

Abstract
Angiogenesis is important in the pathogenesis of systemic inflammatory rheumatic diseases, a family of related disorders that includes rheumatoid arthritis and systemic sclerosis. Rheumatoid arthritis is the rheumatic disease in which the role of angiogenesis has been studied most extensively. However, whereas rheumatoid arthritis is characterized by excessive angiogenesis, the situation is not as clear cut in other rheumatic diseases. For example, systemic sclerosis is characterized by reduced capillary density with insufficient angiogenic responses. Results with angiogenesis inhibitors are controversial, and there is – in parallel – a wide range of upregulated angiogenic factors such as vascular endothelial growth factor. Dysregulation of angiogenesis in systemic sclerosis is accompanied by other pathogenic processes, including fibrosis, autoimmunity and vasculopathy. Animal models with at least partial features of the vasculopathy observed in systemic sclerosis include wound healing models, graft versus host disease models and, in particular, the University of California at Davis line 200 chicken model of systemic sclerosis.
Introduction
Angiogenesis, the process of new blood vessel formation, is controlled by a subtle balance between endogenous stimulators, which induce blood vessel growth, and inhibitors, which prevent growth. In processes such as wound healing, angiogenesis is a well programmed cascade of events that comprises a number of distinct steps. Angiogenic stimuli activate endothelial cells, which produce proteolytic enzymes that degrade the basement membrane and the perivascular extracellular matrix. Endothelial cells proliferate and migrate into the perivascular area, forming 'primary sprouts'. Subsequent lumenation of these primary sprouts leads to formation of capillary loops, which is followed by synthesis of a new basement membrane and blood vessel maturation to complete tube-like structures through which blood can flow [1] (Figure 1).
Figure 1.
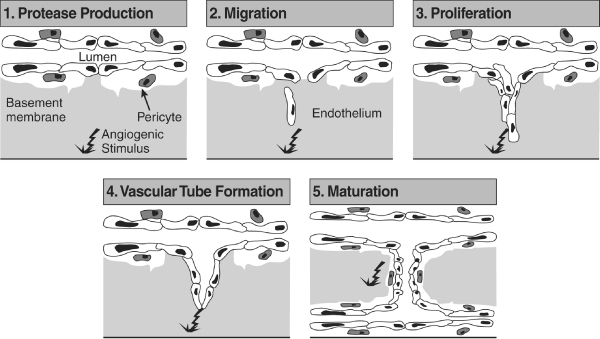
Key stages in the process of angiogenesis. This diagram summarizes the steps involved in the formation of new capillary blood vessels. Steps include protease production, endothelial cell migration and proliferation, vascular tube formation, anastomosis of newly formed tubes, synthesis of a new basement membrane and incorporation of pericytes. Reproduced with permission from Lowe et al. Br J Dermatol 1995 © Blackwell Publishing [1].
Like any biological system, inducers of angiogenesis are counterbalanced by inhibitors. However, in angiogenesis the inhibitors often outweigh the inducers, resulting in a normal physiological balance. When the converse situation occurs, conditions characterized by angiogenesis, such as inflammatory angiogenesis or angiogenesis related to tumour growth, can develop. In this review we explore potential initiators of vascular injury in two example inflammatory rheumatic diseases, namely rheumatoid arthritis (RA) and scleroderma (systemic sclerosis [SSc]), in which the angiogenic process appears to be disrupted very differently. We also discuss how the angiogenic process might be manipulated for therapeutic benefit in the treatment of these debilitating diseases.
Regulation and dysregulation of angiogenesis in rheumatic diseases
The rheumatic diseases are a family of closely related disorders that includes RA, SSc and systemic lupus erythematosus. RA is characterized by excessive angiogenesis [2] and it has been studied extensively in this regard.
Mediators of angiogenesis in rheumatoid arthritis
Proangiogenic mediators associated with RA include the following (Table 1): growth factors such as vascular endothelial growth factor (VEGF); cytokines such as tumour necrosis factor (TNF)-α (which has many effects in addition to angiogenesis); chemokines such as IL-8; and other mediators, including endothelin (ET)-1.
Table 1.
Some proangiogenic mediators involved in the pathogenesis of rheumatoid arthritis
| Type of molecule | Molecule |
| Growth factors | Fibroblast growth factor (FGF)-2 |
| Transforming growth factor (TGF)-β | |
| Hepatocyte growth factor (HGF) | |
| Vascular endothelial growth factors (VEGF, VEGF-C, and VEGF-D) | |
| Cytokines | Tumour necrosis factor (TNF)-α |
| Platelet activating factor (PAF) | |
| Angiopoietin (Ang)-1, Ang-2 | |
| Chemokines | Interleukin (IL)-8 |
| Epithelial neutrophil activating peptide (ENA)-78 | |
| Growth related gene product (GRO)-α | |
| Stromal cell derived factor (SDF)-1 | |
| Fractalkine | |
| Other mediators | Endothelin (ET)-1 |
VEGF, an endothelial selective mitogen that is secreted predominantly by macrophages, is an important cytokine in both angiogenesis and vasculogenesis [3]. There is evidence suggesting that, in RA, VEGF expression is induced by hypoxia. VEGF has a hypoxia-responsive element in its promoter region such that the hypoxic environment of the inflamed RA joint activates the VEGF gene via binding of hypoxia inducible factor. This in turn augments IL-1 or transforming growth factor (TGF)-β induced synovial fibroblast VEGF [4], which contributes significantly to angiogenesis in the synovium and progression of RA.
The limited role played by VEGF in normal human physiology makes it an attractive candidate for therapeutic intervention [5]. Data from both human in vitro and animal in vivo studies show that inhibition of VEGF attenuates arthritis. In one in vitro study VEGF receptor-1 Fc suppressed RA synovial endothelial cell proliferation [6], whereas in a mouse model of collagen-induced arthritis anti-VEGF antibody reduced the onset of angiogenesis as well as onset and severity of arthritis [7,8]. In mouse collagen-induced arthritis, administration of antibodies against VEGF receptor-1 (Flt-1) or soluble VEGF receptor 1 was shown to reduce synovial angiogenesis and inflammatory arthritis [9-11]. By blocking angiogenesis via inhibition of VEGF, it appears that it is possible to block arthritis in these animal models.
Evidence of the importance of TNF-α as a proangiogenic mediator in RA is illustrated by the effect of giving anti-TNF-α to patients with RA. Administration of anti-TNF-α drugs to patients with RA leads to vascular deactivation, including decreased angiogenesis and endothelial cell markers [12].
