interleukin-1 and interleukin-6. Tissue resident T cells also release interferon-γ, which is a key proinflammatory cytokine that has been implicated in the pathogenesis of GCA. Sustained inflammation mediated by T cells, macrophages and the proinflammatory cytokines released by these cells leads to extensive intimal thickening and vessel occlusion (Figure 2). Platelet-derived growth factor and vascular endothelial growth factor play key roles in the subsequent development of the lumen-occlusive arteritis that characterizes GCA.
Figure 2.
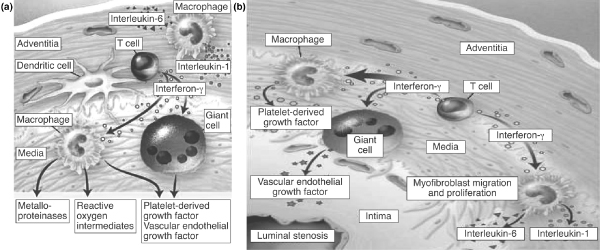
Pathogenesis of giant cell arteritis. (a) Recruitment and stimulation of antigen-specific T cells by dendritic cells. Interferon-γ regulates the differentiation and function of macrophages. Macrophages in the adventitial layer supply interleukin-1 and interleukin-6, and in the media secrete metalloproteinases. (b) Platelet-derived growth factor and vascular endothelial growth factor are responsible for intimal hyperplasia, then favour lumen-occlusive arteritis. Reproduced with permission from Weyland et al. N Engl J Med 2003 Copyright © 2003 Massachusetts Medical Society [5].
In contrast to GCA, which appears to be an antigen-driven disease with local T cell and macrophage activation in the vessel wall, IC formation is usually considered to be the major pathological event in polyarteritis nodosa, cryoglobulinaemia and Henoch-Schönlein purpura. In polyarteritis nodosa, for example, antibody-mediated IC deposition can lead to renal infarction. Micro-aneurisms are another manifestation that can occur as a consequence of IC formation in the medium-sized necrotizing systemic vasculitides, which can also lead to blood vessel occlusion. However, this is mediated to a large extent by circulating antibodies in blood and antibody-dependent effector mechanisms rather than as a result of cell-mediated inflammation. With regard to IC-mediated vascular damage, it has been demonstrated in CD18-/- mice that ICs induce damage from the luminal side but not from perivascular areas. Therefore, in this entity it has been suggested that the interaction of ICs, Fcγ receptors and adhesion molecules leads to disturbances in transmigration and activation of polymorphonuclear neutrophils, with consequent vessel damage [6].
Autoantibodies play a central role in the ANCA-related vasculitis that is characteristic of Wegener's granulomatosis, Churg-Strauss syndrome, microscopic polyangiitis and necrotizing glomerulonephritis. These antibodies are directed against enzymes that are contained in the granules of neutrophils and monocytes. In Wegener's granulomatosis they primarily target proteinase 3, which is a neutral serine protease, and – in microscopic polyangiitis and Churg-Strauss syndrome – they target myeloperoxidase, which is an enzyme that is involved in the generation of reactive oxygen species. Both proteinase 3-ANCA and myeloperoxidase-ANCA can contribute to vasculitis in the small blood vessels, causing adverse clinical sequelae such as pauci-immune glomerulonephritis and pulmonary haemorrhage, as demonstrated in animal models (myeloperoxidase) [7]. An enhancing effect of ANCAs on leucocyte diapedesis caused by their interaction with the endothelium was recently demonstrated in a study involving passive transfer of immunoglobulin [8]; this procedure resulted in microvascular haemorrhage.
Vasculitis in autoimmune diseases
Vasculitis may occur in many autoimmune diseases, including RA and SLE (which are the focus of this review), Sjögren's syndrome, scleroderma and sarcoidosis. Primary Sjögren's syndrome is associated with enhanced risk for B-cell lymphoma [9], in which the presence of purpura or vasculitis are clinical risk indicators for future non-Hodgkin's lymphoma development, as has been identified in a number of studies primarily based on epidemiological data [10]. In association with SLE, for example, distinctive characteristics of coexistent vasculitis include the presence of anti-Ro-SSA, HLA and DR3 antibodies, as well as (more rarely) anti-DNA, anti-Sm and anti-RNP antibodies. Clinical presentation can include purpura, peripheral neuropathy of the lower limbs, and hypergammaglobulinaemia. Many patients will also exhibit evidence of rheumatoid factor activity, cryoglobulinaemia due to monoclonal IgM (kappa) paraprotein, and low complement levels. Pulmonary involvement is uncommon in vasculitis associated with Sjögren's syndrome, although patients may develop interstitial lung disease, pulmonary hypertension, or lymphocytic interstitial pneumonia. Mesenteric vasculitis and biliary cirrhosis are seen in cases with gastrointestinal tract involvement, whereas cognitive dysfunction, infarcts and neuropsychiatric symptoms may indicate central nervous system involvement. Renal involvement is less common in vasculitis related to Sjögren's syndrome, but when it does occur it usually manifests as interstitial nephritis. Vasculitis is extremely rare in scleroderma. If present, it is usually seen in the small blood vessels, sometimes in association with ANCA-related vasculitis. Vasculitis is also rare in sarcoidosis. It may affect the skin as leucocytoclastic granulomatosis often in association with sarcoid necrotizing granulomatosis [11]. It may also occur as synovial vasculitis. Very rarely, the aorta, pulmonary artery and mesenteric arteries are affected, giving rise to a very severe form of large-vessel vasculitis that mimics Takayasu arteritis. Patients with this form of vasculitis develop aortitis with stenoses, aneurisms and dissection, and have a poor prognosis.
