

Abstract
Visual processing deficits are an integral component of schizophrenia and are sensitive predictors of schizophrenic decompensation in healthy adults. The primate visual system consists of discrete subcortical magnocellular and parvocellular pathways, which project preferentially to dorsal and ventral cortical streams. Subcortical systems show differential stimulus sensitivity, while cortical systems, in turn, can be differentiated using surface potential analysis. The present study examined contributions of subcortical dysfunction to cortical processing deficits using high-density event-related potentials. Event-related potentials were recorded to stimuli biased towards the magnocellular system using low-contrast isolated checks in Experiment 1 and towards the magnocellular or parvocellular system using low versus high spatial frequency (HSF) sinusoidal gratings, respectively, in Experiment 2. The sample consisted of 23 patients with schizophrenia or schizoaffective disorder and 19 non-psychiatric volunteers of similar age. In Experiment 1, a large decrease in the P1 component of the visual event-related potential in response to magnocellular-biased isolated check stimuli was seen in patients compared with controls (F = 13.2, P = 0.001). Patients also showed decreased slope of the contrast response function over the magnocellular-selective contrast range compared with controls (t = 9.2, P = 0.04) indicating decreased signal amplification. In Experiment 2, C1 (F = 8.5, P = 0.007), P1 (F = 33.1, P < 0.001) and N1 (F = 60.8, P < 0.001) were reduced in amplitude to magnocellular-biased low spatial frequency (LSF) stimuli in patients with schizophrenia, but were intact to parvocellular-biased HSF stimuli, regardless of generator location. Source waveforms derived from inverse dipole modelling showed reduced P1 in Experiment 1 and reduced C1, P1 and N1 to LSF stimuli in Experiment 2, consistent with surface waveforms. These results indicate pervasive magnocellular dysfunction at the subcortical level that leads to secondary impairment in activation of cortical visual structures within dorsal and ventral stream visual pathways. Our finding of early visual dysfunction is consistent with and explanatory of classic literature showing subjective complaints of visual distortions and is consistent with early visual processing deficits reported in schizophrenia. Although deficits in visual processing have frequently been construed as resulting from failures of top-down processing, the present findings argue strongly for bottom-up rather than top-down dysfunction at least within the early visual pathway. Deficits in magnocellular processing in this task may reflect more general impairments in neuronal systems functioning, such as deficits in non-linear amplification and may thus represent an organizing principle for predicting neurocognitive dysfunction in schizophrenia.
Keywords: event-related potential, schizophrenia, EEG dipole source localization, magnocellular, dorsal stream
Introduction
For decades, schizophrenia has been primarily conceptualized as a ‘top-down’ disorder with deficits in executive brain regions, such as prefrontal cortex, leading to secondary dysfunction elsewhere (Cohen and Servan-Schreiber, 1992; Goldman-Rakic and Selemon, 1997; Weinberger and Gallhofer, 1997; Callicott et al., 2003). Although visual deficits have been well described in the classic literature, these have often been attributed to failures of attention and other top-down mechanisms (Silverstein et al., 1996; Potts et al., 2002; van der Stelt et al., 2004; Rassovsky et al., 2005). Recently, however, a significant paradigm shift has begun to occur with data demonstrating histological, structural and functional deficits even within lower visual regions (Green et al., 1994; Selemon et al., 1995; Slaghuis, 1998; Braus et al., 2002; Ardekani et al., 2003; Keri et al., 2004; Butler et al., 2005, 2006; Kim et al., 2005). These findings suggest dysfunction even within early regions of the visual pathway, which may lead to ‘bottom-up’ dysregulation of higher cortical function. Nevertheless, the loci of dysfunction within the visual system have yet to be clarified.
One method that has proven particularly effective at parsing deficits within the early visual system is that of visual event-related potentials. Visual event-related potentials have been extremely well studied and cross-validated with fMRI and psychophysical studies using stimuli that isolate specific levels of the visual system (Previc, 1988; Martinez et al., 1999; Ellemberg et al., 2001; Martinez et al., 2001b; Di Russo et al., 2002, 2003; Schechter et al., 2005). Specific event-related potential components can provide information about functioning at different levels of the visual system.
The visual system functions at subcortical and cortical levels. Subcortically, the visual system is divided into discrete magnocellular and parvocellular pathways; cortically, the visual system is divided into dorsal and ventral visual streams (Ungerleider and Mishkin, 1982). The subcortical magnocellular and parvocellular pathways begin in the retina and project, via the lateral geniculate nucleus, to different layers of primary visual cortex (V1). Magnocellular neurons project predominantly to layers 4Cα and 4B, which, in turn, send projections to the middle temporal visual area, inferior parietal cortex and other regions within the dorsal visual stream. In contrast, parvocellular neurons project predominantly to layer 4Cβ and superficial layers of V1, which, in turn, send projections primarily to the lateral occipital complex, inferior temporal and other ventral stream areas (Lund, 1973; Merigan and Maunsell, 1993; Schroeder et al., 1998). Processing within streams is highly segregated throughout the initial cortical volley, though there appears to be some convergence of magnocellular and parvocellular input even in V1 (Sawatari and Callaway, 1996; Vidyasagar et al., 2002) and significant interaction between dorsal and ventral streams occurring thereafter.
At subcortical levels of the visual system, pathways are differentiated not only anatomically but also functionally, by preferred stimuli. Magnocellular neurons respond to low-contrast and larger [low spatial frequency (LSF)] stimuli while parvocellular neurons respond to higher-contrast and smaller [high spatial frequency (HSF)] stimuli (Derrington and Lennie, 1984; Tootell et al., 1988a, b; Kaplan, 1991). With regard to contrast, the magnocellular system shows a non-linear contrast response function with high gain to low-contrast stimuli and lower gain to high-contrast stimuli, leading to compression in the contrast response curve at ∼16-32% contrast (Kaplan, 1991) with saturation at the asymptote. The parvocellular system, which does not respond to contrast levels below ∼10%, responds with low gain over a wide range of stimulus contrasts and does not saturate (Tootell et al., 1988a, Kaplan, 1991). With regard to spatial frequency, magnocellular neurons receive input from larger regions of the visual field than parvocellular neurons (Dacey and Petersen, 1992). Thus, the magnocellular system is more sensitive to LSF and the parvocellular system is more sensitive to HSF stimuli. Recent visual evoked potential work in humans with magnocellular- and parvocellular-preferential stimuli demonstrates that the spatial characteristics and contrast dependence of these pathways are similar to those found in monkeys (Zemon and Gordon, 2006). Contrast and spatial frequency can therefore be used to bias processing towards the magnocellular versus parvocellular pathway, and the shape of the contrast response curve can be analysed to provide information about signal amplification.
Patients with schizophrenia show highly reproducible deficits in magnocellular function, as reflected in, for example, reduced contrast sensitivity to magnocellular-biased stimuli (Slaghuis, 1998; Keri et al., 2002, 2004; Butler et al., 2005), or reduced steady state visual evoked potential generation (Butler et al., 2005) suggesting subcortical dysfunction. The present study examined contributions of subcortical dysfunction to cortical processing deficits using high-density event-related potentials.
In response to simple visual stimuli, sensory-evoked event-related potentials index neural activity indicative of primary visual cortical, dorsal or ventral stream activity. Visual stimuli elicit a characteristic sequence of event-related potentials, which, according to common nosology have been termed the C1 (Jeffreys and Axford, 1972), P1 and N1 (Clark and Hillyard, 1996) components. Underlying cortical generators for these components have been mapped using inverse dipole modelling techniques (Mangun et al., 1993; Gomez Gonzalez et al., 1994; Clark and Hillyard, 1996) and, especially, source localization combined with fMRI (Martinez et al., 1999, 2001a; Di Russo et al., 2002, 2003). C1, which has a peak latency of ∼90 ms depending upon stimulus, inverts for stimuli presented to the upper versus lower visual field, consistent with a generator in primary visual cortex (V1) (Gomez Gonzalez et al., 1994; Simpson et al., 1995; Clark and Hillyard, 1996; Martinez et al., 2001a; Di Russo et al., 2002, 2003) and is driven more strongly by parvocellular than magnocellular input (Previc, 1988; Schechter et al., 2005). Visual P1, in contrast, appears to have dual underlying generators, including a dorsal generator within dorsolateral extrastriate cortex (e.g. V3a) and a ventral source within ventrolateral extrastriate cortex (e.g. V4) (Martinez et al., 1999; Di Russo et al., 2002). The dorsal generator is driven predominantly by magnocellular input and the ventral generator by parvocellular input (Martinez et al., 2001a). Finally, N1, which peaks at ∼150–200 ms, appears to reflect primarily ventral stream sources (Allison et al., 1999; Bentin et al., 1999; Doniger et al., 2000, 2001a, 2002).
For the present study, event-related potentials were obtained to two complementary classes of stimuli—isolated checks and sine-wave gratings (Fig. 1)—that have well-characterized psychophysiological properties. Isolated checks were presented across a range of contrasts (Zemon et al., 1988). Parvocellular neurons respond poorly to low-stimulus contrast (<16%), so that cortical responses to low-contrast stimuli reflect activity driven primarily by magnocellular visual pathway input. Activity at higher contrasts reflects additional recruitment of parvocellular input. Sine-wave gratings were presented at both LSF and HSF. LSF stimuli predominantly activate the magnocellular pathway, whereas HSF stimuli predominantly activate the parvocellular pathway. For both types of stimuli, dipole mapping was used to confirm generator localization. On the basis of our prior contrast sensitivity and steady state visual evoked potential findings (Butler et al., 2005; Kim et al., 2005), it was hypothesized that cortically generated potentials would be relatively normal in amplitude when input was driven by the parvocellular system (i.e. HSF gratings), but severely reduced in both of the magnocellular-dependent conditions (i.e. isolated checks, LSF gratings), reflecting impaired subcortical processing but preserved local dorsal and ventral stream cortical responses.
Fig. 1.
(A) Examples of isolated-check stimuli used in Experiment 1. Stimuli were presented at five different contrasts (4–64% contrast). (B) Spatial frequency stimuli used in Experiment 2. Stimuli were presented at spatial frequencies of 1 and 5 c/deg.
Material and methods
Participants
Data were collected in two separate experiments. In Experiment 1 (contrast response) participants were 22 male patients meeting DSM-IV (Diagnostic and Statistical Manual of Mental Disorders) criteria for schizophrenia (n = 19) or schizoaffective (n = 3) disorder and 16 healthy volunteers (12 male) of similar age. In Experiment 2 (spatial frequency) participants were 18 male patients meeting criteria for schizophrenia (n = 16) or schizoaffective (n = 2) disorder and 16 healthy volunteers (12 male) of similar age. Each experiment consisted of a single event-related potential session. Seventeen patients and 13 controls participated in both experiments, so that the total sample included 23 patients and 19 controls.