Chemokines are also important in RA and SSc. Studies have shown that the chemokine IL-8/CXC chemokine ligand (CXCL)8 plays a role in the pathogenesis of RA synovitis. This molecule is angiogenic and appears to be responsible for much of the macrophage-derived angiogenic activity seen in RA [13]. Interestingly, chemokines such as IL-8/CXCL8 that have an ELR motif (glutamic acid-leucine-arginine) are angiogenic, whereas chemokines that lack this motif are angiostatic [14]. Thus, structure clearly plays a role in determining the angiogenic properties of chemokines and thus their involvement in the pathophysiology of RA.
Role of endothelin in angiogenesis
Secreted by endothelial cells, ET-1 has a number of effects on the vascular system, which include vasoconstriction, upregulation of VEGF, cell proliferation and vascular remodelling. With respect to cell proliferation, ET-1 appears not to act alone but rather works in conjunction with various growth factors and cytokines. In combination with platelet-derived growth factor, for example, ET-1 acts synergistically to boost cell proliferation [15].
In addition to its well recognized effects as a potent endogenous vasoconstrictor and smooth muscle mitogen, ET-1 also appears to have proangiogenic effects in some rheumatic diseases. In patients with RA, levels of ET-1 in synovial fluid, serum and plasma are elevated in comparison with those in normal individuals [16-19]. Although this clearly does not demonstrate a causal role for ET-1 in the pathophysiology of RA, it may suggest some degree of involvement.
More robust evidence that ET-1 has proangiogenic effects is derived from the results from several experimental studies, including the rat corneal bioassay, in which its angiogenic effect occurs independently of inflammation [20]. That study compared the angiogenic effects in rat corneas of ET-1 and two positive controls, namely murine VEGF and human IL-8/CXCL8. Murine VEGF and human IL-8/CXCL8 were associated with angiogenesis in 52% and 63% of corneas, respectively, whereas ET-1 resulted in angiogenesis in 71% of corneas. ET-1 has also been shown to be angiogenic in the Matrigel plug model [21] and to stimulate human umbilical vein endothelial cell migration and proliferation, an effect that can be blocked by the action of an ET-1 receptor subtype B antagonist [22]. In combination with VEGF, ET-1 stimulates capillary morphogenesis and activates the hypoxia response pathway, which (as discussed above) leads to VEGF expression [22,23] and angiogenesis in the synovium. Additionally, ET-1 may trigger angiogenesis by stimulating the production of nitric oxide and matrix metalloproteinase (MMP)-2 [22,24].
Role of adhesion molecules in angiogenesis
As mentioned above, adhesion molecules play a pivotal role in angiogenesis, often acting in concert with angiogenic cytokines. Basic fibroblast growth factor, one of the first angiogenic factors to be identified, and TNF-α increase levels of αvβ3 integrin, resulting in angiogenesis (Figure 2). Conversely, MMP-2 is cleaved into a PEX fragment (a noncatalytic MMP fragment with integrin-binding activity) that acts as a negative regulator of angiogenesis [25]. In contrast, the angiogenic growth factors VEGF and TGF-β can elevate protein kinase C and act, in part, via αvβ5 integrin to mediate angiogenesis. Given that cytokines appear to act via integrin intermediaries in the angiogenic process, blocking integrins potentially could be used to regulate angiogenesis. This has in fact been demonstrated in several in vivo animal studies. In a rabbit arthritis model, anti-αvβ3 was found to decrease synovial angiogenesis and inflammation [26], whereas an oral nonpeptide αvβ3 antagonist was shown to ameliorate rat adjuvant-induced arthritis both prophylactically and therapeutically [27]. In another study, a proapoptotic αvβ3 antagonist composed of a RGD peptide linked to a heptapeptide dimer was found to home selectively to arthritic endothelium in the mouse collagen-induced arthritis model when compared with normal joint and control organ endothelium [28]. The results of this study have important implications for RA because they suggest that it is possible to distinguish therapeutically diseased endothelium from normal endothelium.
Figure 2.
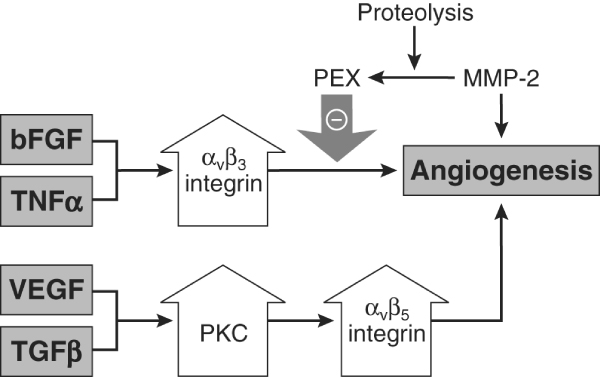
Potential mechanism of action of some adhesion molecules and cytokines involved in angiogenesis in RA. Basic fibroblast growth factor (bFGF) and tumour necrosis factor (TNF)-α act via αvβ3 integrin. In contrast, vascular endothelial growth factor (VEGF) and transforming growth factor (TGF)-β appear to act via increasing protein kinase C (PKC) with subsequent action on αvβ5 integrin. Matrix metalloproteinase (MMP)-2 is proteolytically cleaved into a PEX fragment (a noncatalytic MMP fragment with integrin-binding activity) that acts as a negative regulator of angiogenesis. RA, rheumatoid arthritis. Adapted with permission from Koch. Arthritis Rheum 1998 © John Wiley & Sons/American College of Rheumatology [71].
Despite this promising research, the role of adhesion molecules as angiogenic mediators remains the subject of much debate. The prevailing paradigm was that soluble adhesion molecules are anti-inflammatory. It was believed that adhesion molecules expressed on the wall of endothelial cells could be cleaved so that they bind to leucocytes that would normally bind to endothelial receptors on the endothelial wall and then enter the tissue. In other words, these soluble receptors were thought to act as blocking reagents, binding to leucocytes and so preventing them from binding to their respective receptors on endothelial cells and resulting in inflammation.
An alternative paradigm, and one advocated here, is that endothelial adhesion molecules are in fact shed and have proinflammatory and proangiogenic effects. Evidence for this alternative paradigm comes from experimental work on soluble E-selectin and soluble vascular cell adhesion molecule (VCAM)-1, which induced chemotaxis of human endothelial cells in vitro and were angiogenic in rat corneas. Soluble E-selectin acted on endothelial cells in part through a sialylated Lewisx dependent mechanism [29], whereas soluble VCAM-1 acted on endothelial cells in part through a very late antigen-4 dependent mechanism. An antigen structurally similar to sialylated Lewisx and designated 4A11 is also shed from endothelium and mediates angiogenesis [30]. Using a monoclonal antibody (mAb 4A11) raised in mice to adherent human RA synovial tissue cells, it has been demonstrated that this novel endothelial selective antigen has both proinflammatory and proangiogenic potential. In vitro antibody binding experiments found massive upregulation of 4A11 antigen on the surface of TNF-α and thrombin-stimulated endothelium compared with nonstimulated cells. In contrast to soluble E-selectin and soluble VCAM-1, 4A11 antigen is rapidly cytokine induced in endothelial cells in vitro, within 5 to 20 min, and declines within 2 hours of stimulation. The effects observed in vitro have been confirmed in vivo in a human poison ivy inflammatory model, in which 4A11 antigen exhibited greater basal expression than either soluble E-selectin or soluble VCAM-1, which are known cytokine inducible, inflammatory antigens that are upregulated in this model of contact dermatitis [31].