Atherosclerosis appears to be accelerated in patients with vasculitis, although it is difficult to discern its contributing impact on subsequent vascular occlusion. Moreover, corticosteroids, which are required for the treatment of inflammatory diseases such as RA, chronic obstructive pulmonary disease and inflammatory bowel disease, may also enhance risk for cardiovascular complications, as was recently demonstrated [12].
Vasculitis in rheumatoid arthritis and systemic lupus erythematosus
Vasculitis is a known complication of RA, often occurring several years after the initial onset of disease. Observational studies have shown that, independent of sex, patients with RA have reduced life expectancy in comparison with the non-RA population [13]. Rheumatoid vasculitis is characterized by the occurrence of mononeuritis multiplex, purpura and visceral involvement, with the latter sometimes being severe. This vasculitis is severe and, in its systemic form, should usually be treated with corticosteroids and cyclophosphamide. Despite such treatment, the outcome is worse than with the majority of vasculitides [14].
In addition to this severe inflammatory disease, patients with RA appear to be at significantly increased risk from coronary events in comparison with healthy individuals [15]. Observational studies have shown that, compared with healthy normal individuals, patients with RA are at double the risk for developing coronary artery disease [16,17], as reflected by unrecognized myocardial infarction, as well as congestive heart failure [18] and sudden cardiac death [16] (Figure 3). A recent study that sought to identify demographic and cardiovascular risk factors in RA patients with and without atherosclerotic plaque in the carotid arteries [19] demonstrated that age, hypertension (especially raised systolic blood pressure), raised cholesterol, raised low-density lipoprotein levels and (to a lesser extent) homocysteinaemia were the factors that differed most significantly between the two patient populations.
Figure 3.
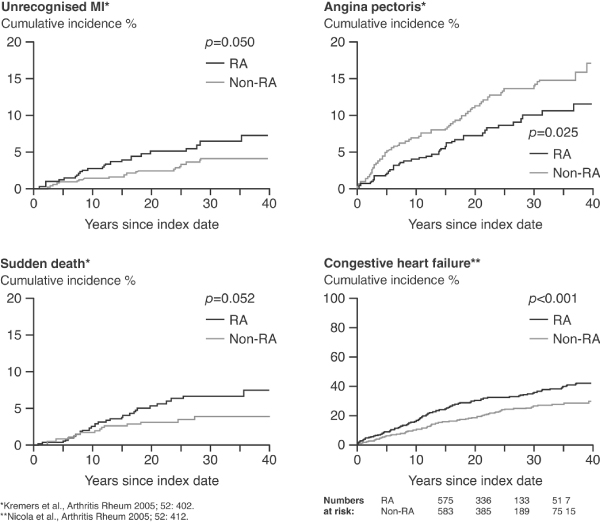
Data showing increased cardiovascular risk in patients with RA versus the normal population. MI, myocardial infarction; RA, rheumatoid arthritis. Reproduced with permission from Nicola et al. Arthritis Rheum 2005 and Maradit-Kremers et al. Arthritis Rheum 2005 © John Wiley & Sons/American College of Rheumatology [16,18].
As data from these and other observational studies have illustrated, RA patients are at significantly increased risk for developing cardiovascular disease relative to the normal population. Studies have also shown that inflammation is a key link between RA and accelerated atherosclerosis. Correlations have been demonstrated between intima-media thickness (a surrogate marker for cardiovascular disease) and measures of inflammation such as erythrocyte sedimentation rate and C-reactive protein [20]. It has also been suggested that corticosteroids may facilitate atherosclerosis [21].
The picture in SLE is similar to that in RA, except that patients with SLE exhibit an even greater incidence of atherosclerotic plaque in carotid arteries [22,23], a higher rate of calcification in the coronary arteries [24], and a correspondingly higher frequency of myocardial infarction and stroke when compared with the normal population. It should be noted that patients with SLE and antiphospholipid syndrome, but also those with subacute cutaneous lupus erythematosus, frequently present with a livedo vasculopathy. This is of particular interest because it reflects a certain type of small-vessel vasculitis. However, in patients with antiphospholipid syndrome that is independent of any underlying disease, livedo may be present in the absence of other signs or findings of vasculitis.