Patients were recruited from in-patient and out-patient facilities associated with the Nathan Kline Institute for Psychiatric Research. Diagnoses were obtained using the Structured Clinical Interview for DSM-IV (First et al., 1997). Healthy volunteers with a history of Structured Clinical Interview for DSM-IV-defined Axis I psychiatric disorder were excluded. Participants were excluded if they had any neurological or ophthalmological disorders that might affect performance or met criteria for alcohol or substance dependence within the last 6 months or abuse within the last month. All participants provided informed consent according to the Declaration of Helsinki. This study was approved by the Nathan Kline Institute for Psychiatric Research/Rockland Psychiatric Center Institutional Review Board.
Patient and control groups did not differ significantly in age [patients, mean ± standard error of the mean (SEM) = 35.9 ± 2.2 years; controls, 33.8 ±1.9 years]. However, there was a significant difference in gender ratio (Fisher's exact test, P = 0.005). Brief Psychiatric Rating Scale total score was 38.3 ± 2.5 (n = 22) and Scale for the Assessment of Negative Symptoms total score (including global scores) was 39.7 ± 2.6 (n = 22). All patients were receiving antipsychotics with 20 patients receiving atypical and 3 receiving typical antipsychotics. Chlorpromazine equivalents were 1365.4 ± 157.7 mg/day. Chlorpromazine equivalents were calculated using conversion factors described previously (Hyman et al., 1995; Peuskens and Link, 1997; Jibson and Tandon, 1998; Woods, 2003). Duration of illness was 16.7 ± 1.8 years.
Participants were tested binocularly. While all participants had at least 20/32 (0.63) corrected visual acuity or better on the Logarithmic Visual Acuity Chart (Precision Vision, LaSalle, IL, USA), the patients had significantly lower visual acuity (vision ratio = 0.9 ± 0.04) than controls (1.1 ± 0.04) for their binocular vision (t = 3.2, d.f. = 40, P = 0.003). Gender and visual acuity were used as covariates in between-group analyses.
Stimuli
Stimuli for both experiments are shown in Fig. 1 and were presented on an Iiyama 502 CRT monitor with a frame rate of ∼100 Hz. For both experiments, subjects were seated in a sound-attenuated, electrically shielded and dimly illuminated recording chamber facing the video monitor.
Stimuli for Experiment I: contrast response to isolated checks
In the contrast response experiment, stimuli consisted of an 8 × 8 array of isolated checks (each check 0.45 × 0.45 degrees of visual angle) at five contrasts (4, 8, 16, 32 and 64% contrast). The luminance of the checks was below that of the background, producing negative contrast (Fig. 1). Line drawings of four different animals were also presented, with one animal type presented per block. With a viewing distance of 114 cm, the stimulus field subtended 5.7 × 5.7 degrees of visual angle. Stimuli were presented in blocks in random order for 60 ms with stimulus-onset asynchronies of 1260, 1460, 1660, 1860 and 2060 ms. Duration of presentation of stimulus-onset asynchronies was also random. Each block contained 12 presentations of each contrast and 6 pictures of the same animal. Blocks were repeated until at least 300 presentations of each contrast were obtained. For the contrast response experiment, to ensure that participants were attending to the stimuli, they were asked to press a button when the animal appeared on the screen. Both groups performed with high accuracy, although patients (86.5 ± 2.3% accuracy) did not perform as well as controls (96.7 ± 1.8% accuracy). When analyses were co-varied for per cent accuracy, all significant results remained.
Stimuli for Experiment 2: spatial frequency
For the spatial frequency experiment, stimuli were black and white sinusoidal horizontal gratings. At a viewing distance of 114 cm, the stimulus field subtended 6.1 × 4.6 degrees of visual angle. The gratings had a spatial frequency of either 1.0 cycle/degree (LSF) or 5 cycles/degree (HSF). The stimuli were presented against a grey field that was isoluminant with the mean luminance of the sine-wave gratings, which had a light/dark contrast of 80%. All stimuli were presented at the centre of gaze for 100 ms durations and stimulus-onset asynchronies of 400 and 600 ms. LSF and HSF stimuli were delivered randomly during runs lasting 3 min. Three runs were administered per subject, yielding ∼540 of each of the two types of stimuli. The subjects were instructed simply to maintain central fixation.
Data acquisition
High-density continuous EEG was acquired from 168 surface electrode sites arranged geodesically using the BioSemi Active II system (BioSemi, Amsterdam, The Netherlands), along with digital stimulus timing tags. Data were digitized online at 512 Hz. All data were recorded relative to a common average and algebraically re-referenced off-line to standard reference before further analyses. The standard reference for Experiment 1 was the average of all the electrodes, and the standard reference for Experiment 2 was a frontal electrode Fpz.
For all subjects, EEG epochs (−200 to 500 ms post-stimulus onset) were constructed off-line. Data were baseline corrected from −100 ms to stimulus onset. Automated artefact rejection was conducted to eliminate trials containing blinks, eye movements or electrical artefact. On average, 28% of all trials were rejected in Experiment 1 and 20% in Experiment 2. There were no significant differences between groups in number of sweeps that survived artefact rejection for Experiment 1 (patients: 415.2 ± 25.8; controls: 438.4 ± 18.0; t = 0.7, d.f. = 36, P = 0.5) and Experiment 2 (patients: 384.4 ± 38.4; controls: 480.9 ± 39.8; t = 1.7, d.f. = 32, P = 0.09). Epochs were then averaged separately for each stimulus type for each subject and subjected to statistical analysis. Grand average waveforms were constructed separately for both patients and controls. Figures show filtered waveform data (45 Hz low pass; 96 dB/octave).
Data analysis
Voltage topography maps were used to select the electrode sites where the prominent event-related potential components, C1, P1 and N1, were largest. The peak amplitude within a specified latency window was obtained for each participant for each event-related potential component. Peak amplitudes averaged across six sites along the midline were used in all subsequent analyses of the C1 component. These six geodesically arranged electrodes for C1 were close to or included midline Oz and P0z, left hemisphere O1 and right hemisphere O2 electrodes of the expanded 10–20 system. Analyses of the P1 and N1 components were carried out on the peak amplitudes averaged across six dorsal-occipital and ventral-occipital sites, respectively, per hemisphere. The electrodes used for each hemisphere were mirror images of each other. On the left hemisphere, the P1 sites were close to or included P3, P5, P7, P07 and TP7. On the right hemisphere, P1 sites included P4, P6, P8, P08 and TP8. On the left hemisphere N1 sites included P5, P7, P03 and P07. On the right hemisphere N1 sites included P6, P8, PO4 and PO8. The same electrode sites were used in both experiments.
The peak amplitude was obtained over specific latency windows corresponding to 20 ms before and after the peak latency in the control participants' waveforms. While latency windows were the same for patients and controls, they differed for each contrast and spatial frequency.
Latency windows for Experiment I: contrast response to isolated checks
For the contrast response experiment, C1 latency windows ranged from 84–124 ms post-stimulus onset at 64% contrast to 112–152 ms at 8% contrast. No C1 was obtained at 4% contrast. P1 latencies were similarly delayed at lower contrasts. At 64% contrast, the P1 latency range was 67–107 ms and at 4% contrast the window was set at 98–138 ms. The N1 component was analysed at 123–163 ms at 64% contrast and at 171–211 ms at 4% contrast.
Latency windows for Experiment 2: spatial frequency
Latency windows were similarly defined for the spatial frequency experiment. The C1 component was analysed over the 70–110 ms epoch for both LSF and HSF stimuli. For P1 the latency window was 115–155 ms for LSF and 130–170 ms for HSF stimuli. For the N1 component the latency ranges were 160–200 and 170–210 ms for LSF and HSF stimuli, respectively.
Statistical analysis
To analyse amplitude and latency effects, separate repeated-measures analyses of covariance (ANCOVAs) with between-group factor of diagnostic group and within-group factors of hemisphere (for P1 and N1 but not C1), contrast (64, 32, 16, 8, 4%; Experiment 1) or spatial frequency (HSF, LSF; Experiment 2) were carried out for each component of interest with gender and vision ratio as covariates.
Results
Experiment 1: contrast response to isolated checks
Figures 2 and 3 show grand average waveforms and surface topographical maps, respectively. C1, P1 and N1 latencies were similar between patients and controls (Table 1). C1 and N1 amplitudes were both somewhat reduced, although between-group differences were not significant (C1: F = 2.56, d.f. = 1, 34, P = 0.12; N1: F = 2.6, d.f. = 1, 34, P = 0.12).
Fig. 2.
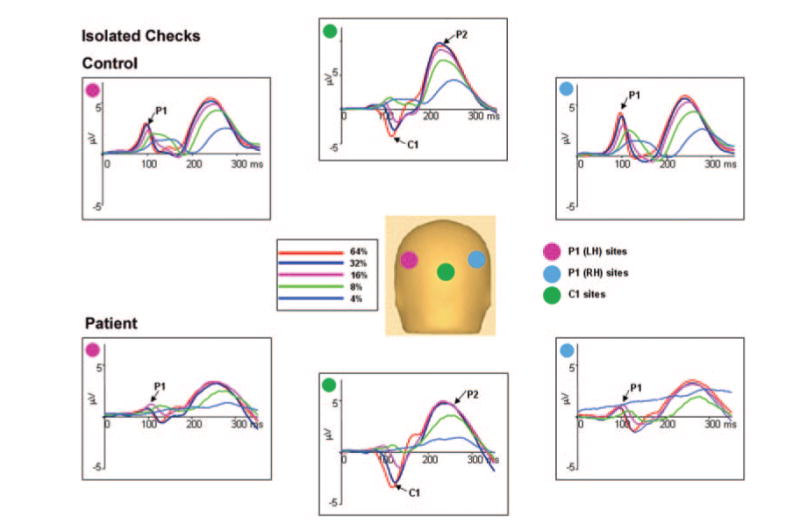
The top plot shows the group-averaged voltage waveforms for controls (n = 16) and the bottom plot for patients (n = 22) in response to isolated checks of different luminance contrasts. Waveforms are the mean of the response at six electrode sites used for P1 in each hemisphere and C1 centrally. A pink disc indicates the location of the six electrode sites in the left hemisphere used for P1. A blue disc indicates location of the six electrodes in the right hemisphere used for P1, and a green disc indicates location of the six central electrodes used for C1.
Fig. 3.
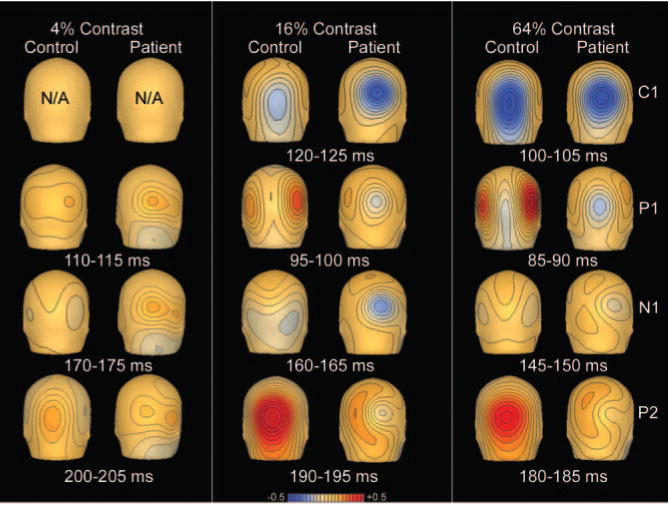
Maps of scalp voltage topography to isolated checks of 4, 16 and 64% contrast for controls (n = 16) and patients (n = 22).