Increased expression of 4A11 antigen in RA synovial tissue compared with normal synovial tissue and synovial fluid from osteoarthritis patients, and a strong, positive angiogenic response in a rat corneal bioassay provide further evidence that this inducible endothelial selective antigen has proinflammatory and angiogenic potential. Structural and histological studies have shown that 4A11 consists of Lewisy and H-2 glycoconjugate antigens (Ley/H). Except for a presence on epithelial cells, such as keratinocytes, Ley/H appear to be endothelium specific and, moreover, are expressed selectively in synovium, lymphoid organs, and skin. This suggests a role in organ-specific homing. It is believed that in the milieu of the inflamed joint in RA, endothelial cells become activated, perhaps by cytokines such as IL-1 and TNF-α, to shed this Ley/H antigen. Subsequently, the Ley/H antigen binds to an as yet unknown receptor that mediates angiogenesis. Interestingly, mAb 4A11 was able to block the strong angiogenic response induced by Ley and H analogues.
The balance of angiogenic factors in rheumatoid arthritis
In RA, the balance of angiogenic inducers and inhibitors appears tipped in favour of inducers, leading to a state of inflammatory angiogenesis. Endogenous inhibitors of angiogenesis with relevance to rheumatic disease have nonetheless been identified. Of these, cartilage-derived factors such as troponin I [32] and endostatin, a fragment of collagen XVIII, appear important. In RA synovial tissue, endostatin can decrease RA synovial tissue volume, inflammation and angiogenesis when this tissue is implanted in a severe combined immunodeficient mouse [33]. Transfer of the endostatin lentiviral gene into TNF-α transgenic mice at the onset of arthritis was found to reduce the arthritis index, a measure of disease severity, as well as reducing the number of synovial blood vessels [34]. To be effective therapeutically, however, strategies to inhibit angiogenesis must block abnormal but not normal blood vessel growth, such as occurs during wound healing or the reproductive cycle. Indeed, excessive vascular regression could compromise delivery of therapeutic agents to the target site and cause unwanted side effects. The use of anti-VEGF therapies in cancer patients, although therapeutically effective, is not without side effects. Hypertension and thromboembolism have occurred following treatment with an anti-VEGF mAb or a small molecule drug that targets VEGF receptor tyrosine kinase [35]. New approaches to angiogenesis inhibition in the therapeutic management of RA must therefore be explored, either by blocking inducers or by enhancing inhibitors. There is also a need for better surrogate markers of angiogenesis and its inhibition.
Dysregulation of angiogenesis in systemic sclerosis
Whereas RA is characteristically a disease of increased angiogenesis, there is evidence for an insufficient angiogenic response in SSc. The dysregulation of angiogenesis leads to failure to replace damaged vessels, resulting in a reduction in capillary density in patients with SSc [36]. Clinically, these changes in the capillary network are evident on nail-fold capillaroscopy and may manifest as ischaemic fingertip ulcers.
Studies in peripheral blood mononuclear cells support the concept of decreased and inefficient angiogenesis in SSc. Isolated peripheral blood mononuclear cells induce less angiogenesis than do peripheral blood mononuclear cells from healthy control individuals [37], whereas supernatants from SSc peripheral blood mononuclear cells decreased endothelial chemotaxis [38], and serum samples from patients with SSc failed to enhance normal mononuclear angiogenic capability [39]. Thus, in contrast to the marked angiogenic response seen in peripheral blood monocytes from normal individuals, those from SSc patients are effectively inert, leading to a defective or dysregulated angiogenic response. Comparisons of skin biopsies from SSc patients with normal skin samples have shown the former to have fewer blood vessels, providing further evidence for decreased angiogenesis in SSc [40].
Although an overall decrease in the angiogenic response is well documented, the mechanisms that lead to the disturbed angiogenesis are much less clear. Several antiangiogenic or angiostatic mediators have been reported to be upregulated in SSc, including endostatin, platelet factor 4, thrombospondin, and IL-4 [41-44] (Table 2). However, results on angiostatic factors in SSc are conflicting [42,45] or flawed by low patient numbers or poor clinical characterization, and heterogeneity of the study population. In addition, the functional contribution of angiostatic factors to the disturbed angiogenesis in SSc has not been addressed.
Table 2.
Some antiangiogenic and proangiogenic mediators in systemic sclerosis
| Type of mediator | Molecule |
| Antiangiogenic | Endostatin (normal in some studies) |
| Platelet factor (PF)4 | |
| Thrombospondin | |
| Interleukin (IL)-4 | |
| Proangiogenic | Soluble vascular cell adhesion molecule (sVCAM)-1 |
| Soluble E-selectin (sE-selectin; normal in some studies) | |
| E-selectin | |
| Soluble P-selectin (sP-selectin) | |
| P-selectin | |
| Soluble intercellular adhesion molecule (sICAM)-1/ICAM-1 | |
| CD44 | |
| Vascular endothelial growth factor (VEGF) | |
| Vascular endothelial growth factor receptors (VEGFRs; VEGFR1 and VEGFR2) | |
| IL-8 | |
| Monocyte chemoattractant protein (MCP)-1/CC chemokine ligand (CCL)2 and CCR2 (receptor for MCP-1) | |
| Macrophage inflammatory protein (MIP)-1α/CCL3 | |
| Fractalkine/CX3 chemokine ligand (CX3CL)1 and CX3 chemokine receptor (CX3CR)1 (receptor for fractalkine) | |
| β-Thromboglobulin | |
| Tumour necrosis factor (TNF)-α | |
| Endothelin (ET)-1 |
Evidence for a mechanism of dysregulated angiogenesis in SSc has emerged from two recent experimental studies. The first of these showed decreased urokinase plasminogen activator dependent invasion, proliferation, and capillary morphogenesis, and increased MMP-12 in SSc endothelial cells [46]. In this study the authors were able to restore the defect in the endothelium with an anti-MMP-12 antibody. They also discovered that urokinase plasminogen activator undergoes truncation between domains 1 and 2, thus impairing its function. The second experimental study showed evidence for decreased expression of tissue kallikreins 9, 11 and 12 in endothelial cells isolated from patients with SSc in comparison with normal endothelial cells [47]. There is also evidence that circulating endothelial cells are increased in SSc as a marker of endothelial cell activation in comparison with normal controls, and that this increase correlates with disease activity scores [48]. Recent studies indicated that, in addition to angiogenesis, the formation of new vessels from precursor cells (vasculogenesis) is impaired in SSc [49]. However, findings regarding the role of vasculogenesis in SSc are inconsistent [50], and this must be addressed by further studies.