Although risk factors for atherosclerosis in SLE patients are similar to those in patients with RA, there are additional SLE-related risk factors (Figure 4). These include the development of autoantibodies to endothelium, high-density lipoprotein and phospholipids, as well as dyslipidaemia [22]. The enhanced frequency of circulating ICs, activated complement products and nephritis are additional SLE-related risk factors [25]. These additional risk factors may explain why SLE patients are at greatly increased cardiovascular risk, even relative to that in patients with RA. Certainly, SLE carries a risk for accelerated atherosclerosis that is greater than the net sum of classical risk factors [26]. Whether accelerated atherosclerosis in SLE is uniquely related to inflammatory activity or to additional factors, or both, remains uncertain. By unravelling the pathogenic mechanisms that are responsible for the vasculitis that occurs in RA and SLE, as well as the interaction of inflammation and procoagulatory mechanisms, we may develop a greater understanding of the contribution of vasculitis to adverse cardiovascular outcomes in these and other CTDs.
Figure 4.
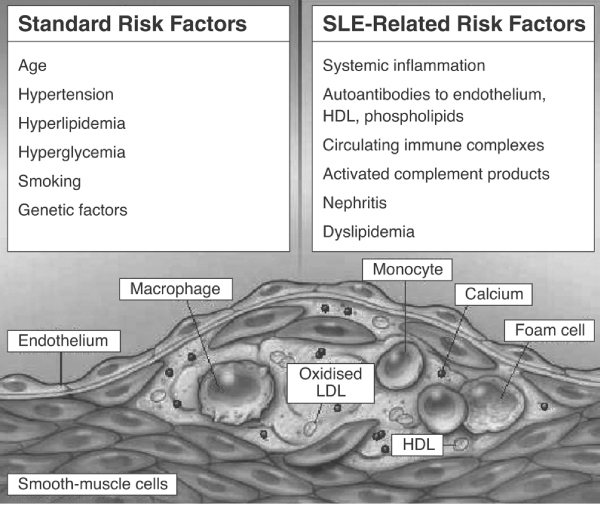
Enhanced cardiovascular risk in SLE and SLE-related risk factors. HDL, high-density lipoprotein; LDL, low-density lipoprotein; SLE, systemic lupus erythematosus. Reproduced with permission from Nicola et al. N Engl J Med 2003 Copyright © 2003 Massachusetts Medical Society [18].
Pathogenic mechanisms of vasculitis in rheumatoid arthritis and systemic lupus erythematosus
Autoantibodies
In autoimmune diseases such as RA and SLE there is evidence of an absolute deficiency in the CD4+ T-cell population, a decreased CD4/CD8 ratio and, most importantly, a diminished relative percentage of CD4+CD25+ regulatory T cells. There is also abundant evidence for induction of autoantibodies, including anti-endothelial cell antibodies (AECAs), antibodies against adhesion molecules on the endothelial cell surface, and antibodies against induced autoantigens on the surface of endothelial cells [27,28]. Although AECAs are a focus of debate and may represent an epiphenomenon, these autoantibodies are thought to activate endothelial cells, leading to (among other things) upregulation of adhesion molecules (intercellular adhesion molecule-1, endothelial leucocyte adhesion molecule-1 and vascular cell adhesion molecule-1) and other major histocompatibility class II molecules [29,30]. In the presence of natural killer cells they may trigger endothelial cell apoptosis [31] or lead to the induction of effector mechanisms via antibody-dependent cellular cytotoxicity and complement activation [32,33]. Of importance, endothelial cells play a crucial role in inflammation and coagulation. A number of heterogeneous factors (antibodies by binding with the variable region to expressed antigens or by their constant region via Fc receptors, ligation of Toll-like receptors on the endothelial cell surface, as well as transmembranous migrating low-density lipoprotein trans fatty acids) can lead to nuclear factor-κB dependent endothelial cell activation (Figure 5). As a result, the endothelial cell achieves a procoagulatory status, which is not obviously dependent on the initial insult.
Figure 5.
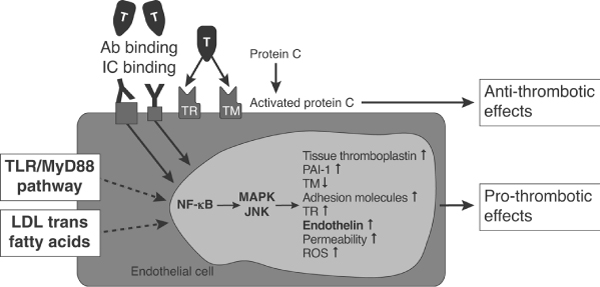
Pathways leading to nuclear factor-κB dependent endothelial cell activation. Ab, antibody; IC, immune complex; JNK, c-Jun amino-terminal kinase; LDL, low-density lipoprotein; MAPK, mitogen-activated protein kinase; MyD88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor-κB; PAI, plasminogen activator inhibitor; ROS, reactive oxygen species; TLR, Toll-like receptor; TM, thrombomodulin; TR, thrombin receptor. Reprinted from Iba et al.: The role of the endothelium in changes in procoagulant activity in sepsis. J Am Coll Surg 1998; 187(3):321–329. © 1998 by the American College of Surgeons, with permission from Elsevier [36].
