

Abstract
Over the past decade, many lines of investigation have shown that receptor-mediated signaling exhibits greater diversity than previously appreciated. Signal diversity arises from numerous factors, which include the formation of receptor dimers and interplay between different receptors. Using adenosine A1 receptors as a paradigm of G protein-coupled receptors, this review focuses on how receptor-receptor interactions may contribute to regulation of the synaptic transmission within the central nervous system. The interactions with metabotropic dopamine, adenosine A2A, A3, neuropeptide Y, and purinergic P2Y1 receptors will be described in the first part. The second part deals with interactions between A1Rs and ionotropic receptors, especially GABAA, NMDA, and P2X receptors as well as ATP-sensitive K+ channels. Finally, the review will discuss new approaches towards treating neurological disorders.
Keywords: Adenosine, G protein-coupled receptors, Receptor interactions, Neurotransmission, Adenosine receptors, Ionotropic receptors
Introduction
The vertebrate central nervous system (CNS) is characterized by a dynamic interplay between signal transduction molecules and their cellular targets. Modulation of synaptic transmission by metabotropic or ionotropic receptors is an important source of control and dynamical adjustment in synaptic activity.
Recent studies have provided new insights into the role of ligand-gated ion channels in modifying synaptic transmission. Along with a growing list of different types of pre- and postsynaptic ionotropic receptors and the cell types that express them, there have also been advances in characterizing the modulatory mechanisms of the receptors that link to receptor activation. This is important due to the convergence of data from biochemical, molecular, and electrophysiological studies, implicating ionotropic receptors in the effects of psychoactive and addictive drugs.
G protein-coupled receptors (GPCRs) make up the largest and most diverse family of membrane receptors in the human genome, relaying information on the presence of diverse extracellular stimuli to the cell interior. An estimated 1% of the mammalian genome encodes for GPCRs, and about 450 of the approximately 950 predicted human GPCRs are thought to be receptors for endogenous ligands [1]. The manipulation of transmembrane signaling by GPCRs may constitute the most important therapeutic target in medicine. Nearly 40% of all current therapeutic drugs target GPCRs [2].
All known GPCRs share a common architecture of seven membrane-spanning helices connected by intracellular and extracellular loops. Drugs acting on GPCRs have been classified as agonists, partial agonists, or antagonists based on a “two-state model of receptor function.” Since experimental evidence pointed out the impossibility of explaining the operation of GPCRs without considering dimers as the minimum structure for many GPCRs the “two-state dimer receptor model” was developed based on the communication between the two subunits of the receptor dimmer [1, 3, 4]. This model is an extension of the “two-state model of receptor function” but considers dimeric structures able to bind one molecule to the orthosteric center in each monomer.
GPCR signaling is subject to extensive negative regulation through receptor desensitization, sequestration, and downregulation, termination of G protein activation by GTPase-activating proteins, and enzymatic degradation of second messengers. Additional protein-protein interactions positively modulate GPCR signaling by influencing ligand binding and specificity.
Multiprotein complexes mediate most cellular functions. In neurons, these complexes are directly involved in the neuronal transmission, which is responsible for learning, memory, and developments. The first publication in this direction came from Hökfelt’s group in 1983. The publication describes how substance P may modulate the high-affinity serotonin (5-HT) binding site in a spinal cord membrane preparation [5]. Over the past decade, the number and outcomes of interactions between receptors have increased continuously [6]. Recent studies have demonstrated close physical interactions where activation of one receptor affects the function of the other.
Adenosine is an endogenous purine nucleoside that has evolved to modulate many physiological processes. Extracellular adenosine mostly originates from release of intracellular adenosine and from release and extracellular breakdown of cAMP and ATP by ecto-5′-nucleotidase and phosphodiesterase [7]. Cellular signaling by adenosine occurs through four known adenosine receptor subtypes (A1Rs, A2ARs, A2BRs, and A3Rs), all of which are seven-transmembrane-spanning GPCRs. Of the four known adenosine receptors, A1Rs and A2ARs are primarily responsible for the central effects of adenosine, especially in modulating synaptic transmission [8]. Adenosine can act on A1Rs to depress transmitter release and neuronal sensitivity to the transmitter [9, 10]. As a result, the A1Rs are important in the regulation of synaptic plasticity, playing a role in determining the amplitude of long-term potentiation or long-term depression [11].
There are numerous reviews that describe regulation of brain adenosine levels, adenosine receptors, their cellular and subcellular localization, signaling pathways, and function in the brain under physiological and pathophysiological conditions as well as selective receptor agonists and antagonists. Using A1Rs as a paradigm of GPCRs, this review focuses on how receptor-receptor interactions contribute to regulatory processes within the central nervous system.
Considering the various types of receptors, one may expect to find three principle paths of receptor interaction: (1) interactions between ionotropic receptors, (2) interactions between a metabotropic receptor and an ionotropic receptor, and (3) interactions between metabotropic receptors. The examples mentioned below stem from the second and third type of interaction. Interactions with metabotropic dopamine receptors as well as A2A, A3, NPY, and P2Y1 receptors will be described in the first part. The second part deals with interactions between A1Rs and ionotropic receptors, especially the GABAA, NMDA, and P2X receptors as well as ATP-sensitive K+ channels. Finally, new approaches for neurological disorders will be discussed.