Paradoxically, there is upregulation or increased expression of a large number of proangiogenic mediators despite the defective angiogenesis in SSc. In addition, the bushy and tortuous capillaries seen on nail-fold capillaroscopy could be interpreted as futile attempts to build new vessels after stimulation with angiogenic factors. Examples of upregulated proangiogenic factors in SSc include soluble VCAM-1 in peripheral blood monocytes, intercellular adhesion molecule-1 in peripheral blood monocytes and skin fibroblasts, E-selectin and P-selectin in skin, ET-1 in monocytes and skin fibroblasts, and monocyte chemoattractant molecule-1/CC chemokine ligand 2 in different cell types in the skin [51-53] (Table 2). Some adhesion molecules such as VCAM-1 and P-selectin, as well as cytokines such as TNF-α and IL-8/CXCL8, have more commonly been found in skin biopsies taken from patients with SSc of duration 1 year or less, whereas other molecules, for example IL-6, are upregulated in late stage disease [54]. Taken together, these findings suggest that certain cellular adhesion molecules and cytokines may play differential roles in the early, inflammatory and later fibrotic stages of SSc.
As an additional example, the potent angiogenic factor VEGF is markedly over-expressed in various cell types in the skin of SSc patients [55,56]. In parallel, VEGF receptors are upregulated on endothelial cells in SSc. In agreement with this finding, a number of studies confirmed that serum levels of VEGF are significantly increased in SSc patients throughout different disease stages [42,57-59]. The serum levels of VEGF correlate significantly with the development of fingertip ulcers. SSc patients with fingertip ulcers have increased serum levels of VEGF as compared with healthy individuals, but they are lower than in SSc patients without fingertip ulcers [42]. These data indicate that there might be a functional deficit of VEGF in SSc patients, which can be overcome if the levels of VEGF exceed an individual threshold.
The mechanisms that lead to increased expression of VEGF in SSc are unclear. In contrast to the situation in RA, hypoxia induced expression of hypoxia inducible factor-1α does not appear to play a major role in the induction of VEGF in SSc [55], whereas induction by cytokines such as platelet-derived growth factor and TGF-β appear to be more important.
A crucial point in the interpretation of the upregulation of angiogenic factors in this disease is that angiogenic factors must be expressed in a strictly controlled temporal and spatial distribution if they are to lead to the development of functionally adequate new vessels. For instance, although the upregulation of VEGF and other angiogenic factors might be a compensatory mechanism for the initial effect of unidentified angiostatic factors, the temporal kinetics of its expression appear to be critical in overcoming the inhibitory effects of angiostatic factors. In this regard, it has been shown that a brief upregulation of VEGF results in instability of newly formed vessels [60]. On the other hand, prolonged over-expression of VEGF, as is seen in SSc patients throughout various disease stages, also has deleterious effects because the vessels fuse in an uncontrolled manner and form a chaotic vessel network that is strikingly similar to the disturbed capillary network observed in SSc [60]. In addition, isolated microvascular endothelial cells from patients exhibit an impaired response to VEGF in the Matrigel capillary morphogenesis assay [46], indicating that VEGF receptor signalling might be impaired in endothelial cells of SSc patients. In contrast, functionally important gene polymorphisms that lead to an impairment in biological properties of VEGF do not exist in SSc patients [61].
The individual functional consequences of the upregulated angiogenic factors for defective angiogenesis in SSc has not yet been addressed in detail. In this regard, it is important to highlight the results of a randomized placebo controlled trial conducted to evaluate the effects of the dual endothelin receptor antagonist bosentan on wound healing of fingertip ulcers in patients with SSc (RAPIDS-2 [RAndomized, double-blind, Placebo-controlled, multi-center study to assess the effect of bosentan on healing and prevention of Ischemic Digital ulcers in patients with systemic Sclerosis-2]) [62]. Wound healing requires angiogenesis, and ET-1 is among the group of angiogenic factors that are upregulated in SSc (Table 2). Interestingly, inhibition of ET-1 did not affect wound healing in this clinical trial, indicating that ET-1 is of little functional importance for the angiogenic responses in patients with SSc.
Models for the vasculopathy that is characteristic of systemic sclerosis
Wound healing as a model for systemic sclerosis
Functional and structural vasculopathy, which manifests as endothelial cell injury, is an early pathological feature of SSc. This is followed, or paralleled, by immunological activation, perivascular inflammation and fibrosis. As a model of matrix alteration and tissue remodelling in SSc, the process of wound healing provides valuable insights. In this context, wound healing, at least in skin, is characterized by two important features: inflammation followed by extracellular matrix synthesis. During wound healing the early inflammatory phase is followed by a proliferative phase and then a tissue remodelling phase, in which TGF-β plays an important role in myofibroblast maturation. The final phase of wound healing is collagen formation. It has been suggested that innate immunity, the body's first line of defence against microbes and mediated via Toll-like receptors (TLRs), is important in wound healing. Myeloid differentiation primary response gene 88 (MyD88), which is one of several molecules that are critical for signalling by most TLRs, provides a convenient tool for determining the importance of TLRs in wound healing and whether the inflammatory response that occurs during wound healing leads to the production of TGF-β and ET-1. Ongoing experimental studies in mechanically injured MyD88 knockout mice, which is a useful model for examining the role of innate immunity in wound healing and tissue remodelling, are examining the rate of wound closure and degree of inflammation compared with those in control mice. The role of TLR signalling in SSc, however, remains to be determined.
Graft versus host disease as a model for systemic sclerosis
Graft versus host disease is another model from which to gain insight into SSc vasculopathy because it mimics all three elements of SSc, with effects on fibrosis, autoimmunity and vascular disease. Murine chronic graft versus host disease models can, for instance, be created by injecting cells from B10.D2 mice into irradiated Balb/C or Balb/C rag2-/- mice, in which there are only minor histocompatibility differences. Within 4 to 6 weeks, this produces a scleroderma-like disease, the characteristic features of which include fibrosis/matrix deposition, a marked increase in myofibroblasts, autoantibody production and dermal infiltration of immune cells, as well as marked upregulation of ET-1 in skin and kidney tissue. It is still unclear by which molecular factors the increase in myofibroblasts is driven. Candidate molecules include TGF-β and ET-1, but conceivably some of these characteristics are due to effects from multiple factors and interactions between them.