Studies in patients with SLE have revealed that as many as 80% of patients produce AECAs, which are heterogeneous antibodies that react with various endothelial antigens. In SLE these antigens include constitutive heparin-like molecules (25 to 200 kDa) and nuclear antigens, of which heat shock protein 60 and the autoantigens Ro(SS-A) and La(SS-B) are the best characterized. These autoantigens can either be expressed on the surface after intracellular translocation or they can adhere to the surface of endothelial cells after release, where they can be recognized by cognate autoantibodies. AECAs are also present in patients with RA and especially in those who present with clinical vasculitis, in which the presence of AECAs correlates with extra-articular manifestations of the disease [34,35].
IC formation and binding to the endothelial cell surface leading to endothelial cell activation plays an important role in the pathogenesis of vasculitis and is a feature of vasculitis in both RA and SLE [35]. In these diseases, ICs can activate endothelial cells simply by binding to Fcγ receptors. This leads to plasma extravasation, recruitment of leucocytes to the perivascular environment, an enhanced inflammatory response from endothelial paracrine mediators and, finally, activation of the coagulation system and development of a prothrombotic state [6,36].
Fcγ receptor binding on endothelial cells is also a critical process after binding of specific autoantibodies to blood cells (for example, ANCA binding to neutrophils) because it initiates binding to the target tissue and subsequent destruction, such as that seen in vasculitis.
Additional processes are involved, such as activation of endothelial cells by shear stress, binding of von Willebrand factor, regulation of ET-1, thrombomodulin and thrombin receptor, adhesion molecules and activation of thrombocytes, the sequence of which is beginning to be understood. Thus, release of these factors leads to increased levels in serum and cannot be considered a primary pathogenic event in vasculitis, whereas, for example, enhanced ET-1 levels in scleroderma and Raynaud's have pathogenic relevance.
Role of endothelin in vasculitides
Binding of autoantibodies and ICs to the surface of endothelial cells results, among other things, in enhanced ET-1 release. ET-1 is known to activate monocytes via activation of the endothelin receptor subtype A [37] and to contribute to matrix remodelling [38]. Raised levels of ET-1 have been described in both RA and SLE patients. Pache and colleagues [39], for example, reported significantly elevated plasma levels of ET-1 in RA patients as compared with control individuals, and suggested that they might be related to vascular dysregulation, as observed in nail-fold capillaroscopy. Raised plasma concentrations of ET-1 have also been described in SLE patients [39]. Again, there is a suggestion that elevated ET-1 may act as a marker of vascular damage [40,41]. There are clear data that ET-1 is enhanced in SLE patients with pulmonary arterial hypertension [42], and usage of the dual endothelin receptor antagonist bosentan resulted in clinical improvements in this subset of patients [43].
Albeit few, there are reports of increased ET-1 in the acute phase of Henoch-Schönlein purpura, with levels returning to normal during remission [44]. Raised levels of ET-1 have also been described in the early stages of GCA and may contribute to the vascular occlusion seen in some patients [45]. Macrophages are known to produce matrix metalloproteinase-2, which is an analogue of endothelin-converting enzyme that cleaves big ET-1 to ET-1; this could account for the high levels of ET-1 seen in the early stage of GCA.
Therapeutic strategies: the potential of endothelin blockade
Immunosuppression
Clinical management of vasculitis associated with CTDs requires a multipronged approach, addressing the underlying disease and its severity as well as the vasculitis itself. Additionally, there may be a need to treat symptomatic manifestations of the disease. Overall, the aim of treatment is to control disease, prevent relapse and increase event-free survival. Preliminary clinical data in RA indicate that controlling disease activity by methotrexate reduces fatal acute myocardial infarction [46] and tumour necrosis factor therapy is able to improve endothelial function [47].
Currently, corticosteroids and immunosuppressants such as cyclophosphamide are the mainstay of treatment for vasculitis. Other drugs such as bisphosphonates and cotrimoxazole may be included as adjunctive treatments. The therapeutic regimen should be carefully chosen and, in cases of primary vasculitis, tailored according to criteria that constitute a five-point prognostic score: proteinuria (>1 g/day), raised creatinine levels (>140 μmol/l), gastrointestinal tract involvement, specific cardiomyopathy and central nervous system involvement. A study examining this scoring system, which has been validated in polyarteritis nodosa, microscopic polyangiitis and Churg-Strauss syndrome, showed that patients fulfilling none of the criteria have only a 12% mortality at 5 years as compared with 50% mortality in those satisfying two of the criteria [48].
A common approach to treatment, and one supported by data from the European Vasculitis Study Group on ANCA-associated vasculitis and Wegener's granulomatosis, is to use short-term cyclophosphamide over a 3-month to 6-month period as induction therapy, and then to switch patients to maintenance therapy with either azathioprine or methotrexate (Figure 6). This is in addition to background corticosteroid therapy [49]. This strategy has also proved effective in vasculitis secondary to RA; patients initially received short-course pulsed cyclophosphamide and were then switched to an alternate immunosuppressant agent. In our experience, azathioprine and methotrexate are equally effective in maintaining remission; for induction, cyclophosphamide remains the best treatment at present.
Figure 6.