Table 1.
Latencies for patients and controls for Experiments 1 and 2
| Experiment 1: contrast response | Experiment 2: spatial frequency | ||||
|---|---|---|---|---|---|
| Controls (n = 16) | Patients (n = 22) | Controls (n = 16) | Patients (n = 18) | ||
| C1 | C1 | ||||
| 64% contrast | 101.6 ± 2.3 | 103.7 ± 1.8 | LSF | 81.5 ± 2.8 | 91.2 ± 3.7* |
| 32% contrast | 108.7 ± 2.3 | 110.7 ± 2.4 | HSF | 91.5 ± 2.2 | 93.3 ± 3.4 |
| 16% contrast | 119.7 ± 2.6 | 119.9 ± 2.1 | |||
| 8% contrast | 130.9 ± 2.9 | 130.2 ± 2.3 | |||
| P1 | P1 | ||||
| 64% contrast | 88.2 ± 1.6 | 88.2 ± 1.8 | R: LSF | 130.8 ± 2.6 | 139.3 ± 3.9 |
| 32% contrast | 92.0 ± 1.6 | 92.0 ± 2.0 | R: HSF | 148.5 ± 2.5 | 158.0 ± 3.1* |
| 16% contrast | 96.8 ± 1.9 | 95.3 ± 1.8 | |||
| 8% contrast | 105.8 ± 2.4 | 106.2 ± 2.3 | L: LSF | 136.5 ± 2.7 | 142.0 ± 3.5 |
| 4% contrast | 114.4 ± 2.2 | 118.7 ± 1.7 | L: HSF | 150.9 ± 2.5 | 157.2 ± 3.0 |
| N1 | N1 | ||||
| 64% contrast | 137.7 ± 2.7 | 142.8 ± 2.4 | R: LSF | 183.5 ± 2.3 | 171.8 ± 3.5 |
| 32% contrast | 152.7 ± 2.6 | 155.9 ± 2.4 | R: HSF | 187.0 ± 4.7 | 183.1 ± 4.8 |
| 16% contrast | 155.0 ± 2.6 | 156.3 ± 2.1 | |||
| 8% contrast | 163.7 ± 1.9 | 163.9 ± 2.4 | L: LSF | 184.1 ± 2.2 | 177.2 ± 3.2 |
| 4% contrast | 187.7 ± 2.4 | 191.8 ± 2.4 | L: HSF | 186.8 ± 4.8 | 192.7 ± 3.8 |
Mean ± SEM. For Experiment 1, latencies for right and left hemispheres were averaged for P1 and N1 because ANCOVA did not show any significant main effect of hemisphere for either component (P1: F = 1.6, d.f. = 1, 36, P = 0.21; N1: F = 1.3, d.f. = 1, 36, P = 0.26). For Experiment 2, latencies for right and left hemisphere are given separately because there was a main effect for hemisphere for the P1 and N1 components.
P < 0.05.
In contrast, isolated checks elicited a robust P1 that was dramatically reduced in amplitude in patients compared with controls. Both the main effect of group (F = 12.5, d.f. = 1, 34, P = 0.001) and group × contrast interaction (F = 6.8, d.f. = 4, 33, P < 0.001) were highly significant. Controls, as expected, had a contrast response curve that was markedly non-linear, with initial steep slope from 4 to 16% contrast and saturation thereafter (Fig. 4B and C). There was a highly significant between-group effect (F = 13.2, d.f. = 1, 34, P = 0.001) and group × contrast interaction (F = 8.3, d.f. = 2, 35, P = 0.001) for magnocellular range stimuli (4–16% contrast), indicating substantial failure of patients to benefit from increased contrast, particularly over the magnocellular-selective range.
Fig. 4.
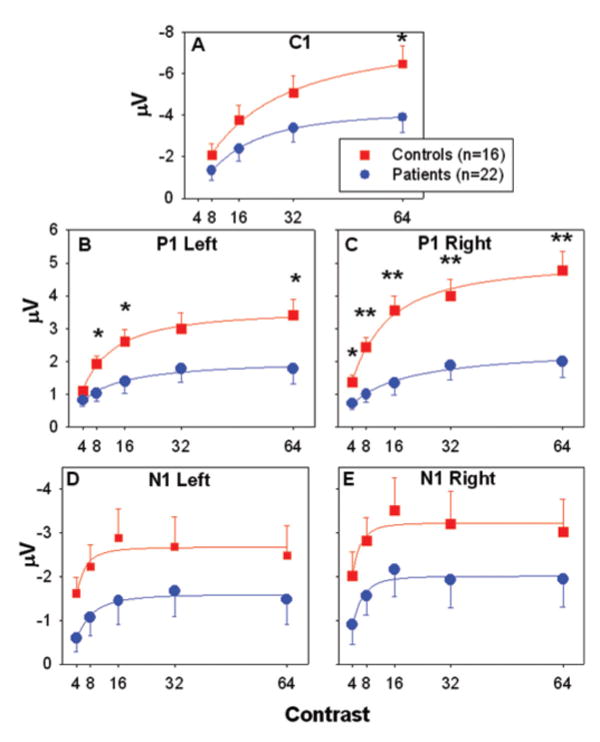
(A–E) C1, P1 and N1 amplitude at each luminance contrast for controls and patients with schizophrenia. Amplitudes are the mean of the response at six electrode sites used for P1 and N1 in each hemisphere and C1 centrally. Error bars represent the standard error of the mean for patients and controls. *P < 0.05, **P < 0.005.
To further analyse contrast functions, the Michaelis– Menten equation was fitted to P1 values, and highly reliable fits were obtained to group mean data (r2 = 0.99). The two groups differed significantly in slope (representing contrast gain; controls: 0.29 ± 0.02; patients: 0.09 ± 0.06, t = 9.2, d.f. = 1, 4, P = 0.04) and plateau (controls: 5.3 ± 0.3; patients: 2.4 ± 0.8, t = 32.1, d.f. = 1, 4, P = 0.005) but not semi-saturation point (controls: 8.9 ± 1.3; patients: 13.8 ± 14.2, t = 0.2, d.f. = 1, 4, P = 0.7), indicating decreased amplification of the contrast response function (Kwon et al., 1992) in patients with schizophrenia, which has been primarily associated with functioning of magnocellular neurons in primates (Kaplan, 1991).
For amplitude, the main effect of hemisphere for P1 (F = 5.1, d.f. = 1, 36, P = 0.03) was significant. There was a significant group × hemisphere (F = 5.3, d.f. = 1, 36, P = 0.03) but no group × hemisphere × contrast (F = 1.0, d.f. = 4, 33, P = 0.43) interaction.
There were no significant relationships between C1, P1 or N1 amplitudes or latencies across contrast levels and chlorpromazine equivalents.
Experiment 2: spatial frequency
Figures 5, 6 and 7 show grand averaged waveforms, peak amplitudes and voltage topography maps, respectively.
Fig. 5.
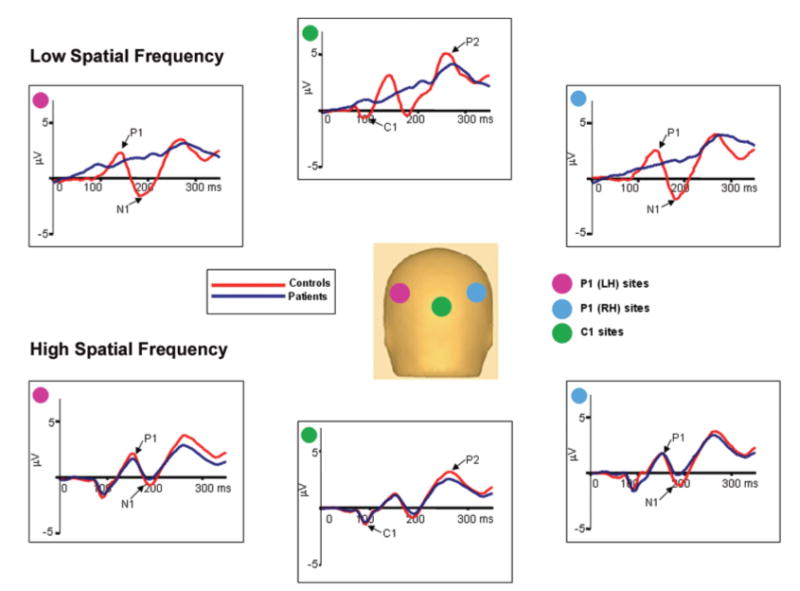
Group-averaged voltage waveforms for controls (n = 16) and patients (n = 18) in response to low (1 c/deg) spatial frequency (top) and high (5 c/deg) spatial frequency (bottom) gratings. Waveforms are the mean of the response at six electrode sites used for P1 and N1 in each hemisphere and C1 centrally. A pink disc indicates the location of the six electrode sites in the left hemisphere used for P1. A blue disc indicates location of the six electrodes in the right hemisphere used for P1 and a green disc indicates location of the six central electrodes used for C1.
Fig. 6.
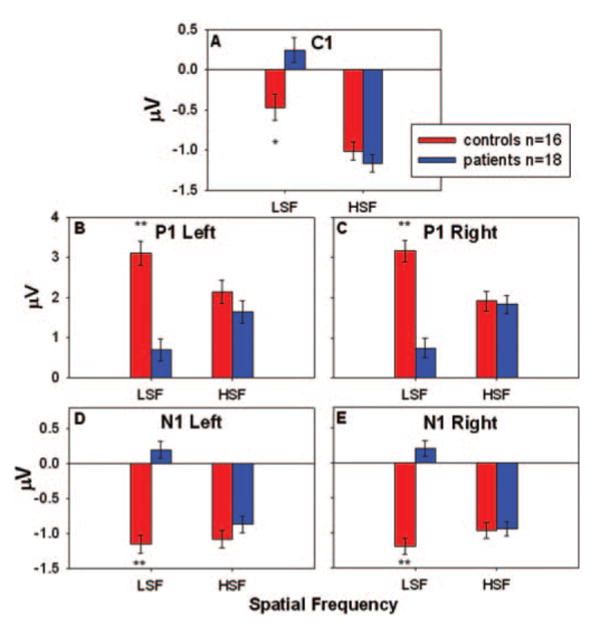
C1, P1 and N1 amplitudes to HSF and LSF stimuli for patients and controls. Amplitudes are the mean of the response at six electrode sites used for P1 and N1 in each hemisphere and C1 centrally. Error bars represent the standard error of the mean for patients and controls. *P < 0.05, **P < 0.001.
Fig. 7.