The University of California at Davis chicken lines 200 and 206
The University of California at Davis (UCD) 200/206 chicken model [63] is an important animal model for vascular changes in SSc. Unlike some animal models, the UCD chicken lines spontaneously develop an inherited scleroderma-like disease exhibiting the entire spectrum of SSc, including vascular occlusion, severe perivascular lymphocytic infiltration of the skin and viscera, fibrosis of skin and internal organs, auto-antibodies against nuclear antigens, anticardiolipin antibodies, anti-endothelial cell antibodies (AECAs), rheumatoid factors and distal polyarthritis [64-66].
Studies of skin lesions from UCD 200/206 chickens found that AECA-dependent cellular cytotoxicity induced endothelial cell apoptosis is a primary event in the pathogenesis of the SSc-like disease in this animal model [67], potentially mediated via the Fas pathway [68]. Similar findings of endothelial cell apoptosis in early disease stages were observed in skin biopsies from human SSc patients. These findings subsequently received support from the observation that AECA-positive serum from UCD 200 chickens injected into normal chicken embryos results in AECA binding to microvascular endothelium in vivo and a significant increase in endothelial cell apoptosis [69]. A further study focused on endothelial cell apoptosis, mononuclear cell infiltration and collagen deposition in the visceral organs of UCD chickens [70]. In this study, apoptotic endothelial cells were found in oesophagus, lung and kidney of UCD 200 chickens at the initial stage of the disease but not in heart or liver. In this study oesophagus was the most affected organ, exhibiting mononuclear cell infiltrations and increased collagen deposition. This supports the hypothesis that endothelial cell apoptosis initiates the disease process, followed by mononuclear cell infiltration and fibrosis.
Conclusion
As this review illustrates, dysregulation of angiogenesis is an important process in the pathogenesis of various rheumatic diseases, including RA and SSc. In RA angiogenesis is central to the formation of the inflammatory pannus and permits leucocyte infiltration into the synovial tissue. Moreover, the extremely hypoxic environment of the arthritic synovium acts as a potent signal for the generation of new blood vessels. In contrast to RA, SSc is characterized by decreased capillary density and insufficient angiogenesis. However, despite the reduction in capillary density in the skin of SSc patients, a number of proangiogenic factors are over-expressed. Therapeutic manipulation of angiogenesis in rheumatic diseases will depend on the net balance of angiogenic and angiostatic events. In RA, in which inducers of angiogenesis outweigh inhibitors, it is clear that blocking angiogenesis blocks arthritis, thereby helping to ameliorate the disease. The picture is less clear for SSc, and functional experiments targeting angiogenic or angiostatic factors are lacking. There are controversial data on upregulation of angiostatic factors, which might serve as targets for therapeutic approaches. In addition, the uncontrolled and chronic expression of angiogenic factors such as VEGF might have deleterious effects on the formation of new vessels per se. The issues that must be addressed in SSc therefore are whether it would be therapeutically useful to augment angiogenesis and whether it is possible to manipulate selectively the positive and negative effects of mediators such as VEGF.
Abbreviations
AECA = anti-endothelial cell antibody; CXCL = CXC chemokine ligand; ET = endothelin; IL = interleukin; Ley/H = Lewisy and H-2 glycoconjugate antigens; mAb = monoclonal antibody; MMP = matrix metalloproteinase; MyD88 = myeloid differentiation primary response gene 88; RA = rheumatoid arthritis; SSc = systemic sclerosis; TGF = transforming growth factor; TLR = Toll-like receptor; TNF = tumour necrosis factor; UCD = University of California at Davis; VCAM = vascular cell adhesion molecule; VEGF = vascular endothelial growth factor.
Competing interests
The authors have received consulting fees from Actelion Pharmaceuticals Ltd, the manufacturer of bosentan. OD has received consulting fees from Encysive Pharmaceuticals Inc, the manufacturer of sitaxentan.
Acknowledgments
Acknowledgements
The authors would like to acknowledge medical writing support funded by an educational grant from Actelion Pharmaceuticals Ltd.
This article is part of Arthritis Research & Therapy Volume 9 Supplement 2: Advances in systemic sclerosis and related fibrotic and vascular conditions, and is based on presentations made at a symposium entitled Advances in systemic sclerosis and connective tissue disease, sponsored by Actelion Pharmaceuticals Ltd, held in Athens, Greece in April 2006. The full contents of the supplement are available online at http://arthritis-research.com/supplements/9/S2. This supplement has been supported by an educational grant from Actelion Pharmaceuticals Ltd.
References
- Lowe PM, Lee ML, Jackson CJ, To SS, Cooper AJ, Schrieber L. The endothelium in psoriasis. Br J Dermatol. 1995;132:497–505. doi: 10.1111/j.1365-2133.1995.tb08702.x. [DOI] [PubMed] [Google Scholar]
- Koch AE. The role of angiogenesis in rheumatoid arthritis: recent developments. Ann Rheum Dis. 2000;(Suppl 1):65–71. doi: 10.1136/ard.59.suppl_1.i65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch AE, Harlow LA, Haines GK, Amento EP, Unemori EN, Wong WL, Pope RM, Ferrara N. Vascular endothelial growth factor. A cytokine modulating endothelial function in rheumatoid arthritis. J Immunol. 1994;152:4149–4156. [PubMed] [Google Scholar]
- Cho ML, Cho CS, Min SY, Kim SH, Lee SS, Kim WU, Min DJ, Min JK, Youn J, Hwang SY, Park SH, Kim HY. Cyclosporine inhibition of vascular endothelial growth factor production in rheumatoid synovial fibroblasts. Arthritis Rheum. 2002;46:1202–1209. doi: 10.1002/art.10215. [DOI] [PubMed] [Google Scholar]
- Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF) J Cell Mol Med. 2005;9:777–794. doi: 10.1111/j.1582-4934.2005.tb00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekimoto T, Hamada K, Oike Y, Matsuoka T, Matsumoto M, Chosa E, Suda T, Tajima N, Yamamura K. Effect of direct angiogenesis inhibition in rheumatoid arthritis using a soluble vascular endothelial growth factor receptor 1 chimeric protein. J Rheumatol. 2002;29:240–245. [PubMed] [Google Scholar]
- Lu J, Kasama T, Kobayashi K, Yoda Y, Shiozawa F, Hanyuda M, Negishi M, Ide H, Adachi M. Vascular endothelial growth factor expression and regulation of murine collagen-induced arthritis. J Immunol. 2000;164:5922–5927. doi: 10.4049/jimmunol.164.11.5922. [DOI] [PubMed] [Google Scholar]
- Sone H, Kawakami Y, Sakauchi M, Nakamura Y, Takahashi A, Shimano H, Okuda Y, Segawa T, Suzuki H, Yamada N. Neutralization of vascular endothelial growth factor prevents collagen-induced arthritis and ameliorates established disease in mice. Biochem Biophys Res Commun. 2001;281:562–568. doi: 10.1006/bbrc.2001.4395. [DOI] [PubMed] [Google Scholar]
- Miotla J, Maciewicz R, Kendrew J, Feldmann M, Paleolog E. Treatment with soluble VEGF receptor reduces disease severity in murine collagen-induced arthritis. Lab Invest. 2000;80:1195–1205. doi: 10.1038/labinvest.3780127. [DOI] [PubMed] [Google Scholar]
- Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, Nagy JA, Hooper A, Priller J, De Klerck B, et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med. 2002;8:831–840. doi: 10.1038/nm731. [DOI] [PubMed] [Google Scholar]
- Afuwape AO, Feldmann M, Paleolog EM. Adenoviral delivery of soluble VEGF receptor 1 (sFlt-1) abrogates disease activity in murine collagen-induced arthritis. Gene Ther. 2003;10:1950–1960. doi: 10.1038/sj.gt.3302104. [DOI] [PubMed] [Google Scholar]
- Paleolog EM, Young S, Stark AC, McCloskey RV, Feldmann M, Maini RN. Modulation of angiogenic vascular endothelial growth factor by tumor necrosis factor alpha and interleukin-1 in rheumatoid arthritis. Arthritis Rheum. 1998;41:258–1265. doi: 10.1002/1529-0131(199807)41:7<1258::AID-ART17>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258:798–1801. doi: 10.1126/science.1281554. [DOI] [PubMed] [Google Scholar]
- Strieter RM, Burdick MD, Gomperts BN, Belperio JA, Keane MP. CXC chemokines in angiogenesis. Cytokine Growth Factor Rev. 2005;16:593–609. doi: 10.1016/j.cytogfr.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Yahiaoui L, Villeneuve A, Valderrama-Carvajal H, Burke F, Fixman ED. Endothelin-1 regulates proliferative responses, both alone and synergistically with PDGF, in rat tracheal smooth muscle cells. Cell Physiol Biochem. 2006;17:37–46. doi: 10.1159/000091462. [DOI] [PubMed] [Google Scholar]
- Haq A, El-Ramahi K, Al-Dalaan A, Al-Sedairy ST. Serum and synovial fluid concentrations of endothelin-1 in patients with rheumatoid arthritis. J Med. 1999;30:51–60. [PubMed] [Google Scholar]
- Miyasaka N, Hirata Y, Ando K, Sato K, Morita H, Shichiri M, Kanno K, Tomita K, Marumo F. Increased production of endothelin-1 in patients with inflammatory arthritides. Arthritis Rheum. 1992;35:397–400. doi: 10.1002/art.1780350406. [DOI] [PubMed] [Google Scholar]
- Pache M, Schwarz HA, Kaiser HJ, Wuest P, Kloti M, Dubler B, Flammer J. Elevated plasma endothelin-1 levels and vascular dysregulation in patients with rheumatoid arthritis. Med Sci Monit. 2002;8:CR616–CR619. [PubMed] [Google Scholar]
- Nahir AM, Hoffman A, Lorber M, Keiser HR. Presence of immunoreactive endothelin in synovial fluid: analysis of 22 cases. J Rheumatol. 1991;18:678–680. [PubMed] [Google Scholar]
- Bek EL, McMillen MA. Endothelins are angiogenic. J Cardiovasc Pharmacol. 2000. pp. S135–S139. [DOI] [PubMed]
- Cruz A, Parnot C, Ribatti D, Corvol P, Gasc JM. Endothelin-1, a regulator of angiogenesis in the chick chorioallantoic membrane. J Vasc Res. 2001;38:536–545. doi: 10.1159/000051089. [DOI] [PubMed] [Google Scholar]
- Salani D, Taraboletti G, Rosano L, Di Castro V, Borsotti P, Giavazzi R, Bagnato A. Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol. 2000;157:1703–1711. doi: 10.1016/S0002-9440(10)64807-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnato A, Spinella F. Emerging role of endothelin-1 in tumor angiogenesis. Trends Endocrinol Metab. 2003;14:44–50. doi: 10.1016/S1043-2760(02)00010-3. [DOI] [PubMed] [Google Scholar]
- Goligorsky MS, Budzikowski AS, Tsukahara H, Noiri E. Co-operation between endothelin and nitric oxide in promoting endothelial cell migration and angiogenesis. Clin Exp Pharmacol Physiol. 1999;26:269–271. doi: 10.1046/j.1440-1681.1999.03029.x. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Kessler T, Silletti S, Cheresh DA, Verma IM. Suppression of angiogenesis by lentiviral delivery of PEX, a noncatalytic fragment of matrix metalloproteinase 2. Proc Natl Acad Sci USA. 2000;97:12227–12232. doi: 10.1073/pnas.220399597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storgard CM, Stupack DG, Jonczyk A, Goodman SL, Fox RI, Cheresh DA. Decreased angiogenesis and arthritic disease in rabbits treated with an alphavbeta3 antagonist. J Clin Invest. 1999;103:47–54. doi: 10.1172/JCI3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badger AM, Blake S, Kapadia R, Sarkar S, Levin J, Swift BA, Hoffman SJ, Stroup GB, Miller WH, Gowen M, Lark MW. Disease-modifying activity of SB 27 an orally active, nonpeptide alphavbeta3 (vitronectin receptor) antagonist, in rat adjuvant-induced arthritis. Arthritis Rheum. 3005;44:128–137. doi: 10.1002/1529-0131(200101)44:1<128::AID-ANR17>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Gerlag DM, Borges E, Tak PP, Ellerby HM, Bredesen DE, Pasqualini R, Ruoslahti E, Firestein GS. Suppression of murine collagen-induced arthritis by targeted apoptosis of synovial neovasculature. Arthritis Res. 2001;3:357–361. doi: 10.1186/ar327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch AE, Halloran MM, Haskell CJ, Shah MR, Polverini PJ. Angiogenesis mediated by soluble forms of E-selectin and vascular cell adhesion molecule-1. Nature. 1995;376:517–519. doi: 10.1038/376517a0. [DOI] [PubMed] [Google Scholar]