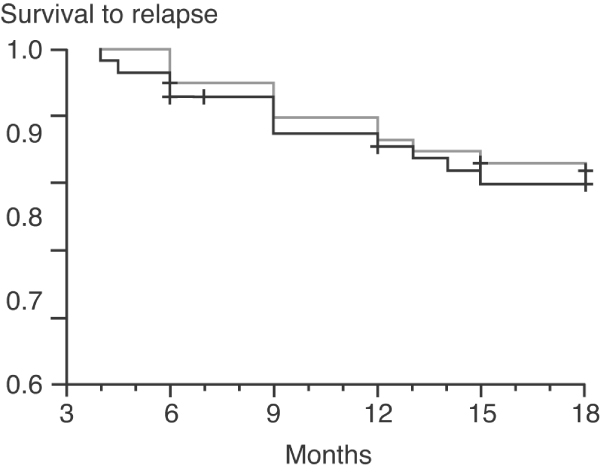
Comparison of maintenance therapy with cyclophosphamide (grey line) or azathioprine (black line) in ANCA-associated vasculitis. ANCA, anti-neutrophil cytoplasmic antibody. Reproduced with permission from Jayne et al. N Engl J Med 2003 © 2003 Massachusetts Medical Society [49].
Biological therapies
The advent of biological immunomodulatory therapies, such as rituximab and infliximab, has potentially increased therapeutic options in systemic vasculitis. Promising results for anti-CD20 therapy with rituximab have been described in patients with Wegener's granulomatosis and ANCA-associated vasculitis, including refractory ANCA vasculitis [50,51]. In a small, open-label study [51], treatment with rituximab depleted circulating B cells while also suppressing ANCA antibody titres and inducing longstanding remission. Treatment of necrotizing vasculitides, such as Wegener's granulomatosis, with the anti-tumour necrosis factor-α agent infliximab has also shown early promise. Encouragingly, efficacy was seen in patients who were unresponsive to standard therapies, rituximab and intravenous immune globulin.
The potential for endothelin-1 blockade
Although data are limited, there is evidence suggesting that ET-1 plays some role in the clinical manifestations of vasculitis; blockade of ET-1 could therefore be of therapeutic benefit, albeit probably as add-on therapy. From a clinical perspective, there is a lack of longitudinal data on ET-1 levels in relation to disease activity and of ET-1 levels in response to standard therapies. It is also unclear whether elevated ET-1 is a risk factor for cardiovascular complications in CTDs, although it undoubtedly is for pulmonary arterial hypertension. ET-1 antagonism with drugs such as bosentan is an established treatment for pulmonary arterial hypertension. In addition to its established indications, it is possible that drugs such as bosentan might be helpful in specific vascular manifestations, such as renal vasculitis, ischaemia, and ischaemia complicating severe Raynaud's phenomenon. Recently bosentan was approved in Europe to reduce the number of new digital ulcers in patients with SSc and ongoing digital ulcer disease.
Conclusion
Systemic vasculitis is characterized by multiple phenotypes that reflect the different pathogenic mechanisms that are responsible for inducing inflammation in the large, medium and small blood vessels (arterioles, venules and capillaries) of the body's tissues and organs. Although cell-mediated inflammation is characteristic of GCA, a systemic vasculitis of the large blood vessels, IC-mediated inflammation and ANCA-mediated inflammation are seen in the necrotizing vasculitides that affect medium-sized and small blood vessels.
Systemic vasculitis, a rare complication of autoimmune disorders, can nevertheless occur in most if not all CTDs. Evidence suggests that inflammation is a key link between vasculitis and accelerated atherosclerosis in these patients. Certainly, patients with RA and SLE are at significantly increased risk for cardiovascular morbidity and mortality in comparison with the normal population. Although preliminary data point to effects of immunosuppression on cardiovascular complications, this must be substantiated. There is a clear need for prevention studies (lipid and blood pressure control) in conjunction with control of disease activity, which requires delineation of the factors that drive vasculitis and endothelial cell activation.
Endothelial cell activation via autoantibody or IC binding is a common pathogenic pathway in systemic vasculitis associated with RA and SLE, superimposing on 'baseline risks' (blood pressure, Toll-like receptor ligands, enhanced low-density lipoproteins and smoking), leading among other things to enhanced release of ET-1. There is evidence, albeit limited, suggesting that ET-1 plays a role in some clinical manifestations of vasculitis. Potentially, endothelin receptor antagonism may have a place in the treatment of specific vascular manifestations of vasculitis as an adjunct to standard therapy with corticosteroids and immunosuppressant agents. Effective ET-1 blockade has been demonstrated in SLE patients with pulmonary arterial hypertension.
Abbreviations
AECA = anti-endothelial cell antibody; ANCA = anti-neutrophil cytoplasmic antibody; CTD = connective tissue disease; ET = endothelin; GCA = giant cell arteritis; IC = immune complex; RA = rheumatoid arthritis; SLE = systemic lupus erythematosus.
Competing interests
The authors have received speaker fees and reimbursement for travel expenses from Actelion Pharmaceuticals Ltd.
Acknowledgments
Acknowledgements
The authors would like to acknowledge medical writing support funded by an educational grant from Actelion Pharmaceuticals Ltd.