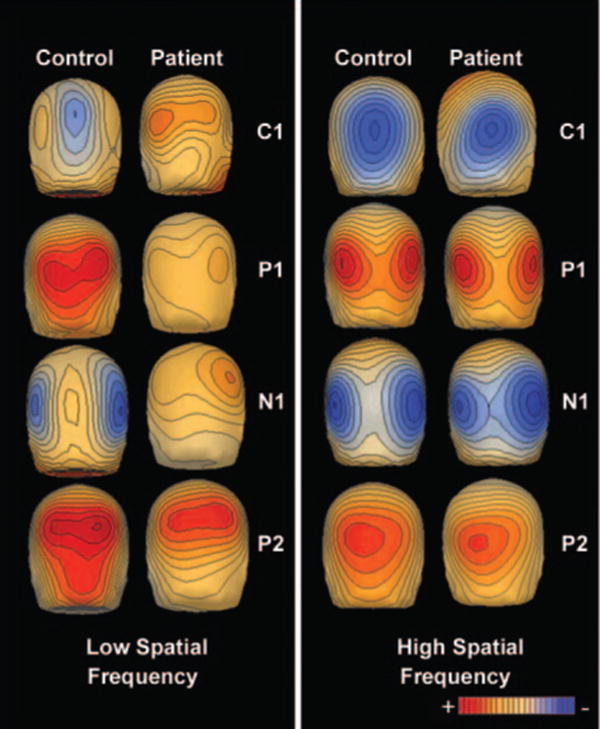
Voltage topography maps of responses of controls (n = 16) and patients (n = 18) to low (1 c/deg) and high (5 c/deg) spatial frequency gratings. The amplitude of the C1 component was maximal at 94 ms, the P1 component at 138 ms, the N1 component at 186 ms and the P2 component at 262 ms.
For amplitude, for the C1 component, there was a significant group × spatial frequency interaction (F = 10.3, d.f. = 1, 32, P = 0.003), a main effect of spatial frequency (F = 67.5, d.f. = 1, 32, P < 0.001), but no significant main effect of group (F = 3.5, d.f. = 1, 30, P = 0.07). Separate follow-up ANCOVAs of the group × spatial frequency interaction showed a significant main effect of group for the LSF condition (F = 8.5, d.f. = 1, 30, P = 0.007) but not for the HSF condition (F = 0.8, d.f. = 1, 30, P = 0.38).
As in Experiment 1, P1 amplitude was dramatically reduced in patients compared with controls (F = 12.6, d.f. = 1, 30, P = 0.001). In addition, the group × spatial frequency interaction was highly significant (F = 73.4, d.f. = 1, 32, P < 0.001), owing to a significant P1 reduction in patients in the LSF (F = 33.1, d.f. = 1, 30, P < 0.001), but not the HSF (F = 0.5, d.f. = 1, 30, P = 0.47), condition. There was a significant group × hemisphere × spatial frequency interaction in the overall ANCOVA (F = 5.2, d.f. = 1, 32, P = 0.03), but no main effect of hemisphere (F = 0.2, d.f. = 1, 32, P = 0.7) or group × hemisphere interaction (F = 3.0, d.f. = 1, 32, P = 0.09).
N1 amplitude was relatively large compared with the contrast response experiment and there was a significant main effect of group (F = 28.2, d.f. = 1, 30, P < 0.001). There was also a significant group × spatial frequency interaction (F = 35.7, d.f. = 1, 32, P < 0.001) indicating, as with the P1 component, that patients had a significant decrease in N1 amplitude compared with controls in the LSF (F = 60.8, d.f. = 1, 30, P < 0.001) but not in the HSF (F = 0.4, d.f. = 1, 30, P = 0.51) condition. There were no significant main effects of hemisphere in the overall ANCOVA (F = 0.0, d.f. = 1, 32, P = 0.98), group × hemisphere (F = 2.0, d.f. = 1, 32, P = 0.17) or group × hemisphere × spatial frequency interactions (F = 3.7, d.f. = 1, 32, P = 0.06).
There were no significant relationships between C1, P1 or N1 amplitudes across spatial frequencies and chlorpromazine equivalents.
C1, P1 and N1 latencies are reported in Table 1. For C1 latency, there were significant main effects of group (F = 5.2, d.f. = 1, 30, P = 0.03) and spatial frequency (F = 5.7, d.f. = 1, 32, P = 0.02) but no significant group × spatial frequency interaction (F = 2.4, d.f. = 1, 32, P = 0.13). Follow-up t-tests showed that patients had significantly longer latencies than controls in the LSF condition (t = 2.1, d.f. = 32, P = 0.05), but not the HSF condition (t = 0.4, d.f. = 32, P = 0.66).
For P1 latency there were also significant main effects of group (F = 4.8, d.f. = 1, 30, P = 0.04) and spatial frequency (F = 48.4, d.f. = 1, 32, P < 0.001) but no significant group × spatial frequency interaction (F = 0.03, d.f. = 1, 32, P = 0.85). Separate ANCOVAs showed a significant increase in P1 latency in patients compared with controls in the HSF (F = 6.2, d.f. = 1, 30, P = 0.02), but not in the LSF (F = 1.7, d.f. = 1, 30, P = 0.21), condition. Follow-up t-tests showed that the right hemisphere HSF P1 was significantly longer in patients than controls (t = 2.3, d.f. = 32, P = 0.03). In addition, there was a significant main effect of hemisphere (F = 5.0, d.f. = 1, 32, P = 0.03) but no significant group × hemisphere interaction (F = 1.9, d.f. = 1, 32, P = 0.18).
For N1 latency, there were no significant main effects of group (F = 0.12, d.f. = 1, 30, P = 0.73) or spatial frequency (F = 3.6, d.f. = 1, 32, P = 0.07) and no significant group × spatial frequency interaction (F = 1.4, d.f. = 1, 32, P = 0.25). However, both the main effect of hemisphere (F = 6.3, d.f. = 1, 32, P = 0.02) and the group × hemisphere interaction (F = 5.6, d.f. = 1, 32, P = 0.02) were significant.
Source localization for Experiment 1 and 2
Inverse dipole modelling of the early event-related potential components was carried out on the grand average waveforms using the brain electrical source algorithm (Scherg, 1990). For both experiments, models were constructed using three sets of paired dipoles with each set constrained to be mirror-symmetrical in location but not orientation, over restricted time intervals corresponding to the components of interest (Di Russo et al., 2002). An additional, ocular dipole was added to absorb ocular-movement-related noise. For both experiments, dipole locations (Table 2) were calculated on the basis of control data, and subsequently applied to corresponding patient results.
Table 2.
Latency windows, residual variances and Talairach coordinates for each pair of dipoles
| Latency (ms) | x | y | z | Control RV (%) | Patient RV (%) | |
|---|---|---|---|---|---|---|
| C1 | ||||||
| 64% contrast | 84–124 | ±2 | −86 | 1 | 2.55 | 3.66 |
| Low SF | 67–107 | ±4 | −81 | 1 | 5.96 | 7.18 |
| High SF | 70–110 | ±3 | −89 | −2 | 1.57 | 2.50 |
| P1 | ||||||
| 64% contrast | 67–107 | ±31 | −72 | 23 | 2.31 | 3.00 |
| Low SF | 115–155 | ±41 | −71 | 23 | 1.62 | 7.82 |
| High SF | 130–170 | ±36 | −77 | 16 | 3.34 | 4.93 |
| N1 | ||||||
| 64% contrast | 123–163 | ±46 | −76 | −4 | 4.79 | 4.13 |
| Low SF | 160–200 | ±36 | −64 | −4 | 3.19 | 8.39 |
| High SF | 170–210 | ±36 | −68 | −4 | 3.33 | 4.11 |
RV = residual variance.
In both experiments, the C1 distribution was accounted for well by a pair of dipoles situated in medial occipital cortex in or near the calcarine fissure. Another pair of dipoles located in dorsolateral extrastriate cortex accounted for the P1 component, while a dipole pair in ventrolateral cortex accounted for the N1 distribution (Fig. 8). Together, the three dipole pairs for both controls and schizophrenia patients accounted for >96% of the total variance in the modelled latency window. Consistent with the surface components, source waveforms from the dorsolateral electrodes in patients showed a dramatic reduction in amplitude in both the isolated check (30% of control) and LSF conditions (10% of control), but not HSF condition. The C1 response was much larger in the isolated check and HSF conditions relative to LSF for both groups. In both the isolated check and HSF conditions, this component was relatively normal for patients (77 and 74% of control, respectively) but was absent for patients in the LSF condition (2% of control).
Fig. 8.
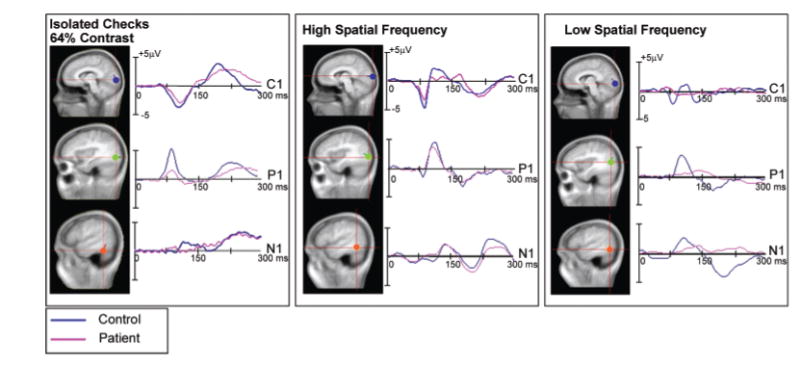
Anatomical localization of modelled dipoles and derived source waveforms. The dipoles and derived source waveforms were obtained at 64% contrast for Experiment 1 and at LSF and HSF for Experiment 2 in patients and controls.
Discussion
Schizophrenia is associated with deficits in visual processing, which represent a critical component of the disorder. Mechanisms underlying such deficits remain controversial, with competing theories emphasizing top-down (Cohen and Servan-Schreiber, 1992; Silverstein et al., 1996; Weinberger and Gallhofer, 1997; van der Stelt et al., 2004) versus bottom-up (Doniger et al., 2002; Gonzalez-Hernandez et al., 2003; Butler et al., 2005; Johnson et al., 2005) mechanisms. The present study evaluated function of the early visual system using high-density event-related potentials and well-characterized components to evaluate function within dorsal and ventral stream visual regions in response to stimuli biased towards the magnocellular or parvocellular visual pathways. Patients showed deficits in the generation of all cortical potentials, including C1, P1 and N1, when stimuli were biased toward the magnocellular system, but intact event-related potential generation when stimuli were biased toward the parvocellular system (Table 3). These findings indicate first that schizophrenia is associated with severe deficits even in response to very primitive visual stimuli and, second, that deficits in cortical response reflect primarily disturbed input to cortex emanating from the magnocellular visual system.
Table 3.
Summary of results for ERP components reflecting dorsal and ventral stream activity as a function of stimulus properties
| Cortical response | |||
|---|---|---|---|
| C1 | Dorsal (P1 ampl) | Ventral (N1 ampl) | |
| Stimulus property | |||
| Magnocellular (16% isolated checks) | ↔ | ↓↓↓ | N.D. |
| Magnocellular (LSF) | ↓↓↓ | ↓↓↓ | ↓↓↓ |
| Parvocellular (HSF) | ↔ | ↔ | ↔ |
↓↓↓ = decreased P < 0.007 patients versus controls; ↔ = no significant change; N.D. = not determined.