- Halloran MM, Carley WW, Polverini PJ, Haskell CJ, Phan S, Anderson BJ, Woods JM, Campbell PL, Volin MV, Backer AE, et al. Ley/H: an endothelial-selective, cytokine-inducible, angiogenic mediator. J Immunol. 2000;164:868–4877. doi: 10.4049/jimmunol.164.9.4868. [DOI] [PubMed] [Google Scholar]
- Koch AE, Nickoloff BJ, Holgersson J, Seed B, Haines GK, Burrows JC, Leibovich SJ. 4A11, a monoclonal antibody recognizing a novel antigen expressed on aberrant vascular endothelium. Upregulation in an in vivo model of contact dermatitis. Am J Pathol. 1994;144:244–259. [PMC free article] [PubMed] [Google Scholar]
- Moses MA, Wiederschain D, Wu I, Fernandez CA, Ghazizadeh V, Lane WS, Flynn E, Sytkowski A, Tao T, Langer R. Troponin I is present in human cartilage and inhibits angiogenesis. Proc Natl Acad Sci USA. 1999;96:2645–2650. doi: 10.1073/pnas.96.6.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno H, Yudoh K, Uzuki M, Nakazawa F, Sawai T, Yamaguchi N, Olsen BR, Kimura T. Treatment with the angiogenesis inhibitor endostatin: a novel therapy in rheumatoid arthritis. J Rheumatol. 2002;29:890–895. [PubMed] [Google Scholar]
- Yin G, Liu W, An P, Li P, Ding I, Planelles V, Schwarz EM, Min W. Endostatin gene transfer inhibits joint angiogenesis and pannus formation in inflammatory arthritis. Mol Ther. 2002;5:547–554. doi: 10.1006/mthe.2002.0590. [DOI] [PubMed] [Google Scholar]
- Ranieri G, Patruno R, Ruggieri E, Montemurro S, Valerio P, Ribatti D. Vascular endothelial growth factor (VEGF) as a target of bevacizumab in cancer: from the biology to the clinic. Curr Med Chem. 2006;13:1845–1857. doi: 10.2174/092986706777585059. [DOI] [PubMed] [Google Scholar]
- Distler JH, Gay S, Distler O. Angiogenesis and vasculogenesis in systemic sclerosis. Rheumatology (Oxford) 2006;(Suppl 3):iii26–iii27. doi: 10.1093/rheumatology/kel295. [DOI] [PubMed] [Google Scholar]
- Marczak M, Majewski S, Skopinska-Rozewska E, Polakowski I, Jablonska S. Enhanced angiogenic capability of monocyte-enriched mononuclear cell suspensions from patients with systemic scleroderma. J Invest Dermatol. 1986;86:355–358. doi: 10.1111/1523-1747.ep12285572. [DOI] [PubMed] [Google Scholar]
- Kahaleh MB, DeLustro F, Bock W, LeRoy EC. Human monocyte modulation of endothelial cells and fibroblast growth: possible mechanism for fibrosis. Clin Immunol Immunopathol. 1986;39:242–255. doi: 10.1016/0090-1229(86)90088-7. [DOI] [PubMed] [Google Scholar]
- Majewski S, Skopinska-Rozewska E, Jablonska S, Polakowski I, Pawinska M, Marczak M, Szmurlo A. Modulatory effect of sera from scleroderma patients on lymphocyte-induced angiogenesis. Arthritis Rheum. 1985;28:1133–1139. doi: 10.1002/art.1780281009. [DOI] [PubMed] [Google Scholar]
- Konttinen YT, Mackiewicz Z, Ruuttila P, Ceponis A, Sukura A, Povilenaite D, Hukkanen M, Virtanen I. Vascular damage and lack of angiogenesis in systemic sclerosis skin. Clin Rheumatol. 2003;22:196–202. doi: 10.1007/s10067-003-0698-1. [DOI] [PubMed] [Google Scholar]
- Hebbar M, Peyrat JP, Hornez L, Hatron PY, Hachulla E, Devulder B. Increased concentrations of the circulating angiogenesis inhibitor endostatin in patients with systemic sclerosis. Arthritis Rheum. 2000;43:889–893. doi: 10.1002/1529-0131(200004)43:4<889::AID-ANR21>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Distler O, Del Rosso A, Giacomelli R, Cipriani P, Conforti ML, Guiducci S, Gay RE, Michel BA, Bruhlmann P, Muller-Ladner U, et al. Angiogenic and angiostatic factors in systemic sclerosis: increased levels of vascular endothelial growth factor are a feature of the earliest disease stages and are associated with the absence of fingertip ulcers. Arthritis Res. 2002;4:R11. doi: 10.1186/ar596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macko RF, Gelber AC, Young BA, Lowitt MH, White B, Wigley FM, Goldblum SE. Increased circulating concentrations of the counteradhesive proteins SPARC and thrombospondin-1 in systemic sclerosis (scleroderma). Relationship to platelet and endothelial cell activation. J Rheumatol. 2002;29:2565–2570. [PubMed] [Google Scholar]
- Needleman BW, Wigley FM, Stair RW. Interleukin-1, interleukin-2, interleukin-4, interleukin-6, tumor necrosis factor alpha, and interferon-gamma levels in sera from patients with scleroderma. Arthritis Rheum. 1992;35:67–72. doi: 10.1002/art.1780350111. [DOI] [PubMed] [Google Scholar]
- Dziankowska-Bartkowiak B, Waszczykowska E, Zalewska A, Sysa-Jedrzejowska A. Correlation of endostatin and tissue inhibitor of metalloproteinases 2 (TIMP2) serum levels with cardiovascular involvement in systemic sclerosis patients. Mediators Inflamm. 2005;3:144–149. doi: 10.1155/MI.2005.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alessio S, Fibbi G, Cinelli M, Guiducci S, Del Rosso A, Margheri F, Serrati S, Pucci M, Kahaleh B, Fan P, et al. Matrix metalloproteinase 12-dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. 2004;50:3275–3285. doi: 10.1002/art.20562. [DOI] [PubMed] [Google Scholar]
- Giusti B, Serrati S, Margheri F, Papucci L, Rossi L, Poggi F, Magi A, Del Rosso A, Cinelli M, Guiducci S, et al. The antiangiogenic tissue kallikrein pattern of endothelial cells in systemic sclerosis. Arthritis Rheum. 2005;52:3618–3628. doi: 10.1002/art.21383. [DOI] [PubMed] [Google Scholar]
- Del Papa N, Colombo G, Fracchiolla N, Moronetti LM, Ingegnoli F, Maglione W, Comina DP, Vitali C, Fantini F, Cortelezzi A. Circulating endothelial cells as a marker of ongoing vascular disease in systemic sclerosis. Arthritis Rheum. 2004;50:1296–1304. doi: 10.1002/art.20116. [DOI] [PubMed] [Google Scholar]
- Kuwana M, Okazaki Y, Yasuoka H, Kawakami Y, Ikeda Y. Defective vasculogenesis in systemic sclerosis. Lancet. 2004;364:603–610. doi: 10.1016/S0140-6736(04)16853-0. [DOI] [PubMed] [Google Scholar]
- Del Papa N, Quirici N, Soligo D, Scavullo C, Cortiana M, Borsotti C, Maglione W, Comina DP, Vitali C, Fraticelli P, et al. Bone marrow endothelial progenitors are defective in systemic sclerosis. Arthritis Rheum. 2006;54:2605–2615. doi: 10.1002/art.22035. [DOI] [PubMed] [Google Scholar]