Functional interactions with metabotropic receptors
Two forms of GPCR classification exist. There is the historical division into three main families: (1) rhodopsin-like family which includes adenosine receptors, (2) secretin-like family, and (3) metabotropic glutamate receptor-like family. The families share some basic similarities—the seven-transmembrane-spanning domains, intracellularly located C terminus, and extracellularly residing N terminus. Differences between the families arise in the length of the intracellular and extracellular termini and amino acid sequences, disulfide bridge linking, and conserved domains. Five different groups can be classified by applying phylogenetic analyses. The GRAFS system distinguishes between glutamate, rhodopsin, adhesion, fizzled/taste, and secretin-like GPCRs [12].
The agonist binding on the receptor results in coupling to heterotrimeric G proteins and regulates a variety of cell responses. In brief, an exchange of G protein-bound GDP to GTP occurs, and the heterotrimer dissociates into the α subunit and the βχ dimer. The resulting products activate or inhibit effectors independently from each other. Currently, 16 different genes encode G protein α subunits, five genes encode β subunits, while 14 genes encode χ subunits [13]. The α subunits can be categorized into four basic groups: the stimulatory 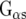 family couples to adenylate cyclase and increases cAMP levels, whereas the inhibitory
family couples to adenylate cyclase and increases cAMP levels, whereas the inhibitory 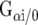 family acts in the opposite way. Moreover, the
family acts in the opposite way. Moreover, the 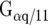 family activates the phospholipase
family activates the phospholipase 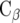 (
(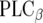 ), and lastly, the
), and lastly, the 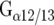 family which regulates Rho proteins.
family which regulates Rho proteins. 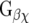 dimers are capable of triggering effects on inward rectifier K+ channels (GIRK1–4), voltage-dependent Ca2+ channels (VDCC), and phospholipase A2 (PLA2),
dimers are capable of triggering effects on inward rectifier K+ channels (GIRK1–4), voltage-dependent Ca2+ channels (VDCC), and phospholipase A2 (PLA2), 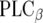 , and the Na+/H+ exchanger (NHE1). Thus, it is not surprising that GPCRs are such interesting candidates in current drug research with their amazing potential in affecting signaling events. A single GPCR possesses the potential to activate more than just one signaling pathway [12]; for example, activation of A1Rs includes coupling to
, and the Na+/H+ exchanger (NHE1). Thus, it is not surprising that GPCRs are such interesting candidates in current drug research with their amazing potential in affecting signaling events. A single GPCR possesses the potential to activate more than just one signaling pathway [12]; for example, activation of A1Rs includes coupling to 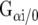 and increasing of IP3 level [14, 15]. Furthermore, homodimerization and heterodimerization are common paths of interaction and have been described in A1Rs and the A2Rs several times [16–18]. In addition, functional interactions on the A1R without receptor assembling have already been revealed [19, 20], or are currently being elucidated. The next paragraphs will deal with a few selected examples of this limitlessly wide topic.
and increasing of IP3 level [14, 15]. Furthermore, homodimerization and heterodimerization are common paths of interaction and have been described in A1Rs and the A2Rs several times [16–18]. In addition, functional interactions on the A1R without receptor assembling have already been revealed [19, 20], or are currently being elucidated. The next paragraphs will deal with a few selected examples of this limitlessly wide topic.
Relationship between A1Rs and A3Rs
The A3R was the latest receptor subtype of the adenosine receptor family to be identified [21], and its functional role is still controversially discussed. Several findings indicate neuroprotective as well as neurotoxic action depending on experimental approach [22–29]. A3Rs couple to inhibition of adenylyl cyclase as well as to activation of PLC, and to elevation of inositol triphosphate levels [30, 31]. Furthermore, an increase in intracellular Ca2+ levels due to release from intracellular stores and Ca2+ influx has been described [32, 33]. One interesting example of A3Rs’ functional role is their involvement in acute neurotoxic situations and interplay with A1Rs. Dunwiddie et al. [34] reported on the potential of A3Rs to modify responses via A1Rs in the hippocampus. The activation of hippocampal A3Rs induced a desensitization of A1Rs on combined superfusion of Cl-IB-MECA and adenosine. This phenomenon was thought to reduce the protective effects of endogenous adenosine caused by the lack of sensitivity of A1Rs. Further investigations on pyramidal cells of the rat cingulate cortex did not confirm Dunwiddie et al.’s assumption [35]. In this brain area, A1Rs and A3Rs did not show any interaction. The receptor subtypes were unable to affect each other. The discrepancy was taken to be a genetic phenomenon, such as alternative splicing of the rat A3R transcript causing distinguished pharmacological and functional properties in the brain. Furthermore, Hentschel et al. [36] demonstrated the involvement of A3Rs in inhibition of excitatory neurotransmission during hypoxic conditions, indicating a neuroprotective action of endogenously released adenosine on A3Rs in addition to A1Rs. Lastly, Lopes et al. [37] attempted to define the possible role of A3Rs in the rat hippocampus using experiments similar to those of Dunwiddie et al. in non-stressful and stressful situations, with particular attention to whether A3Rs control A1Rs. These data suggested that no interaction between the two receptor subtypes exist, but confirm that A3Rs do not affect synaptic transmission on superfusion with A3R agonist Cl-IB-MECA or A3R antagonist MRS 1191. The authors pointed out that Cl-IB-MECA binds to A1Rs even at low nanomolar concentrations. Thus, the existence of an interaction between A1Rs and A3Rs has to wait for reliable ligands.