This article is part of Arthritis Research & Therapy Volume 9 Supplement 2: Advances in systemic sclerosis and related fibrotic and vascular conditions, and is based on presentations made at a symposium entitled Advances in systemic sclerosis and connective tissue disease, sponsored by Actelion Pharmaceuticals Ltd, held in Athens, Greece in April 2006. The full contents of the supplement are available online at http://arthritis-research.com/supplements/9/S2. This supplement has been supported by an educational grant from Actelion Pharmaceuticals Ltd.
References
- Hunder GG, Arend WP, Bloch DA, Calabrese LH, Fauci AS, Fries JF, Leavitt RY, Lie JT, Lightfoot RW, Jr, Masi AT, et al. The American College of Rheumatology 1990 crtieria for the classification of vasculitis: introduction. Arthritis Rheum. 1990;33:1065–1076. doi: 10.1002/art.1780330802. [DOI] [PubMed] [Google Scholar]
- Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, Hagen EC, Hoffman GS, Hunder GG, Kallenberg CG, et al. Nomenclature of systemic vasculitides: the proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–192. doi: 10.1002/art.1780370206. [DOI] [PubMed] [Google Scholar]
- Sunderkötter C, Sindrilaru A. Clinical classification of vasculitis. Eur J Dermatol. 2006;16:114–124. [PubMed] [Google Scholar]
- Lentsch AB, Ward PA. Regulation of inflammatory vascular damage. J Pathol. 2000;190:343–348. doi: 10.1002/(SICI)1096-9896(200002)190:3<343::AID-PATH522>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med. 2003;349:160–169. doi: 10.1056/NEJMra022694. [DOI] [PubMed] [Google Scholar]
- Sindrilaru A, Seeliger S, Ehrchen JM, Peters T, Roth J, Scharffetter-Kochanek K, Sunderkotter CH. Site of blood vessel damage and relevance of CD18 in a murine model of immune complex-mediated vasculitis. J Invest Dermatol. 2007;127:447–454. doi: 10.1038/sj.jid.5700563. [DOI] [PubMed] [Google Scholar]
- Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, Maeda N, Falk RJ, Jennette JC. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110:955–963. doi: 10.1172/JCI200215918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little MA, Smyth CL, Yadav R, Ambrose L, Cook HT, Nourshargh S, Pusey CD. Antineutrophil cytoplasm antibodies directed against myeloperoxidase augment leukocyte-microvascular interactions in vivo. Blood. 2005;106:2050–2058. doi: 10.1182/blood-2005-03-0921. [DOI] [PubMed] [Google Scholar]
- Kassan SS, Thomas TL, Moutsopoulos HM, Hoover R, Kimberly RP, Budman DR, Costa J, Decker JL, Chused TM. Increased risk of lymphoma in sicca syndrome. Ann Intern Med. 1978;89:888–892. doi: 10.7326/0003-4819-89-6-888. [DOI] [PubMed] [Google Scholar]
- Kassan SS, Moutsopoulos HM. Clinical manifestations and early diagnosis of Sjogren syndrome. Arch Intern Med. 2004;164:1275–1284. doi: 10.1001/archinte.164.12.1275. [DOI] [PubMed] [Google Scholar]
- Winkelmann RK, Frigas E. Eosinophilic panniculitis: a clinicopathologic study. J Cutan Pathol. 1986;13:1–12. doi: 10.1111/j.1600-0560.1986.tb00455.x. [DOI] [PubMed] [Google Scholar]
- Frostegard J. SLE, atherosclerosis and cardiovascular disease. J Intern Med. 2005;257:485–495. doi: 10.1111/j.1365-2796.2005.01502.x. [DOI] [PubMed] [Google Scholar]
- Symmons DP, Jones MA, Scott DL, Prior P. Longterm mortality outcome in patients with rheumatoid arthritis: early presenters continue to do well. J Rheumatol. 1998;25:1072–1077. [PubMed] [Google Scholar]
- Scott DG, Bacon PA, Elliott PJ, Tribe CR, Wallington TB. Systemic vasculitis in a district general hospital 1972–1980: clinical and laboratory features, classification and prognosis of 80 cases. QJM. 1982;51:292–311. [PubMed] [Google Scholar]
- Turesson C, Jarenros A, Jacobsson L. Increased incidence of cardiovascular disease in patients with rheumatoid arthritis: results from a community based study. Ann Rheum Dis. 2004;63:952–955. doi: 10.1136/ard.2003.018101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maradit-Kremers H, Crowson CS, Nicola PJ, Ballman KV, Roger VL, Jacobsen SJ, Gabriel SE. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum. 2005;52:402–411. doi: 10.1002/art.20853. [DOI] [PubMed] [Google Scholar]
- Solomon DH, Karlson EW, Rimm EB, Cannuscio CC, Mandl LA, Manson JE, Stampfer MJ, Curhan GC. Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis. Circulation. 2003;107:1303–1307. doi: 10.1161/01.CIR.0000054612.26458.B2. [DOI] [PubMed] [Google Scholar]