In the present study, two separate manipulations were used to isolate the magnocellular system. First, in the isolated-check condition, stimuli were presented at both low (4–16%) and high (32–64%) contrast. Low-contrast stimuli selectively activate the magnocellular system, with additional activation at higher contrasts reflecting activation of both magnocellular and parvocellular neurons. Second, in the sine-wave grating condition, LSF and HSF stimuli were used to bias stimuli towards the magnocellular and parvocellular pathways, respectively. Convergent data were obtained using the two paradigms, supporting the magnocellular/ parvocellular distinction.
In the isolated-check condition, the P1 and N1 components were reduced in amplitude under conditions (e.g. 8 and 16% contrast) that emphasize activity in the magnocellular system, with parallel increases in amplitude occurring in both groups thereafter. In the sine-wave conditions, significant group × HSF/LSF effects were observed for C1, P1 and N1 amplitude, with deficits being observed only in the magnocellular-biased conditions. Thus, in addition to showing early visual deficits, the present study demonstrates differential involvement of the magnocellular system.
Event-related potential generator locations in this study were extremely similar to those reported previously, including a C1 located within the calcarine fissure and P1 and N1 in extrastriate visual areas (Gomez Gonzalez et al., 1994; Clark and Hillyard, 1996; Bentin et al., 1999; Doniger et al., 2000; Martinez et al., 2001a; Di Russo et al., 2002, 2003). As with surface waveforms, derived source waveforms in patients showed reduced C1, P1 and N1 amplitudes in the LSF, but not HSF, condition and reduced P1 amplitude in the isolated check condition compared with controls.
In studies involving traditional stimuli (e.g. sine-wave gratings), P1 onset and peak latency typically follows that of C1. Interestingly, in this study, P1 preceded C1 in response to the isolated checks, but followed C1 in response to sine-wave gratings. Isolated checks have been used extensively to date in steady state visual evoked potential studies but, to our knowledge, have not been studied previously with transient event-related potentials. Thus, the basis for the early P1, relative to C1, with these stimuli remains to be determined.
One possible explanation for the differential timing of the components is that we may have misidentified P1 and/or C1 in the isolated check condition. However, we feel, on the basis of surface topography and source localization, that this is not the case. Instead, the differential latencies of these components may reflect differential engagement of magnocellular and parvocellular processes under different stimulation conditions, and are consistent with primate studies showing rapid transmission of information particularly through the magnocellular visual system (Schmolesky et al., 1998; Schroeder et al., 1998; Lamme and Roelfsema, 2000; Foxe and Simpson, 2002). In our study, the C1 response to isolated checks, as expected, had a posterior midline maximum (Fig. 3) similar to that observed with sine-wave gratings. Further, in both conditions, C1 generators were located in the medial occipital cortex in or near the calcarine fissure (Fig. 8 and Table 2). In addition, when isolated checks were presented to the upper and lower visual fields, polarity inversion of the C1 was seen (A. Martinez, unpublished data) as expected for a source with a primary generator in V1. This polarity inversion also argues against C1 being driven in the isolated check condition by feedback from higher visual regions. Thus, in both conditions, what we identify as C1 appears to represent a similar underlying process. Similarly, scalp topography and source localization for the P1 component were similar in the isolated check (Experiment 1) and sine-wave grating (Experiment 2) conditions, despite the shorter latency observed to isolated check stimuli, again suggesting that a similar process is being measured in both conditions.
The present results suggest that, for reasons still to be determined, different stimulus types can differentially affect the relative timing of the P1 and C1 components. This concept, while discussed to only a limited degree in the human literature, is entirely consistent with relative timing experiments of P1 and C1 generation in monkeys. P1, as shown both in this and prior (Martinez et al., 1999; Di Russo et al., 2002, 2003) studies, reflects primary activation within dorsal stream structures such as V3a. C1, in contrast, reflects activity within primary visual cortex (V1, V2). In monkey studies, activation of dorsal areas such as the middle temporal visual area may occur even before activation of layer 4 of V1 in response to light flashes (Schroeder et al., 1998). This may be because dorsal stream areas are largely activated by the fast magnocellular pathway, while primary visual cortex (V1) is activated by both magnocellular as well as a larger number of slower parvocellular neurons that produce slower net activation of V1. Monkey studies have not yet been conducted with the types of stimuli used here (isolated checks, sine-wave gratings). However, a clear prediction of our present studies is that the relative activation latencies of V1 and middle temporal visual area may depend upon the class of stimulus used. It should be noted that in human flash-evoked potentials, scalp recordings show a P1 followed by an N1, with no evident C1 component (Kutas et al., 1994). The dissociability of C1 and P1 latency as a function both of stimulus type and contrast thus supports the concept that magnocellular and parvocellular systems may differentially contribute to the generation of these components, consistent with the present observation of larger C1 responses to parvocellular-biased stimuli and larger P1 responses to magnocellular-biased stimuli.
Our finding of early visual dysfunction in schizophrenia is consistent with, and explanatory of, a considerable but underappreciated literature concerning visual dysfunction in schizophrenia. Thus, in first person accounts dating back decades (e.g. McGhie and Chapman, 1961), patients frequently complained of visual distortions as among the earliest symptoms of schizophrenia. Similarly, in symptom inventories, subjective complaints regarding visual disturbances were as common as complaints regarding higher-order processes such as attention or memory (McGhie and Chapman, 1961; Cutting and Dunne, 1989; Klosterkotter et al., 2001). Complaints included changes in visual perception such that vision is less clear, muted or blurry (Freedman and Chapman, 1973) or more acute (Freedman, 1974), that ‘everything is in bits’ (McGhie and Chapman, 1961) and that ‘the organization of things was different’ (Cutting and Dunne, 1989). Consistent with these subjective reports, patients show contrast sensitivity (Slaghuis, 1998; Keri et al., 2002; Butler et al., 2005), vernier threshold (Keri et al., 2004) and spatial frequency discrimination (O'Donnell et al., 2002) deficits, particularly to magnocellular-biased stimuli, as well as deficits in ability to discriminate changes in velocity (Chen et al., 1999b), detect location of stimuli (Cadenhead et al., 1998), detect coherent motion (Stuve et al., 1997; Li, 2002; Brenner et al., 2003; Chen et al., 2003) or match coherent and incoherent motion or trajectory across brief delays (O'Donnell et al., 1996; Kim et al., 2006). Indeed, it has recently been found that anomalous visual experiences in schizophrenia are related to magnocellular dysfunction (Keri et al., 2005) supporting the hypothesis that early stage visual dysfunction is related to visual perceptual disturbances. The present study argues for greater appreciation of sensory-level impairment when designing stimuli for cognitive performance tasks in schizophrenia.
As in the present study, the magnocellular pathway plays a critical role in detection of specific classes of stimuli, including primitive LSF and motion stimuli (Legge, 1978; Ungerleider and Mishkin, 1982; Tootell et al., 1988b). In addition, the magnocellular system plays a critical role in processing of complex visual stimuli. Most ‘real-world’ stimuli contain a mixture of low and high spatial frequencies and low- and high-contrast components. For such stimuli, the primary role of the magnocellular system may be considered within ‘frame and fill’ models of magnocellular and parvocellular functioning (Schroeder et al., 1998; Vidyasagar, 1999; Bar, 2003). Information is transmitted through the magnocellular pathway/dorsal stream system significantly more rapidly than through the parvocellular system, permitting the dorsal stream to exert an organizing, cross-over influence on ventral stream processing (Nowak and Bullier, 1997; Foxe et al., 1998; Schmolesky et al., 1998; Schroeder et al., 1998; Vidyasagar, 1999). The cross-over magnocellular input provides a low-resolution template of the visual image, which is then ‘filled in’ by slower information conveyed via the parvocellular pathway.
The present finding of impaired dorsal stream activation to magnocellular input suggests that, in schizophrenia, the organizing or ‘frame’ function of the dorsal stream may be severely impaired. Such a theory would be consistent with behavioural deficits in gestalt processing (Place and Gilmore, 1980; Silverstein et al., 1996; Johnson et al., 2005), object recognition (Schwartz et al., 1999) and perceptual closure (Doniger et al., 2002) that have been observed in schizophrenia by ourselves and others. Further, in our prior studies of ventral-stream processing, we and others (Foxe et al., 2005; Spencer et al., 2003) have observed normal ventral-stream-related activity for processes such as illusory contour recognition that occur during the initial feed-forward sweep within the ventral stream (∼170 ms peak), but impaired generation of later potentials such as closure-related negativity (Ncl), that occur during the recursive-processing window (∼270 ms) (Doniger et al., 2002). In both our studies (Doniger et al., 2001b) and those of others (Carter et al., 1996; Silverstein et al., 1996; Granholm et al., 1999), patients showed intact ability to utilize explicit instructions, hints or prior exposure to improve object recognition performance, despite impaired perceptual organization overall. Preserved ability to utilize ‘high-level’ information is consistent with relatively intact performance at the cortical level, along with impaired sensory input into cortex.
In contrast to the present study, which suggests impaired function within the early visual pathway, several functional brain imaging studies have reported intact V1 activation to visual stimulation, leading at least some authors to conclude that primary sensory cortical areas are unaffected in schizophrenia (Braus et al., 2002; Barch et al., 2003). The present study suggests caution in interpreting results of such fMRI studies, especially when relatively non-selective stimuli are used. If stimuli produce strong activation of the parvocellular pathway in addition to the magnocellular pathway, preserved V1 activation in fMRI studies of schizophrenia is likely to reflect intact parvocellular function, and may have limited implications for the functional status of the magnocellular system.
Further evidence for the bottom-up nature of the present deficits comes from diffusion tensor imaging studies in schizophrenia. Diffusion tensor imaging changes are wide-spread in schizophrenia, reflecting distributed deficits in white matter function. Deficits are thus present in white matter tracts projecting from lateral geniculate nucleus to primary visual cortex (Ardekani et al., 2003; Butler et al., 2006) as well as in higher brain regions. Using a region of interest approach and tracing white matter integrity from primary visual sensory to association areas, fractional anisotropy was reduced in the optic radiations, but not in primary visual cortex or dorsal or ventral stream projection areas, suggesting that in the visual system integrity of the earliest pathway is most affected (Butler et al., 2006). Magnocellular neurons, being more heavily myelinated than parvocellular neurons, would be expected to be affected preferentially by dysmyelinating pathology similar to that observed in schizophrenia. The pattern of event-related potential reduction we have observed in schizophrenia is also consistent with patterns of visual dysfunction typically observed following infusion of N-methyl-d-aspartate antagonists into lateral geniculate nucleus or primary visual cortex (Fox et al., 1990; Kwon et al., 1992). Thus, deficits in magnocellular processing might be indicative of dysfunctional N-methyl-d-aspartate-dependent processing, consistent with recent neurochemical theories of schizophrenia (Javitt and Zukin, 1991; Olney and Farber, 1995; Tsai and Coyle, 2002).