Antagonistic interaction between A1Rs and A2ARs
A2ARs are widely distributed in the CNS, but local and subcellular differences in allocation exist. They show high levels in all subregions of the striatum and in the globus pallidus. A2ARs are also expressed in neurons in the neocortex and limbic cortex, but at a density a twentieth of that found in basal ganglia [38]. Colocalization of A1Rs and A2ARs was approved for glutamatergic nerve terminals in the hippocampus [39]. In the striatum, A1R/A2AR heteromers were found on synapses with spines of medium spiny neurons and integrated in the presynaptic membrane of glutamatergic terminals that represent the cortical-limbic-thalamic input [18]. A1Rs and A2ARs modulate excitatory synaptic transmission, albeit in an opposite manner. A1R activation inhibited glutamatergic synaptic transmission mainly through presynaptic inhibition of glutamate release, while A2ARs have been shown to facilitate glutamatergic synaptic transmission [40–42]. At first sight, stimulating A1Rs and inhibiting A2ARs may have a neuroprotective influence on the mature CNS. However, problems arise due to long-term desensitization of A1Rs. A2ARs do not upregulate after antagonist administration, but have a low abundance in hippocampal and cortical areas compared with A1Rs [40, 43, 44]. A1Rs and A2ARs cannot be regarded in isolation from one another since cross talk between the subtypes has been described several times [16, 17, 45–47]. A2AR activation by agonists caused A1R desensitization resulting in decreased binding affinity for CPA in the hippocampus in young adult rats. Controlling A1Rs by A2ARs was mediated by protein kinase C in a cAMP-independent manner. A2AR activation was seen to play a role in fine-tuning A1Rs by attenuating the tonic effect of presynaptic A1Rs located on glutamatergic nerve terminals [46, 47]. In the striatal system, A1R/A2AR heteromers became prominent to show an antagonistic reciprocal interaction [18]. As in the hippocampus, A2AR stimulation decreased the affinity of A1Rs for agonists. The A1R/A2AR heteromer allows adenosine to perform a detailed modulation of glutamate release [16, 48]. Regarding A1Rs and A2ARs, basal conditions generate a low tone of endogenous adenosine and cause A1R activation, in contrast to situations of increased adenosine where A2AR activation becomes dominant. When adenosine concentrations rise, as during anoxia, likely also time appears to be important in regulating A2A receptor activity, which means A2A receptors are “active” under prolonged stimulation [49]. Finally, activation of the A1R/A2AR heteromer contributes to A2AR signaling when adenosine levels are elevated and may provide a mechanism to facilitate plastic changes in the excitatory synapse [18].
Interactions between adenosine and dopaminergic system
Dopamine is an important transmitter in basal ganglia and is noted for influencing motor activity, playing an important role in Parkinson’s disease. Adenosine-dopamine interactions are complex and cannot be limited on functional considerations of A1Rs. Intramembrane heteromeric receptor-receptor interactions and the involvement of A2ARs in influencing dopaminergic signaling have to be mentioned due to the implications in the treatment of Parkinson’s disease. Ginés et al. [50] described the formation of functionally interacting heteromeric complexes between dopamine D1 receptors (D1Rs) and A1Rs in mouse fibroblast Ltk− cells cotransfected with respective cDNAs. Coaggregation occurred when cells were pretreated with R-PIA as A1R agonist, but was decreased by combined pretreatment with R-PIA and SKF-38393, a D1R agonist. Furthermore, the D1R agonist-induced cAMP accumulation was reduced by combined pretreatment of D1R- and A1R agonist, but remained unaffected when given alone, respectively. The results confirmed an antagonistic interaction between A1Rs and D1Rs that had already been observed by Ferré et al. [51] in behavioral studies using reserpinized mice and rabbits. In vivo and in vitro data on adenosine-dopamine interactions were mostly obtained from investigations in the basal ganglia and limbic regions [52, 53] due to the high abundance of A1Rs, A2ARs, D1Rs, and D2Rs in these areas and their involvement in the pathology of Parkinson’s disease. The antagonistic interaction of combined receptor activation seems to distinguish between adenosine and dopamine receptor subtypes. While A1Rs communicate mainly with the D1R subtype in strionigral-strioentopenduncular neurons, A2AR and D2R interaction occurs in striopallidal neurons. Studies on mice and monkeys pretreated with MPTP suggest that some degree of dopaminergic activity is needed to obtain adenosine antagonistic-induced motor activity. Furthermore, blockade of dopaminergic neurotransmission counteracts the antagonistic effect induced by adenosine [54]. Sufficient endogenous adenosine is present interstitially in the substantia nigra pars reticulata to control dopaminergic effects. The effects of adenosine are absent when dopaminergic influence is suppressed [53]. Thus, it seems that monotherapy with A2AR antagonists may only be useful in the early stages of Parkinson’s disease, but could support a therapeutic treatment with dopamine agonists in advanced stages. A promising approach using these therapeutic strategies can be seen in istradefylline, an A2AR antagonist that has since successfully passed clinical trials [55]. However, A1R blockade may also contribute to an increased dopamine release but this effect seems without clinical relevance.