- Nicola PJ, Maradit-Kremers H, Roger VL, Jacobsen SJ, Crowson CS, Ballman KV, Gabriel SE. The risk of congestive heart failure in rheumatoid arthritis: a population-based study over 46 years. Arthritis Rheum. 2005;52:412–420. doi: 10.1002/art.20855. [DOI] [PubMed] [Google Scholar]
- Roman MJ, Moeller E, Davis A, Paget SA, Crow MK, Lockshin MD, Sammaritano L, Devereux RB, Schwartz JE, Levine DM, Salmon JE. Preclinical carotid atherosclerosis in patients with rheumatoid arthritis. Ann Intern Med. 2006;144:249–256. doi: 10.7326/0003-4819-144-4-200602210-00006. [DOI] [PubMed] [Google Scholar]
- Del Rincon I, Williams K, Stern MP, Freeman GL, O'Leary DH, Escalante A. Association between carotid atherosclerosis and markers of inflammation in rheumatoid arthritis patients and healthy subjects. Arthritis Rheum. 2003;48:1833–1840. doi: 10.1002/art.11078. [DOI] [PubMed] [Google Scholar]
- Roman MJ, Shanker BA, Davis A, Lockshin MD, Sammaritano L, Simantov R, Crow MK, Schwartz JE, Paget SA, Devereux RB, Salmon JE. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2399–2406. doi: 10.1056/NEJMoa035471. [DOI] [PubMed] [Google Scholar]
- Maxwell SR, Moots RJ, Kendall MJ. Corticosteroids: do they damage the cardiovascular system? Postgrad Med J. 1994;70:863–870. doi: 10.1136/pgmj.70.830.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manger K, Manger B, Repp R, Geisselbrecht M, Geiger A, Pfahlberg A, Harrer T, Kalden JR. Definition of risk factors for death, end stage renal disease, and thromboembolic events in a monocentric cohort of 338 patients with systemic lupus erythematosus. Ann Rheum Dis. 2002;61:1065–1070. doi: 10.1136/ard.61.12.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma Y, Oeser A, Shintani AK, Turner E, Olsen N, Fazio S, Linton MF, Raggi P, Stein CM. Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2407–2415. doi: 10.1056/NEJMoa035611. [DOI] [PubMed] [Google Scholar]
- Hahn BH. Systemic lupus erythematosus and accelerated atherosclerosis. N Engl J Med. 2003;349:2379–2380. doi: 10.1056/NEJMp038168. [DOI] [PubMed] [Google Scholar]
- Bruce IN. 'Not only ... but also': factors that contribute to accelerated atherosclerosis and premature coronary heart disease in systemic lupus erythematosus. Rheumatology (Oxford) 2005;44:1492–1502. doi: 10.1093/rheumatology/kei142. [DOI] [PubMed] [Google Scholar]
- Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26:1110–1118. doi: 10.1183/09031936.05.00045705. [DOI] [PubMed] [Google Scholar]
- Jiang H, Chess L. Regulation of immune responses by T cells. N Engl J Med. 2006;354:1166–1176. doi: 10.1056/NEJMra055446. [DOI] [PubMed] [Google Scholar]
- Carvalho D, Savage CO, Black CM, Pearson JD. IgG antiendothelial cell autoantibodies from scleroderma patients induce leukocyte adhesion to human vascular endothelial cells in vitro. Induction of adhesion molecule expression and involvement of endothelium-derived cytokines. J Clin Invest. 1996;97:111–119. doi: 10.1172/JCI118377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okawa-Takatsuji M, Aotsuka S, Fujinami M, Uwatoko S, Kinoshita M, Sumiya M. Up-regulation of intercellular adhesion molecule-1 (ICAM-1), endothelial leucocyte adhesion molecule-1 (ELAM-1) and class II MHC molecules on pulmonary artery endothelial cells by antibodies against U1-ribonucleoprotein. Clin Exp Immunol. 1999;116:174–180. doi: 10.1046/j.1365-2249.1999.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordron A, Dueymes M, Levy Y, Jamin C, Leroy JP, Piette JC, Shoenfeld Y, Youinou PY. The binding of some human antiendothelial cell antibodies induces endothelial cell apoptosis. J Clin Invest. 1998;101:2029–2035. doi: 10.1172/JCI2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse J, Barst R, Horn E, Cuervo N, Deng Z, Knowles J. Pulmonary hypertension in scleroderma spectrum of disease: lack of bone morphogenetic protein receptor 2 mutations. J Rheumatol. 2002;29:2379–2381. [PubMed] [Google Scholar]
- Mouthon L, Guillevin L, Humbert M. Pulmonary arterial hypertension: an autoimmune disease? Eur Respir J. 2005;26:986–988. doi: 10.1183/09031936.05.00112105. [DOI] [PubMed] [Google Scholar]