Although deficits in visual tasks in schizophrenia are frequently attributed to ‘attentional’ deficits, the attentional load in the present study was extremely modest. Further, in Experiment 2 LSF and HSF stimuli were intermixed, making it unlikely that attentional disturbances could lead to the prominent group × spatial frequency interaction observed in the present study. Thus, deficits in the present study cannot be easily attributed to impairments in attention, cognition or overall cooperation levels.
Our findings of reduced P1 strongly support prior studies showing P1 deficits in patients with schizophrenia (Matsuoka et al., 1996; Basinska, 1998; Foxe et al., 2001; Doniger et al., 2002; Spencer et al., 2003; Foxe et al., 2005; Schechter et al., 2005) and first-degree relatives (Yeap et al., 2006). Other studies have not found deficits (Strandburg et al., 1994; Alain et al., 1998; Bruder et al., 1998; van der Stelt et al., 2004). Studies to date frequently used stimuli chosen for cognitive, rather than psychophysical, content. The present study suggests that choice of stimulus may be critical in demonstrating P1 deficits.
A limitation of the study is that all patients were receiving medication at the time of testing. However, visual processing deficits have been found in both medicated and unmedicated patients (Braff and Saccuzzo, 1982; Harvey et al., 1990; Butler et al., 1996; Cadenhead et al., 1997; Butler et al., 2002), as well as in (unmedicated) first-degree relatives of patients with schizophrenia (Green et al., 1997; Chen et al., 1999a; Keri et al., 2004). In addition, no significant correlations were found between chlorpromazine equivalents and P1 or N1 amplitude across contrasts. Further, while dopamine may play a role within the early visual system (Bodis-Wollner and Tzelepi, 1998), no differential effects of dopamine upon magnocellular versus parvocellular processing have been reported to date.
In conclusion, the present study demonstrates that patients with schizophrenia show significant and substantial deficits in early visual processing affecting the subcortical magnocellular pathway that lead to secondary impairment in activation of cortical visual structures within both the dorsal and ventral stream pathways. Although deficits in visual processing have frequently been construed as resulting from failures of top-down processing, the present findings argue strongly for bottom-up rather than top-down dysfunction at least within the early visual pathway. Further, conflicting literature in this area may reflect inadequate attention to the psychophysical properties of stimuli used in visual activation tasks. Deficits in magnocellular processing in this task may reflect more general impairments in neuronal systems functioning, such as deficits in non-linear amplification (Butler et al., 2005), and may thus represent an organizing principle for predicting neurocognitive dysfunction in schizophrenia.
Acknowledgments
This study was supported in part by a Lieber Young Investigator Award from the National Alliance for Research on Schizophrenia and Depression (P.D.B.); USPHS grants RO1 MH66374 (P.D.B.), R01 MH65350 (J.J.F.), R03 MH067579 (A.M.), R37 MH49334 and K02 MH01439 (D.C.J.); and a Burroughs Wellcome Translational Scientist Award (to D.C.J.). This work was presented in part at the Society of Biological Psychiatry Annual Meeting, New York, NY, April 29–May 1, 2004 and Society for Neuroscience Meeting, San Diego, CA, October 23–27, 2004. We would like to thank Dr Charles E. Schroeder for helpful discussions of the manuscript.
Abbreviations
- HSF
high spatial frequency
- LSF
low spatial frequency
References
- Alain C, Hargrave R, Woods DL. Processing of auditory stimuli during visual attention in patients with schizophrenia. Biol Psychiatry. 1998;44:1151–9. doi: 10.1016/s0006-3223(97)00478-2. [DOI] [PubMed] [Google Scholar]
- Allison T, Puce A, Spencer D, McCarthy G. Electrophysiological studies of human face perception I: potentials generated in occipitotemporal cortex by face and non-face stimuli. Cereb Cortex. 1999;9:415–30. doi: 10.1093/cercor/9.5.415. [DOI] [PubMed] [Google Scholar]
- Ardekani BA, Nierenberg J, Hoptman MJ, Javitt DC, Lim KO. MRI study of white matter diffusion anisotropy in schizophrenia. Neuroreport. 2003;14:2025–9. doi: 10.1097/00001756-200311140-00004. [DOI] [PubMed] [Google Scholar]
- Bar M. A cortical mechanism for triggering top-down facilitation in visual object recognition. J Cogn Neurosci. 2003;15:600–9. doi: 10.1162/089892903321662976. [DOI] [PubMed] [Google Scholar]
- Barch DM, Mathews JR, Buckner RL, Maccotta L, Csernansky JG, Snyder AZ. Hemodynamic responses in visual, motor, and somatosensory cortices in schizophrenia. Neuroimage. 2003;20:1884–93. doi: 10.1016/s1053-8119(03)00449-x. [DOI] [PubMed] [Google Scholar]
- Basinska A. Altered electrophysiological pattern of target detection in schizophrenia in the continuous attention test. Acta Neurobiol Exp. 1998;58:207–20. doi: 10.55782/ane-1998-1275. [DOI] [PubMed] [Google Scholar]
- Bentin S, Mouchetant-Rostaing Y, Giard MH, Echallier JF, Pernier J. ERP manifestations of processing printed words at different psycholinguistic levels: time course and scalp distribution. J Cogn Neurosci. 1999;11:235–60. doi: 10.1162/089892999563373. [DOI] [PubMed] [Google Scholar]
- Bodis-Wollner I, Tzelepi A. The push-pull action of dopamine on spatial tuning of the monkey retina: the effects of dopaminergic deficiency and selective D1 and D2 receptor ligands on the pattern electroretinogram. Vision Res. 1998;38:1479–87. doi: 10.1016/s0042-6989(98)00028-5. [DOI] [PubMed] [Google Scholar]
- Braff DL, Saccuzzo DP. Effect of antipsychotic medication on speed of information processing in schizophrenic patients. Am J Psychiatry. 1982;139:1127–30. doi: 10.1176/ajp.139.9.1127. [DOI] [PubMed] [Google Scholar]
- Braus DF, Weber-Fahr W, Tost H, Ruf M, Henn FA. Sensory information processing in neuroleptic-naive first-episode schizophrenic patients: a functional magnetic resonance imaging study. Arch Gen Psychiatry. 2002;59:696–701. doi: 10.1001/archpsyc.59.8.696. [DOI] [PubMed] [Google Scholar]
- Brenner CA, Wilt MA, Lysaker PH, Koyfman A, O'Donnell BF. Psychometrically matched visual-processing tasks in schizophrenia spectrum disorders. J Abnorm Psychol. 2003;112:28–37. [PubMed] [Google Scholar]
- Bruder G, Kayser J, Tenke C, Rabinowicz E, Friedman M, Amador X, et al. The time course of visuospatial processing deficits in schizophrenia: an event-related brain potential study. J Abnorm Psychol. 1998;107:399–411. doi: 10.1037//0021-843x.107.3.399. [DOI] [PubMed] [Google Scholar]
- Butler PD, Harkavy-Friedman JM, Amador XF, Gorman JM. Backward masking in schizophrenia: relationship to medication status, neuropsychological functioning, and dopamine metabolism. Biol Psychiatry. 1996;40:295–8. doi: 10.1016/0006-3223(96)00007-8. [DOI] [PubMed] [Google Scholar]
- Butler PD, DeSanti LA, Maddox J, Harkavy-Friedman JM, Amador XF, Goetz RR, et al. Visual backward-masking deficits in schizophrenia: relationship to visual pathway function and symptomatology. Schizophr Res. 2002;59:199–209. doi: 10.1016/s0920-9964(01)00341-3. [DOI] [PubMed] [Google Scholar]
- Butler PD, Zemon V, Schechter I, Saperstein AM, Hoptman MJ, Lim KO, et al. Early-stage visual processing and cortical amplification deficits in schizophrenia. Arch Gen Psychiatry. 2005;62:495–504. doi: 10.1001/archpsyc.62.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler PD, Hoptman MJ, Nierenberg J, Foxe JJ, Javitt DC, Lim KO. Visual white matter integrity in schizophrenia. Am J Psychiatry. 2006 doi: 10.1176/appi.ajp.163.11.2011. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenhead KS, Geyer MA, Butler RW, Perry W, Sprock J, Braff DL. Information processing deficits of schizophrenia patients: relationship to clinical ratings, gender and medication status. Schizophr Res. 1997;28:51–62. doi: 10.1016/s0920-9964(97)00085-6. [DOI] [PubMed] [Google Scholar]
- Cadenhead KS, Serper Y, Braff DL. Transient versus sustained visual channels in the visual backward masking deficits of schizophrenia patients. Biol Psychiatry. 1998;43:132–8. doi: 10.1016/S0006-3223(97)00316-8. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Mattay VS, Verchinski BA, Marenco S, Egan MF, Weinberger DR. Complexity of prefrontal cortical dysfunction in schizophrenia: more than up or down. Am J Psychiatry. 2003;160:2209–15. doi: 10.1176/appi.ajp.160.12.2209. [DOI] [PubMed] [Google Scholar]
- Carter CS, Robertson LC, Nordahl TE, Chaderjian M, Oshora-Celaya L. Perceptual and attentional asymmetries in schizophrenia: further evidence for a left hemisphere deficit. Psychiatry Res. 1996;62:111–9. doi: 10.1016/0165-1781(96)02849-1. [DOI] [PubMed] [Google Scholar]
- Chen Y, Nakayama K, Levy DL, Matthysse S, Holzman PS. Psychophysical isolation of a motion-processing deficit in schizophrenics and their relatives and its association with impaired smooth pursuit. Proc Natl Acad Sci USA. 1999a;96:4724–9. doi: 10.1073/pnas.96.8.4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Palafox GP, Nakayama K, Levy DL, Matthysse S, Holzman PS. Motion perception in schizophrenia. Arch Gen Psychiatry. 1999b;56:149–54. doi: 10.1001/archpsyc.56.2.149. [DOI] [PubMed] [Google Scholar]
- Chen Y, Nakayama K, Levy D, Matthysse S, Holzman P. Processing of global, but not local, motion direction is deficient in schizophrenia. Schizophr Res. 2003;61:215–27. doi: 10.1016/s0920-9964(02)00222-0. [DOI] [PubMed] [Google Scholar]
- Clark VP, Hillyard SA. Spatial selective attention affects early extrastriate but not striate components of the visual evoked potential. J Cogn Neurosci. 1996;8:387–402. doi: 10.1162/jocn.1996.8.5.387. [DOI] [PubMed] [Google Scholar]