Relationship between A1Rs and NPY
Neuropeptide Y (NPY) is one of the most abundant neuropeptides and exerts various functions on at least six GPCR subtypes (Y1Rs-Y5Rs, y6Rs). Immunohistochemical investigations revealed the appearance of the Y1R and Y5R subtypes in the rat frontal cortex [56–58]. Activation of NPY receptors results in an inhibition of excitatory synaptic transmission, while a presynaptic influence on cortical neurons has been postulated [59]. NPY receptors affect pertussis toxin-sensitive G proteins, which inhibit adenylyl cyclase and decrease cAMP levels. Inhibitory and facilitating effects on K+ and Ca2+ mobilization have also been observed [60]. Receptor-receptor interactions between Y1Rs have already been described, such as the antagonistic interaction with galanin receptors in the hypothalamus of the rat and their functional relevance for food intake. In contrast, a facilitatory interaction between the two receptors exists in the amygdala which may be of relevance for fear-related behavior [61].
In the CNS, A1Rs and NPY receptors share some similarities in distribution. Both A1Rs and Y1Rs are located on neurons of the prefrontal cortex, and their activation inhibits glutamatergic neurotransmission [62]. This is evidence for potential interaction between A1Rs and Y1Rs that may modulate long-term desensitization of A1Rs during pathophysiological situations. To investigate possible functional interactions, postsynaptic potentials (PSPs) were generated by electrical field stimulation on pyramidal neurons of layer V in the rat cingulate cortex as described by Brand et al. [35] and Hentschel et al. [36]. The Y1R agonist [F7,P34]pNPY inhibited the amplitude of PSPs. The inhibitory effect was reversible and reproducible, indicating that no desensitization appeared (Fig. 1a). An additional decrease in PSP amplitude was observed when NPY was superfused in combination with the A1R agonist CPA (Fig. 1b).
Fig. 1.
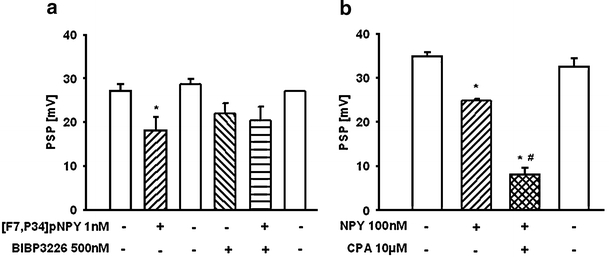
Effect of the selective Y1 agonist [F7,P34]pNPY as well as neuropeptide Y (NPY) alone and in combination with selective A1R agonist N6-cyclopentyladenosine (CPA) on the amplitude of postsynaptic potentials (PSPs) evoked by electrical field stimulation (0.2 Hz, 2 ms) in layer I of the rat cingulate cortex (Sichardt et al., unpublished results). Intracellular recordings were performed in rat brain slices using glass microelectrodes placed in pyramidal cells of layer V. a [F7,P34]pNPY superfused for 5 min inhibits reversibly the PSPs by 34.6 ± 8%. Y1 antagonist BIBP3226 itself reduces the PSP by 23 ± 10%, whereas [F7,P34]pNPY has no inhibitory effect in the presence of the antagonist. b NPY depresses PSPs by 28.6 ± 0.7%. The combined superfusion of NPY and CPA resulted in an additional depression of the PSPs by 48.1 ± 5%. The depressant effects of the two agonists were reversible during washout. Data are expressed as mean ± SEM from n = 3 independent experiments. *p < 0.05 significant vs control; #p < 0.05 significant vs NPY alone
The additional inhibition induced by CPA was in the same range as that found with CPA alone (48.1 ± 5% vs 55 ± 3%). NPY inhibited PSPs after blockading CPA-mediated inhibitory effects by DPCPX. No significant changes existed before and after blockading A1Rs (Fig. 2). The results suggest that no interaction between A1Rs and Y1Rs exist. Each neuromodulator contributes to inhibitory regulation of excitatory neurotransmission. Regarding the desensitization of A1Rs but not of Y1Rs, this may be important under pathophysiological conditions with increased adenosine concentration in the synaptic cleft.
Fig. 2.