- Dörner T, Hucko M, Mayet WJ, Trefzer U, Burmester GR, Hiepe F. Enhanced membrane expression of the 52 kDa Ro(SS-A) and La(SS-B) antigens by human keratinocytes induced by TNF alpha. Ann Rheum Dis. 1995;54:904–909. doi: 10.1136/ard.54.11.904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández N, Jancar S, Sánchez Crespo M. Blood and endothelium in immune complex-mediated tissue injury. Trends Pharmcol Sci. 2004;25:512–517. doi: 10.1016/j.tips.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Iba T, Kidokoro A, Yagi Y. The role of the endothelium in changes in procoagulant activity in sepsis. J Am Coll Surg. 1998;187:321–329. doi: 10.1016/S1072-7515(98)00177-X. [DOI] [PubMed] [Google Scholar]
- Zouki C, Baron C, Fournier A, Filep JG. Endothelin-1 enhances neutrophil adhesion to human coronary artery endothelial cells: role of ETA receptors and platelet-activating factor. Br J Pharmacol. 1999;127:969–979. doi: 10.1038/sj.bjp.0702593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman M, Thannickal VJ, Pardo A, Zisman DA, Martinez FJ, Lynch JP., III Idiopathic pulmonary fibrosis: pathogenesis and therapeutic approaches. Drugs. 2004;64:405–430. doi: 10.2165/00003495-200464040-00005. [DOI] [PubMed] [Google Scholar]
- Pache M, Schwarz HA, Kaiser HJ, Wuest P, Kloti M, Dubler B, Flammer J. Elevated plasma endothelin-1 levels and vascular dysregulation in patients with rheumatoid arthritis. Med Sci Monit. 2002;8:CR616–CR619. [PubMed] [Google Scholar]
- Julkunen H, Saijonmaa O, Gronhagen-Riska C, Teppo AM, Fyhrquist F. Raised plasma concentrations of endothelin-1 in systemic lupus erythematosus. Ann Rheum Dis. 1991;50:526–527. doi: 10.1136/ard.50.7.526-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri C, Latorraca A, Catapano G, Greco F, Mazzoni A, Clerico A, Pedrinelli R. Increased plasma endothelin-1 immunoreactive levels in vasculitis: a clue to the use of endothelin-1 as a marker of vascular damage? J Hypertens Suppl. 1993;11:S142–S143. [PubMed] [Google Scholar]
- Shen JY, Chen SL, Wu YX, Tao RQ, Gu YY, Bao CD, Wang Q. Pulmonary hypertension in systemic lupus erythematosus. Rheumatol Int. 1999;18:147–151. doi: 10.1007/s002960050074. [DOI] [PubMed] [Google Scholar]
- Sharma S, Kashour T, Philipp R. Secondary pulmonary arterial hypertension: treated with endothelin receptor blockade. Tex Heart Inst J. 2005;32:405–410. [PMC free article] [PubMed] [Google Scholar]
- Muslu A, Islek I, Gok F, Aliyazicioglu Y, Dagdemir A, Dundaroz R, Kucukoduk S, Sakarcan A. Endothelin levels in Henoch-Schonlein purpura. Pediatr Nephrol. 2002;17:920–925. doi: 10.1007/s00467-002-0885-3. [DOI] [PubMed] [Google Scholar]
- Pache M, Kaiser HJ, Haufschild T, Lubeck P, Flammer J. Increased endothelin-1 plasma levels in giant cell arteritis: a report on four patients. Am J Ophthalmol. 2002;133:160–162. doi: 10.1016/S0002-9394(01)01202-8. [DOI] [PubMed] [Google Scholar]
- Krishnan E, Lingala VB, Singh G. Declines in mortality from acute myocardial infarction in successive incidence and birth cohorts of patients with rheumatoid arthritis. Circulation. 2004;110:1774–1179. doi: 10.1161/01.CIR.0000142864.83780.81. [DOI] [PubMed] [Google Scholar]
- Hurlimann D, Forster A, Noll G, Enseleit F, Chenevard R, Distler O, Bechir M, Spieker LE, Neidhart M, Michel BA, et al. Anti-tumor necrosis factor-alpha treatment improves endothelial function in patients with rheumatoid arthritis. Circulation. 2002;106:2184–2187. doi: 10.1161/01.CIR.0000037521.71373.44. [DOI] [PubMed] [Google Scholar]
- Guillevin L, Lhote F, Gayraud M, Cohen P, Jarrousse B, Lortholary O, Thibult N, Casassus P. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore) 1996;75:17–28. doi: 10.1097/00005792-199601000-00003. [DOI] [PubMed] [Google Scholar]
- Jayne D, Rasmussen N, Andrassy K, Bacon P, Tervaert JW, Dadoniene J, Ekstrand A, Gaskin G, Gregorini G, de Groot K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med. 2003;349:36–44. doi: 10.1056/NEJMoa020286. [DOI] [PubMed] [Google Scholar]
- Specks U, Fervenza FC, McDonald TJ, Hogan MC. Response of Wegener's granulomatosis to anti-CD20 chimeric monoclonal antibody therapy. Arthritis Rheum. 2001;44:2836–2840. doi: 10.1002/1529-0131(200112)44:12<2836::AID-ART471>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Keogh KA, Wylam ME, Stone JH, Specks U. Induction of remission by B lymphocyte depletion in eleven patients with refractory antineutrophil cytoplasmic antibody-associated vascu litis. Arthritis Rheum. 2005;52:262–268. doi: 10.1002/art.20718. [DOI] [PubMed] [Google Scholar]