- Cohen JD, Servan-Schreiber D. Context, cortex, and dopamine: a connectionist approach to behavior and biology in schizophrenia. Psychol Rev. 1992;99:45–77. doi: 10.1037/0033-295x.99.1.45. [DOI] [PubMed] [Google Scholar]
- Cutting J, Dunne F. Subjective experience of schizophrenia. Schizophr Bull. 1989;15:217–31. doi: 10.1093/schbul/15.2.217. [DOI] [PubMed] [Google Scholar]
- Dacey DM, Petersen MR. Dendritic field size and morphology of midget and parasol ganglion cells of the human retina. Proc Natl Acad Sci USA. 1992;89:9666–70. doi: 10.1073/pnas.89.20.9666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrington AM, Lennie P. Spatial and temporal contrast sensitivities of neurones in lateral geniculate nucleus of macaque. J Physiol. 1984;357:219–40. doi: 10.1113/jphysiol.1984.sp015498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Russo F, Martinez A, Sereno MI, Pitzalis S, Hillyard SA. Cortical sources of the early components of the visual evoked potential. Hum Brain Mapp. 2002;15:95–111. doi: 10.1002/hbm.10010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Russo F, Martinez A, Hillyard SA. Source analysis of event-related cortical activity during visuo-spatial attention. Cereb Cortex. 2003;13:486–99. doi: 10.1093/cercor/13.5.486. [DOI] [PubMed] [Google Scholar]
- Doniger GM, Foxe JJ, Murray MM, Higgins BA, Snodgrass JG, Schroeder CE, et al. Activation timecourse of ventral visual stream object-recognition areas: high density electrical mapping of perceptual closure processes. J Cogn Neurosci. 2000;12:615–21. doi: 10.1162/089892900562372. [DOI] [PubMed] [Google Scholar]
- Doniger GM, Foxe JJ, Schroeder CE, Murray MM, Higgins BA, Javitt DC. Visual perceptual learning in human object recognition areas: a repetition priming study using high-density electrical mapping. Neuroimage. 2001a;13:305–13. doi: 10.1006/nimg.2000.0684. [DOI] [PubMed] [Google Scholar]
- Doniger GM, Silipo G, Rabinowicz EF, Snodgrass JG, Javitt DC. Impaired sensory processing as a basis for object-recognition deficits in schizophrenia. Am J Psychiatry. 2001b;158:1818–26. doi: 10.1176/appi.ajp.158.11.1818. [DOI] [PubMed] [Google Scholar]
- Doniger GM, Foxe JJ, Murrary MM, Higgins BA, Javitt DC. Impaired visual object recognition and dorsal/ventral stream interaction in schizophrenia. Arch Gen Psychiatry. 2002;59:1011–20. doi: 10.1001/archpsyc.59.11.1011. [DOI] [PubMed] [Google Scholar]
- Ellemberg D, Hammarrenger B, Lepore F, Roy MS, Guillemot JP. Contrast dependency of VEPs as a function of spatial frequency: the parvocellular and magnocellular contributions to human VEPs. Spat Vis. 2001;15:99–111. doi: 10.1163/15685680152692042. [DOI] [PubMed] [Google Scholar]
- First MB, Spitzer RL, Gibbon M, Williams JBW. Structured clinical interview for DSM-IV axis I disorders-patient edition. New York: New York State Psychiatric Institute; 1997. [Google Scholar]
- Fox K, Sato H, Daw N. The effect of varying stimulus intensity on NMDA-receptor activity in cat visual cortex. J Neurophysiol. 1990;64:1413–28. doi: 10.1152/jn.1990.64.5.1413. [DOI] [PubMed] [Google Scholar]
- Foxe JJ, Simpson GV, Ahlfors SP. Parieto-occipital approximately 10 Hz activity reflects anticipatory state of visual attention mechanisms. Neuroreport. 1998;9:3929–33. doi: 10.1097/00001756-199812010-00030. [DOI] [PubMed] [Google Scholar]
- Foxe JJ, Doniger GM, Javitt DC. Early visual processing deficits in schizophrenia: impaired PI generation revealed by high-density electrical mapping. Neuroreport. 2001;12:3815–20. doi: 10.1097/00001756-200112040-00043. [DOI] [PubMed] [Google Scholar]
- Foxe JJ, Simpson GV. Flow of activation from V1 to frontal cortex in humans. A framework for defining ‘early’ visual processing. Exp Brain Res. 2002;142:139–50. doi: 10.1007/s00221-001-0906-7. [DOI] [PubMed] [Google Scholar]
- Foxe JJ, Murray MM, Javitt DC. Filling-in in schizophrenia: a high-density electrical mapping and source-analysis investigation of illusory contour processing. Cereb Cortex. 2005;15:1914–27. doi: 10.1093/cercor/bhi069. [DOI] [PubMed] [Google Scholar]
- Freedman B, Chapman LJ. Early subjective experience in schizophrenic episodes. J Abnorm Psychol. 1973;82:46–54. doi: 10.1037/h0034952. [DOI] [PubMed] [Google Scholar]
- Freedman BJ. The subjective experience of perceptual and cognitive disturbances in schizophrenia. Arch Gen Psychiatry. 1974;30:333–40. doi: 10.1001/archpsyc.1974.01760090047008. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Selemon LD. Functional and anatomical aspects of prefrontal pathology in schizophrenia. Schizophr Bull. 1997;23:437–58. doi: 10.1093/schbul/23.3.437. [DOI] [PubMed] [Google Scholar]
- Gomez Gonzalez CM, Clark VP, Fan S, Luck SJ, Hillyard SA. Sources of attention-sensitive visual event-related potentials. Brain Topogr. 1994;7:41–51. doi: 10.1007/BF01184836. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Hernandez JA, Pita-Alcorta C, Cedeno I, Dias-Comas L, Figueredo-Rodriguez P. Abnormal functional asymmetry in occipital areas may prevent frontotemporal regions from achieving functional laterality during the WCST performance in patients with schizophrenia. Schizophr Res. 2003;61:229–33. doi: 10.1016/s0920-9964(02)00236-0. [DOI] [PubMed] [Google Scholar]
- Granholm E, Perry W, Filoteo JV, Braff D. Hemispheric and attentional contributions to perceptual organization deficits on the global-local task in schizophrenia. Neuropsychology. 1999;13:271–81. doi: 10.1037//0894-4105.13.2.271. [DOI] [PubMed] [Google Scholar]
- Green MF, Nuechterlein KH, Mintz J. Backward masking in schizophrenia and mania. II. Specifying the visual channels. Arch Gen Psychiatry. 1994;51:945–51. doi: 10.1001/archpsyc.1994.03950120017004. [DOI] [PubMed] [Google Scholar]
- Green MF, Nuechterlein KH, Breitmeyer B. Backward masking performance in unaffected siblings of schizophrenic patients. Evidence for a vulnerability indicator. Arch Gen Psychiatry. 1997;54:465–72. doi: 10.1001/archpsyc.1997.01830170091012. Published erratum appears in Arch Gen Psychiatry 1997; 54: 846. [DOI] [PubMed] [Google Scholar]
- Harvey PD, Keefe RS, Moskowitz J, Putnam KM, Mohs RC, Davis KL. Attentional markers of vulnerability to schizophrenia: performance of medicated and unmedicated patients and normals. Psychiatry Res. 1990;33:179–88. doi: 10.1016/0165-1781(90)90072-d. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Arana GW, Rosenbaum JF. Handbook of psychiatric drug therapy. Boston: Little, Brown and Company; 1995. [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–8. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Jeffreys DA, Axford JG. Source locations of pattern-specific components of human visual evoked potentials. I. Component of striate cortical origin. Exp Brain Res. 1972;16:1–21. doi: 10.1007/BF00233371. [DOI] [PubMed] [Google Scholar]
- Jibson MD, Tandon R. New atypical antipsychotic medications. J Psychiatr Res. 1998;32:215–28. doi: 10.1016/s0022-3956(98)00023-5. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Lowery N, Kohler C, Turetsky BI. Global-local visual processing in schizophrenia: evidence for an early visual processing deficit. Biol Psychiatry. 2005;58:937–46. doi: 10.1016/j.biopsych.2005.04.053. [DOI] [PubMed] [Google Scholar]
- Kaplan E. The receptive field structure of retinal ganglion cells in cat and monkey. In: Leventhal AG, editor. Vision and visual dysfunction. Boston: CRC Press; 1991. pp. 10–40. [Google Scholar]
- Keri S, Antal A, Szekeres G, Benedek G, Janka Z. Spatiotemporal visual processing in schizophrenia. J Neuropsychiatry Clin Neurosci. 2002;14:190–6. doi: 10.1176/jnp.14.2.190. [DOI] [PubMed] [Google Scholar]
- Keri S, Kelemen O, Benedek G, Janka Z. Vernier threshold in patients with schizophrenia and in their unaffected siblings. Neuropsychology. 2004;18:537–42. doi: 10.1037/0894-4105.18.3.537. [DOI] [PubMed] [Google Scholar]
- Keri S, Kiss I, Kelemen O, Benedek G, Janka Z. Anomalous visual experiences, negative symptoms, perceptual organization and the magnocellular pathway in schizophrenia: a shared construct? Psychol Med. 2005;35:1445–55. doi: 10.1017/S0033291705005398. [DOI] [PubMed] [Google Scholar]
- Kim D, Zemon V, Saperstein A, Butler PD, Javitt DC. Dysfunction of early-stage visual processing in schizophrenia: harmonic analysis. Schizophr Res. 2005;76:55–65. doi: 10.1016/j.schres.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Kim D, Wylie G, Pasternak R, Butler PD, Javitt DC. Magnocellular contributions to impaired motion processing in schizophrenia. Schizophr Res. 2006;82:1–8. doi: 10.1016/j.schres.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klosterkotter J, Hellmich M, Steinmeyer EM, Schultze-Lutter F. Diagnosing schizophrenia in the initial prodromal phase. Arch Gen Psychiatry. 2001;58:158–64. doi: 10.1001/archpsyc.58.2.158. [DOI] [PubMed] [Google Scholar]
- Kutas M, Iragui V, Hillyard SA. Effects of aging on event-related brain potentials (ERPs) in a visual detection task. Electroencephalogr Clin Neurophysiol. 1994;92:126–39. doi: 10.1016/0168-5597(94)90053-1. [DOI] [PubMed] [Google Scholar]
- Kwon YH, Nelson SB, Toth LJ, Sur M. Effect of stimulus contrast and size on NMDA receptor activity in cat lateral geniculate nucleus. J Neurophysiol. 1992;68:182–96. doi: 10.1152/jn.1992.68.1.182. [DOI] [PubMed] [Google Scholar]
- Lamme VA, Roelfsema PR. The distinct modes of vision offered by feedforward and recurrent processing. Trends Neurosci. 2000;23:571–9. doi: 10.1016/s0166-2236(00)01657-x. [DOI] [PubMed] [Google Scholar]
- Legge G. Sustained and transient mechanisms in human vision: temporal and spatial properties. Vision Res. 1978;18:69–82. doi: 10.1016/0042-6989(78)90079-2. [DOI] [PubMed] [Google Scholar]