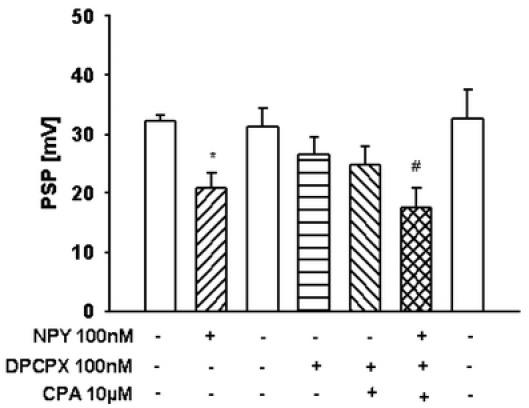
Effect of neuropeptide Y (NPY) on the PSPs alone and in combination with the selective A1R agonist N6-cyclopentyladenosine (CPA) after preincubation with the selective A1R antagonist 1,3-dipropyl-8-cyclopentylxantine (DPCPX) (Sichardt et al., unpublished results). The experimental procedure was similar to that shown in Fig. 1. In the presence of NPY, PSPs were decreased by 35.4 ± 7%. Combined superfusion of NPY and CPA after preincubation with DPCPX decreased PSPs by 32.7 ± 8%. The depression was reversible during washout. Data are expressed as mean ± SEM from n = 5 independent experiments. *p < 0.05 significant vs control; #p < 0.05 significant vs superfusion of CPA and DPCPX
Interaction of A1Rs and P2Y1Rs
P2Y1Rs have been cloned and characterized in several species including human and rat, whereas mRNA was detected in various regions of the brain. The receptor subtype can be activated by ATP, but ADP as a degradation product of ATP is a more potent endogenous agonist. Cellular signaling differs between A1Rs and P2Y1Rs since A1Rs couple to Gi/0 and P2Y1Rs to Gq/G11. In fact, P2Y1Rs can be assumed to exert stimulatory effects in cells. P2Y1R signaling occurs in non-neuronal and non-muscular cell types, as well as on neurons in the CNS [63] where a colocalization of A1Rs and P2Y1Rs was demonstrated immunohistochemically in rat brain cortex, hippocampus, and cerebellum [64]. In 1996, Ikeuchi et al. reported the activation of an undefined P2YR by adenosine in patch clamp and calcium imaging experiments on hippocampal neurons [65]. Furthermore, extensive heteromerization experiments have been conducted on cotransfected HEK293 cells using immunoprecipitation, Western blotting, and bioluminescence resonance energy transfer (BRET). Receptor binding experiments in combination with cAMP assays have also been described [64, 66–70]. These respective studies confirmed heteromerization associated with changes in the agonist binding and signaling compared to monomer properties. The binding for selective A1R agonists was decreased while the A1R antagonist binding remained unaffected. Interestingly, ADP binding was blocked by DPCPX but not by the P2Y1R antagonist, suggesting an altered binding pocket on the A1R/P2Y1R complex. The G protein-coupling was sensitive to pertussis toxin and revealed a Gi/0 status for the heteromer. Although colocalization of A1Rs and P2Y1Rs in several brain areas has been demonstrated, there is still a lack of functional investigations. Nevertheless, the physiological relevance of this interaction has been postulated as follows: costorage and release of ATP with neurotransmitters, such as glutamate or noradrenaline, occurs in the CNS [71–73]. ADP, the degradation product of ATP, acts as an A1R agonist due to the activation of the A1R/P2Y1R complex and contributes to the inhibitory modulation of excitatory synaptic transmission via adenosine acting on A1Rs. This interaction can be assumed as an additional mechanism for influencing and fine-tuning synaptic neurotransmission.
Functional interaction with ionotropic receptors
Neuronal excitability is regulated by voltage and ligand-gated ion channels. Ionotropic receptors also referred to as ligand-gated ion channels (LGICs) are a group of intrinsic transmembrane ion channels that open and close in response to binding of a chemical messenger, as opposed to voltage-gated ion channels or stretch-activated ion channels. Ion channels are regulated by a ligand and are usually very selective to one or more ions such as Na+, K+, Ca2+, or Cl−. These receptors located at synapses convert the chemical signal of presynaptically released neurotransmitter directly and very quickly into a postsynaptic electrical signal. Many LGICs are additionally modulated by allosteric ligands by channel blockers, ions, or membrane potential.
Nicotinic acetylcholine receptor serves as the prototypical LGIC [74] and consists of a pentamer of protein subunits with two binding sites, which, when bound, alter the receptor configuration and cause an internal pore to open. This pore, permeable to Na+ ions, allows them to flow down their electrochemical gradient into the cell. With a sufficient number of channels opening at once, the intracellular Na+ concentration rises to the point at which the positive charge within the cell is sufficient to depolarize the membrane, and an action potential is initiated [75]. Many important ion channels are ligand-gated, and they show a great degree of homology at the genetic level. The LGICs are classified into three superfamilies; the first—the Cys-loop receptor family—is subdivided into the anionic GABAA and glycine receptors on the one hand, and cationic 5-HT3 serotonin and nicotinic acetylcholine receptors on the other. The second group—ionotropic glutamate receptors—consists of NMDA, kainate, and AMPA receptors. The third group covers the ATP-gated channels—the P2X receptors [76].
Adenosine is known to inhibit glutamatergic neurotransmission by activation of presynaptic A1Rs [35]. This is probably due to reduction of the calcium influx, possibly by modulating both P/Q- and N-type presynaptic voltage-dependent calcium channels, which in turn controls transmitter release [77]. Furthermore, A1Rs have long been known to mediate neuroprotection by reduction of excitatory effects at the postsynaptic level [10, 78, 79]. In addition to its direct presynaptic and postsynaptic actions on neurons, A1R interaction with NMDA [80–82, 84], GABAA [85–88], and P2X receptors [80, 89–91] contributes to fine-tuning neuromodulation via adenosine.