- Distler O, Pap T, Kowal-Bielecka O, Meyringer R, Guiducci S, Landthaler M, Scholmerich J, Michel BA, Gay RE, Matucci-Cerinic M, et al. Overexpression of monocyte chemoattractant protein 1 in systemic sclerosis: role of platelet-derived growth factor and effects on monocyte chemotaxis and collagen synthesis. Arthritis Rheum. 2001;44:2665–2678. doi: 10.1002/1529-0131(200111)44:11<2665::AID-ART446>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Eckes B, Krieg T. High expression and autoinduction of monocyte chemoattractant protein-1 in scleroderma fibroblasts. Eur J Immunol. 2001;31:2936–2941. doi: 10.1002/1521-4141(2001010)31:10<2936::AID-IMMU2936>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Galindo M, Santiago B, Rivero M, Rullas J, Alcami J, Pablos JL. Chemokine expression by systemic sclerosis fibroblasts: abnormal regulation of monocyte chemoattractant protein 1 expression. Arthritis Rheum. 2001;44:1382–1386. doi: 10.1002/1529-0131(200106)44:6<1382::AID-ART231>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Koch AE, Kronfeld-Harrington LB, Szekanecz Z, Cho MM, Haines GK, Harlow LA, Strieter RM, Kunkel SL, Massa MC, Barr WG, et al. In situ expression of cytokines and cellular adhesion molecules in the skin of patients with systemic sclerosis. Their role in early and late disease. Pathobiology. 1993;61:239–246. doi: 10.1159/000163802. [DOI] [PubMed] [Google Scholar]
- Distler O, Distler JH, Scheid A, Acker T, Hirth A, Rethage J, Michel BA, Gay RE, Muller-Ladner U, Matucci-Cerinic M, et al. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ Res. 2004;95:109–116. doi: 10.1161/01.RES.0000134644.89917.96. [DOI] [PubMed] [Google Scholar]
- Davies CA, Jeziorska M, Freemont AJ, Herrick AL. The differential expression of VEGF, VEGFR-2, and GLUT-1 proteins in disease subtypes of systemic sclerosis. Hum Pathol. 2006;37:190–197. doi: 10.1016/j.humpath.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Choi JJ, Min DJ, Cho ML, Min SY, Kim SJ, Lee SS, Park KS, Seo YI, Kim WU, Park SH, et al. Elevated vascular endothelial growth factor in systemic sclerosis. J Rheumatol. 2003;30:1529–1533. [PubMed] [Google Scholar]
- Allanore Y, Borderie D, Lemarechal H, Ekindjian OG, Kahan A. Nifedipine decreases sVCAM-1 concentrations and oxidative stress in systemic sclerosis but does not affect the concentrations of vascular endothelial growth factor or its soluble receptor 1. Arthritis Res Ther. 2004;6:R309–R314. doi: 10.1186/ar1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryliszyn-Moskal A, Klimiuk PA, Sierakowski S. Soluble adhesion molecules (sVCAM-1, sE-selectin), vascular endothelial growth factor (VEGF) and endothelin-1 in patients with systemic sclerosis: relationship to organ systemic involvement. Clin Rheumatol. 2005;24:111–116. doi: 10.1007/s10067-004-0987-3. [DOI] [PubMed] [Google Scholar]
- Dor Y, Djonov V, Abramovitch R, Itin A, Fishman GI, Carmeliet P, Goelman G, Keshet E. Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. EMBO J. 2002;21:1939–1947. doi: 10.1093/emboj/21.8.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allanore Y, Borderie D, Airo P, Guiducci S, Czirjak L, Nassonov EL, Riemekasten G, Caramaschi P, Majdan M, Krasowska D, et al. Lack of association between 3 vascular endothelial growth factor gene polymorphisms and systemic sclerosis: results from a multicenter EUSTAR study of European Caucasian patients. Ann Rheum Dis. 2007;66:257–259. doi: 10.1136/ard.2006.054346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibold JR, Matucci-Cerinic M, Denton CP, Furst DE, Mayes MD, Kramer F, Morganti A, Korn JH. Bosentan reduces the number of new digital ulcers in patients with systemic sclerosis. Ann Rheum Dis. 2006;65:S90. [Google Scholar]
- Gershwin ME, Abplanalp H, Castles JJ, Ikeda RM, van der Water J, Eklund J, Haynes D. Characterization of a spontaneous disease of white leghorn chickens resembling progressive systemic sclerosis (scleroderma) J Exp Med. 1981;153:1640–1659. doi: 10.1084/jem.153.6.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruschwitz MS, Shoenfeld Y, Krupp M, Gershwin ME, Penner E, Brezinschek HP, Wick G. Antinuclear antibody profile in UCD line 200 chickens: a model for progressive systemic sclerosis. Int Arch Allergy Immunol. 1993;100:307–313. doi: 10.1159/000236430. [DOI] [PubMed] [Google Scholar]
- Sgonc R, Wick G. What can we learn from an avian model for scleroderma? In: Schoenfeld Y, editor. The Decade of Autoimmunity. Amsterdam, The Netherlands: Elsevier; 1999. pp. 209–217. [Google Scholar]
- Wick G, Sgonc R, Kroemer G. Autoimmune disease, spontaneous animal models. In: Delves PJ, Roitt IM, editor. Encyclopaedia of Immunology. London, UK: Academic Press; 1998. pp. 280–287. [Google Scholar]
- Sgonc R, Gruschwitz MS, Dietrich H, Recheis H, Gershwin ME, Wick G. Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J Clin Invest. 1996;98:785–792. doi: 10.1172/JCI118851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgonc R, Gruschwitz MS, Boeck G, Sepp N, Gruber J, Wick G. Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via CD95. Arthritis Rheum. 2000;43:2550–2562. doi: 10.1002/1529-0131(200011)43:11<2550::AID-ANR24>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Worda M, Sgonc R, Dietrich H, Niederegger H, Sundick RS, Gershwin ME, Wick G. In vivo analysis of the apoptosis-inducing effect of anti-endothelial cell antibodies in systemic sclerosis by the chorionallantoic membrane assay. Arthritis Rheum. 2003;48:2605–2614. doi: 10.1002/art.11179. [DOI] [PubMed] [Google Scholar]
- Nguyen VA, Sgonc R, Dietrich H, Wick G. Endothelial injury in internal organs of University of California at Davis line 200 (UCD 200) chickens, an animal model for systemic sclerosis (scleroderma) J Autoimmun. 2000;14:143–149. doi: 10.1006/jaut.1999.0355. [DOI] [PubMed] [Google Scholar]
- Koch AE. Review: angiogenesis: implications for rheumatoid arthritis. Arthritis Rheum. 1998;41:951–962. doi: 10.1002/1529-0131(199806)41:6<951::AID-ART2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]