- Li CS. Impaired detection of visual motion in schizophrenia patients. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:929–34. doi: 10.1016/s0278-5846(02)00207-5. [DOI] [PubMed] [Google Scholar]
- Lund JS. Organization of neurons in the visual cortex, area 17, of the monkey (Macaca mulatta) J Comp Neurol. 1973;147:455–96. doi: 10.1002/cne.901470404. [DOI] [PubMed] [Google Scholar]
- Mangun GR, Hillyard SA, Luck SJ. Electrocortical substrates of visual selective attention. In: Meyer DE, Kornblum S, editors. Attention and performance XIV: synergies in experimental psychology, artificial intelligence, and cognitive neuroscience. Cambridge, MA: MIT Press; 1993. pp. 219–43. [Google Scholar]
- Martinez A, Anllo-Vento L, Sereno MI, Frank LR, Buxton RB, Dubowitz DJ, et al. Involvement of striate and extrastriate visual cortical areas in spatial attention. Nat Neurosci. 1999;2:364–9. doi: 10.1038/7274. [DOI] [PubMed] [Google Scholar]
- Martinez A, Di Russo F, Anllo-Vento L, Hillyard SA. Electrophysiological analysis of cortical mechanisms of selective attention to high and low spatial frequencies. Clin Neurophysiol. 2001a;112:1980–98. doi: 10.1016/s1388-2457(01)00660-5. [DOI] [PubMed] [Google Scholar]
- Martinez A, DiRusso F, Anllo-Vento L, Sereno MI, Buxton RB, Hillyard SA. Putting spatial attention on the map: timing and localization of stimulus selection processes in striate and extrastriate visual areas. Vision Res. 2001b;41:1437–57. doi: 10.1016/s0042-6989(00)00267-4. [DOI] [PubMed] [Google Scholar]
- Matsuoka H, Saito H, Ueno T, Sato M. Altered endogenous negativities of the visual event-related potential in remitted schizophrenia. Electroencephalogr Clin Neurophysiol. 1996;100:18–24. doi: 10.1016/0168-5597(95)00212-x. [DOI] [PubMed] [Google Scholar]
- McGhie A, Chapman J. Disorders of attention and perception in early schizophrenia. Br J Med Psychol. 1961;34:103–16. doi: 10.1111/j.2044-8341.1961.tb00936.x. [DOI] [PubMed] [Google Scholar]
- Merigan WH, Maunsell JHR. How parallel are the primate visual pathways? Annu Rev Neurosci. 1993;16:369–402. doi: 10.1146/annurev.ne.16.030193.002101. [DOI] [PubMed] [Google Scholar]
- Nowak LG, Bullier J. The timing of information transfer in the visual system. In: Rockland K, Kass J, Peters A, editors. Cerebral cortex Vol 12 extrastriate cortex. New York: Plenum Press; 1997. pp. 205–41. [Google Scholar]
- O'Donnell BF, Swearer JM, Smith LT, Nestor PG, Shenton ME, McCarley RW. Selective deficits in visual perception and recognition in schizophrenia. Am J Psychiatry. 1996;153:687–92. doi: 10.1176/ajp.153.5.687. [DOI] [PubMed] [Google Scholar]
- O'Donnell BF, Potts GF, Nestor PG, Stylianopoulos KC, Shenton ME, McCarley RW. Spatial frequency discrimination in schizophrenia. J Abnorm Psychol. 2002;111:620–5. doi: 10.1037//0021-843x.111.4.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- Peuskens J, Link CGG. A comparison of quetiapine and chlorpromazine in the treatment of schizophrenia. Acta Psychiatr Scand. 1997;96:265–73. doi: 10.1111/j.1600-0447.1997.tb10162.x. [DOI] [PubMed] [Google Scholar]
- Place EJ, Gilmore GC. Perceptual organization in schizophrenia. J Abnorm Psychol. 1980;89:409–18. doi: 10.1037//0021-843x.89.3.409. [DOI] [PubMed] [Google Scholar]
- Potts GF, O'Donnell BF, Hirayasu Y, McCarley RW. Disruption of neural systems of visual attention in schizophrenia. Arch Gen Psychiatry. 2002;59:418–24. doi: 10.1001/archpsyc.59.5.418. [DOI] [PubMed] [Google Scholar]
- Previc FH. The neurophysiological significance of the N1 and P1 components of the visual evoked potential. Clin Vis Sci. 1988;3:195–202. [Google Scholar]
- Rassovsky Y, Green MF, Nuechterlein KH, Breitmeyer B, Mintz J. Modulation of attention during visual masking in schizophrenia. Am J Psychiatry. 2005;162:1533–5. doi: 10.1176/appi.ajp.162.8.1533. [DOI] [PubMed] [Google Scholar]
- Sawatari A, Callaway EM. Convergence of magno- and parvocellular pathways in layer 4B of macaque primary visual cortex. Nature. 1996;380:442–6. doi: 10.1038/380442a0. [DOI] [PubMed] [Google Scholar]
- Schechter I, Butler PD, Zemon VM, Revheim N, Saperstein AM, Jalbrzikowski M, et al. Impairments in generation of early-stage transient visual evoked potentials to magno- and parvocellular-selective stimuli in schizophrenia. Clin Neurophysiol. 2005;116:2204–15. doi: 10.1016/j.clinph.2005.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherg M. Fundamental of dipole source analysis. In: Grandori F, Hoke M, Romani GL, editors. Auditory evoked magnetic fields and electric potentials. Basel: Karger; 1990. pp. 40–69. [Google Scholar]
- Schmolesky MT, Wang Y, Hanes DP, Thompson KG, Leutgeb S, Schall JD, et al. Signal timing across the macaque visual system. J Neurophysiol. 1998;79:3272–8. doi: 10.1152/jn.1998.79.6.3272. [DOI] [PubMed] [Google Scholar]
- Schroeder CE, Mehta AD, Givre SJ. A spatiotemporal profile of visual system activation revealed by current source density analysis in the awake macaque. Cereb Cortex. 1998;8:575–92. doi: 10.1093/cercor/8.7.575. [DOI] [PubMed] [Google Scholar]
- Schwartz BD, Maron BA, Evans WJ, Winstead DK. High velocity transient visual processing deficits diminish ability of patients with schizophrenia to recognize objects. Neuropsychiatry Neuropsychol Behav Neurol. 1999;12:170–7. [PubMed] [Google Scholar]
- Selemon LD, Rajkowska G, Goldman-Rakic PS. Abnormally high neuronal density in the schizophrenic cortex. A morphometric analysis of prefrontal area 9 and occipital area 17. Arch Gen Psychiatry. 1995;52:805–18. doi: 10.1001/archpsyc.1995.03950220015005. discussion 819–20. [DOI] [PubMed] [Google Scholar]
- Silverstein SM, Knight RA, Schwarzkopf SB, West LL, Osborn LM, Kamin D. Stimulus configuration and context effects in perceptual organization in schizophrenia. J Abnorm Psychol. 1996;105:410–20. doi: 10.1037//0021-843x.105.3.410. [DOI] [PubMed] [Google Scholar]
- Simpson GV, Foxe JJ, Vaughan HG, Jr, Mehta AD, Schroeder CE. Integration of electrophysiological source analyses, MRI and animal models in the study of visual processing and attention. EEG Clin Neurophysiol. 1995;44(Suppl):76–92. [PubMed] [Google Scholar]
- Slaghuis WL. Contrast sensitivity for stationary and drifting spatial frequency gratings in positive- and negative-symptom schizophrenia. J Abnorm Psychol. 1998;107:49–62. doi: 10.1037//0021-843x.107.1.49. [DOI] [PubMed] [Google Scholar]
- Spencer KM, Nestor PG, Niznikiewicz MA, Salisbury DF, Shenton ME, McCarley RW. Abnormal neural synchrony in schizophrenia. J Neurosci. 2003;23:7407–11. doi: 10.1523/JNEUROSCI.23-19-07407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strandburg RJ, Marsh JT, Brown WS, Asarnow RF, Guthrie D, Higa J, et al. Reduced attention-related negative potentials in schizophrenic adults. Psychophysiology. 1994;31:272–81. doi: 10.1111/j.1469-8986.1994.tb02216.x. [DOI] [PubMed] [Google Scholar]
- Stuve TA, Friedman L, Jesberger JA, Gilmore GC, Strauss ME, Meltzer HY. The relationship between smooth pursuit performance, motion perception and sustained visual attention in patients with schizophrenia and normal controls. Psychol Med. 1997;27:143–52. doi: 10.1017/s0033291796004230. [DOI] [PubMed] [Google Scholar]
- Tootell RB, Hamilton SL, Switkes E. Functional anatomy of macaque striate cortex. IV. Contrast and magno-parvo streams. J Neurosci. 1988a;8:1594–609. doi: 10.1523/JNEUROSCI.08-05-01594.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tootell RB, Silverman MS, Hamilton SL, Switkes E, De Valois RL. Functional anatomy of macaque striate cortex. V. Spatial frequency. J Neurosci. 1988b;8:1610–24. doi: 10.1523/JNEUROSCI.08-05-01610.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol. 2002;42:165–79. doi: 10.1146/annurev.pharmtox.42.082701.160735. [DOI] [PubMed] [Google Scholar]
- Ungerleider LG, Mishkin M. Two cortical visual systems. In: Ingle DJ, Mansfield RJW, Goodale MS, editors. The analysis of visual behavior. Cambridge, MA: MIT Press; 1982. pp. 549–86. [Google Scholar]
- van der Stelt O, Frye J, Lieberman JA, Belger A. Impaired P3 generation reflects high-level and progressive neurocognitive dysfunction in schizophrenia. Arch Gen Psychiatry. 2004;61:237–48. doi: 10.1001/archpsyc.61.3.237. [DOI] [PubMed] [Google Scholar]
- Vidyasagar TR. A neuronal model of attentional spotlight: parietal guiding the temporal. Brain Res Brain Res Rev. 1999;30:66–76. doi: 10.1016/s0165-0173(99)00005-3. [DOI] [PubMed] [Google Scholar]
- Vidyasagar TR, Kulikowski JJ, Lipnicki DM, Dreher B. Convergence of parvocellular and magnocellular information channels in the primary visual cortex of the macaque. Eur J Neurosci. 2002;16:945–56. doi: 10.1046/j.1460-9568.2002.02137.x. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Gallhofer B. Cognitive function in schizophrenia. Vol. 12. 1997. pp. S29–36. [DOI] [PubMed] [Google Scholar]
- Woods SW. Chlorpromazine equivalent doses for the newer atypical antipsychotics. J Clin Psychiatry. 2003;64:663–7. doi: 10.4088/jcp.v64n0607. [DOI] [PubMed] [Google Scholar]
- Yeap S, Kelly SP, Sehatpour P, Magno E, Javitt DC, Garavan H, et al. Are early visual sensory deficits endophenotypic for schizophrenia? High-density electrical mapping in clinically unaffected first-degree relatives. Arch Gen Psychiatry. 2006 doi: 10.1001/archpsyc.63.11.1180. In press. [DOI] [PubMed] [Google Scholar]
- Zemon V, Gordon J, Welch J. Asymmetries in ON and OFF visual pathways of humans revealed using contrast-evoked cortical potentials. Vis Neurosci. 1988;1:145–50. doi: 10.1017/s0952523800001085. [DOI] [PubMed] [Google Scholar]
- Zemon V, Gordon J. Luminance contrast mechanisms in humans: visual evoked potentials and a nonlinear model. Vision Res. 2006 doi: 10.1016/j.visres.2006.07.007. In press. [DOI] [PubMed] [Google Scholar]