Interaction between A1Rs and NMDA receptors
Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system. In most brain areas, glutamate mediates fast synaptic transmission by activating ionotropic receptors of the AMPA, kainate, and NMDA type. Additionally, NMDA receptors play a critical role in synaptic plasticity, synaptic development, and neurotoxicity. Recent studies suggest that some NMDA-mediated actions are altered or mediated by adenosine. Synaptic currents mediated by glutamate in rat substantia nigra pas reticulata neurons were reduced by adenosine acting via A1Rs. The inhibitory action was not mediated by a postsynaptic site since adenosine did not block currents evoked by local application of glutamate [86].
NMDA is known to increase the extracellular level of adenosine via bidirectional adenosine transporters or from released adenine nucleotides degraded by a chain of ectonucleotidases [92, 93]. On the other hand, endogenous adenosine present in the extracellular fluid of hippocampal slices tonically inhibits NMDA receptor-mediated dendritic spikes as well as AMPA/kainate receptor-mediated synchronized EPSPs by activation of A1Rs in CA1 pyramidal cells [81]. In line with these results, it has been shown that the tonic activation of A1Rs by ambient adenosine depressed field potentials in the striatum. The effect of adenosine in the striatum [84] or hippocampus [94] has not been found in A1R knockout mice and clearly demonstrates the involvement of A1Rs. The involvement of A1Rs was also supported by experiments using the selective receptor ligand 2-CA. In isolated rat hippocampal pyramidal cells [95] and in bipolar cells of the retina [96], 2-CA decreased inward currents induced by iontophoretic application of NMDA.
Another interesting interaction concerns NMDA preconditioning to protect against glutamate neurotoxicity. The A1R antagonist 8-CPT has been shown to prevent neuroprotection evoked by NMDA preconditioning against glutamate-induced cellular damage in cerebellar granule cells [83]. In this study, the functionality of A1Rs was not affected by NMDA preconditioning, but this treatment promoted A2AR desensitization in concert with A1R activation [83]. These results are in line with other studies indicating that adenosine downregulates excitatory and inhibitory synaptic transmission in several brain areas through activation of A1Rs and A2AR [97, 98]. Furthermore, activation of A1Rs mediates reversal of long-term potentiation (LTP) produced by brief application of NMDA in hippocampal CA1 neurons [99].
Taken together, there are several ways in which adenosine may interact with NMDA-induced cellular events. Adenosine can affect glutamatergic transmission via both presynaptic and postsynaptic mechanisms by activating A1Rs [78]. NMDA receptors and A1Rs interact to downregulate glutamate release presynaptically in pyramidal cells of the cingulate cortex [35], neurons of the hippocampus [100], and striatal neurons [84]. Another putative mechanism is related to postsynaptic A1Rs; adenosine elevates the threshold to open NMDA receptor-operated channels by antagonizing membrane depolarization [101].
Interaction between A1Rs and GABAA receptors
Fast synaptic inhibition in the brain and spinal cord is largely mediated by GABAA receptors that are also targeted by drugs such as benzodiazepines, barbiturates, neurosteroids, and some anesthetics. The modulation of their function will have important consequences for neuronal excitation [102]. One accepted means of modifying the efficacy is a functional interaction with adenosine. Adenosine may have an effect on either presynaptic GABA release in interneurons and/or on postsynaptic GABAA receptors in projection neurons. The site of action may be studied electrophysiologically by inducing fast inhibitory postsynaptic potentials (IPSPs) or application of GABA directly onto the cell. Adenosine and selective A1R agonist CHA reduced the amplitude of the fast IPSP in lateral amygdala slice preparations. The effect of CHA was blocked by DPCPX, indicating the involvement of A1Rs. Additionally, adenosine did not block currents evoked by local application of GABA [85]. Thus, the modulatory effect of adenosine on the GABAergic neurotransmission appears to take place on a presynaptic site by inhibiting GABA release from nerve terminals [85, 86]. The assumption that the activation of A1Rs can presynaptically modulate inhibitory postsynaptic responses agrees with findings in several brain areas, such as the thalamus [87], suprachiasmatic and arcuate nucleus [88], and substantia nigra pars compacta [86].
There is some evidence that activation of A1Rs is also involved in GABAA receptor downregulation, implying a facilitation of the neurotransmission on a postsynaptic site. GABA but not adenosine evoked an inward current in rat sacral dorsal commissural neurons (SDCN). The GABA-induced current was significantly reduced be adenosine. CHA and DPCPX, but not selective ligands for A2ARs, mimicked or blocked the inhibitory effect of adenosine, respectively [103]. Adenosine and muscimol induced a concentration-dependent reduction in the amplitude of population potentials in hippocampal slices. Additionally, adenosine potentiated the ability of muscimol to inhibit evoked potentials, which were blocked by the A1R-selective antagonist 8-CPT. The effects of adenosine as well as muscimol were reduced by the chloride channel blocker DIDS, indicating the ability of adenosine to regulate the GABAA chloride channel by activation of A1Rs [104].
Sebastiao’s group studied the mechanisms by which GABA modulates adenosine-mediated effects and found that endogenous GABA exerts an inhibitory effect through GABAA receptors via a predominant adenosine-mediated action in the hippocampus. The authors concluded that there is an A1R-mediated ability to inhibit synaptic transmission [19]. Further, this study showed that the blockade of GABAergic inhibition induced the release of NO, which was able to potentiate the inhibitory action of adenosine. They therefore suggested that the modulation of the A1R-mediated response by activation of GABAA receptors occurs indirectly via NO [19].
Activation of GABAA receptors is effective in limiting neuronal ischemic damage [105] and endogenous adenosine that arises during hypoxia, and acts neuroprotectively partly by activating A1Rs [36]. Therefore, the contribution and potential interactions of GABA and adenosine as modulators of synaptic transmission during hypoxia has been investigated. Activation of A1Rs inhibits the release of GABA from the ischemic cerebral cortex in vivo [106]. In contrast, the administration of an A1R agonist in the hippocampus failed to affect the release of GABA during ischemia [107]. In the light of these controversial results, the role of the two neuromodulators during hypoxia was investigated in the CA1 area of rat hippocampal slices using selective A1R antagonists [108]. Indeed, activation of A1R and GABAA receptors is partly involved in the inhibition of synaptic transmission during hypoxia. The action of GABA becomes evident when A1Rs are blocked. Regarding the desensitization of A1Rs during hypoxia [109, 110], it may be assumed that GABAA-mediated inhibition of the synaptic transmission is evident when the A1R is desensitized or downregulated [108].
Comodulation by A1Rs and GABAA receptors was also suggested in acute cerebellar ethanol-induced ataxia. Using GABAA and A1R agonists and antagonists, respectively, a functional similarity between GABAA receptors and A1Rs has been shown even though both receptor types are known to couple to different signaling systems [111]. This provides conclusive evidence that A1Rs and GABAA receptors both play a comodulatory role in ethanol-induced cerebellar ataxia without any direct interaction.
Functional interaction between A1Rs and P2X receptors
P2X receptors are ligand-gated ion channel receptors; seven subunits (P2X1-P2X7) have been identified [63]. The P2X receptor subunits show many differences in localization, pharmacology, kinetics, and signaling pathways [112, 113]. The P2X1 to P2X6 receptors have 379–472 amino acids, with a predicted tertiary structure of transmembrane segments, a large extracellular loop and intracellular C and N termini. The P2X2, P2X4, and P2X4/P2X6 receptors appear to be the predominant neuronal types [91]. These subunits may occur as homooligomers or as heterooligomeric assemblies of more than one subunit. The P2X7 receptor has a similar structure, but with a much larger intracellular C terminus. This contrasts strikingly with any of the other known ligand-gated ionotropic receptors [114]. P2X7 subunits do not form heterooligomeric assemblies, but are involved in mediating apoptosis and necrosis in glial cells and possibly neurons.
Interaction between adenosine receptor-mediated and P2 receptor-mediated effects have been shown to occur in neuronal and non-neuronal cells [80]. Both adenosine and ATP induce astroglial cell proliferation and formation of reactive astrocytes [89]. In hippocampus, adenosine and ATP are released on stimulation and are potent neuronal transmission inhibitors [115, 116]. It should be pointed out that the interpretation of effects induced by both is difficult since ATP is degraded enzymatically to adenosine [38]. Adenosine is formed by extracellular catabolism of released ATP via the ectonucleotidase pathway [90, 117]. The role of the ectonucleotidases in forming adenosine is difficult to study since this system is extremely efficient, and it is difficult to block an enzyme system. The experimental paradigm used by Cunha et al. [118] demonstrates that ATP has to be converted outside the cell into adenosine to exert its inhibitory effects on hippocampal synaptic transmission. The inhibitory effect of ATP was not modified by the P2 receptor antagonist suramin, but was attenuated by the ecto-5’-nucleotidase inhibitor and was nearly prevented by the adenosine A1R antagonist DPCPX, whereas dipyridamole, an inhibitor of adenosine uptake, potentiated the inhibitory effect of ATP [118]. These results offer evidence for localized catabolism of adenine nucleotides followed by substrate channeling to A1Rs. This localized catabolism may mask the adenosine-mediated ATP effect [119]. Recently it was demonstrated that the exogenous application of ATP or ATPχS reduced the hippocampal neurotransmission. The inhibitory effect was blocked by the selective A1R antagonist DPCPX and was potentiated by different ecto-ATPase inhibitors [120]. These results suggest that the synaptic inhibition may consist of an inhibitory purinergic component of ATP itself in addition to degradation to adenosine.
Interaction with neuronal ATP-sensitive K+ channels
ATP-sensitive K+ channels (KATP) are widely expressed in the cytoplasmic membrane of neurons and couple cell metabolism to excitability [121]. These channels are regulated by the intracellular ATP/ADP ratio [122] and modulated by many endogenous mediators, including adenosine, via A1Rs. Activation of A1Rs inhibited the activity of inspiratory neurons in the brainstem by opening KATP in neonatal mice [123]. A1R stimulation promotes KATP activity in principal dopamine neurons in the substantia nigra pars compacta [124] and hippocampus [125]. In contrast, one recent study has demonstrated that adenosine induces internalization of KATP, resulting in a decrease in KATP-mediated response in the hippocampus [126]. The discrepancy might be due to the additional activation of A2ARs by adenosine located in hippocampus, but not in the substantia nigra pars compacta [127]. In addition to the inhibitory effect on the presynaptic site, the activation of A1Rs acts as an inhibitory modulator to electrical activity on the postsynaptic site, and this effect has been attributed to enhancement of KATP activity. The modulating effect on the membrane potential may differ depending on the brain regions, as neuronal KATP is heterogeneous in different neurons.
A1R interactions—new approaches for neurological disorders
By activation of its receptors, adenosine regulates many pathophysiological processes, particularly in excitable tissues of the brain (Fig. 3). Its widespread functions in the body include regulation of seizure susceptibility [128, 129], neuroprotection [40], regulation of pain perception [130], sleep induction [131], and involvement in Parkinson’s disease [132]. There is increasing evidence that the functional interaction of A1Rs with other neuronal receptors may contribute to fine tuning in synaptic transmission, and A1R agonists may represent a useful therapeutic approach for the treatment of some neurological disorders by regulating homeostasis in transmitter systems. However, the use of A1R agonists has not proved clinically useful due to mainly cardiovascular side effects as well as low brain permeability. Pioneering experimental approaches have been evaluated using focal drug delivery in epilepsy models. One experimental study has used intraventricular implantation of an adenosine-releasing synthetic polymer [128]. In a later study, Guttinger et al. [133] used encapsulated C2C12 myoblasts that were engineered to release adenosine by disruption of their adenosine kinase gene [133]. The local delivery of adenosine by implanted cells appears to be a promising strategy for the control not only for affecting seizure activity but also other neurodegenerative diseases with dysregulated synaptic neurotransmission.
Fig. 3.
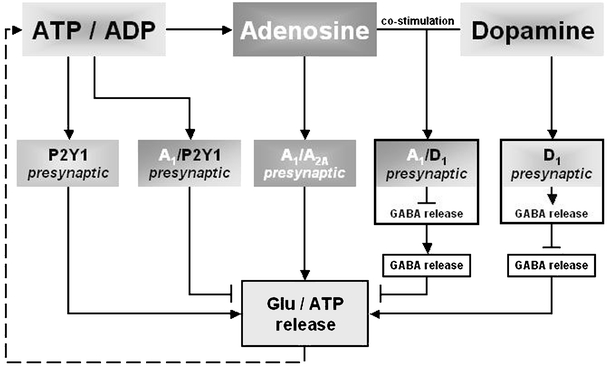
Schematic representation of possible interactions of A1Rs with metabotropic receptors. Heteromerization between presynaptically located A1Rs and A2ARs, D1Rs, and P2Y1Rs causes changes in influencing glutamate release by adenosine. Cross talk between A1Rs and P2Y1Rs contributes mainly to triggering fast attenuation of transmitter release, whereas ADP acts as a ligand on the heteromer. During elevated adenosine levels, A2AR signaling becomes dominant in the A1R/A2AR complex, providing enhancement of glutamate release. The A1R/D1R heteromer requires both adenosine and dopamine to be activated and inhibits transmitter release
Concluding remarks
As a consequence of its ubiquitous distribution and because of its linkage to the energy pool, adenosine has evolved as an important messenger in extracellular signaling. Modifications in extracellular adenosine levels with subsequent alterations in the activation of its receptors interferes with the action of other receptor systems. Figure 4 summarizes possible interactions of A1Rs with metabotropic receptors, Fig. 5 shows the interactions between A1Rs and ionotropic receptors as well as with KATP, and Fig. 3 shows the neurological disorders where A1R interactions may play a role.
Fig. 4.
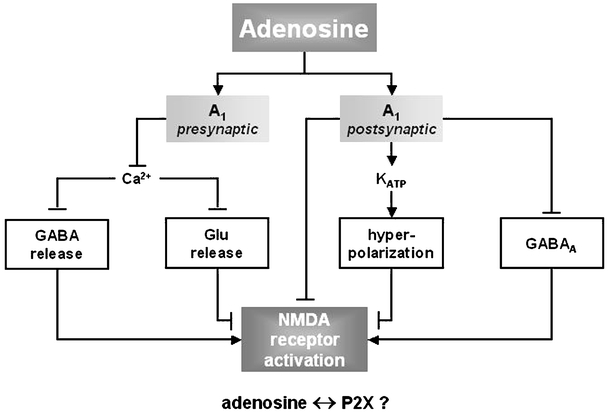
Schematic representation of possible interactions of A1Rs with ionotropic receptors contributing to the fine-tuning of neurotransmission. Adenosine acting via presynaptic A1Rs may attenuate the influx of Ca2+ through voltage-dependent calcium channels and thus decrease the release of glutamate and GABA, which inhibits or facilitates the activation of postsynaptically located NMDA receptors, respectively. Adenosine acting through postsynaptic A1Rs may activate KATP, which leads to hyperpolarization of postsynaptic neurons and inhibits directly the activity of NMDA and GABAA receptors
Fig. 5.
Neurological disorders where A1R interactions may play a role
There is evidence that various regulatory mechanisms exist as well as multiple mechanisms that act independently of each other on the same cell depending on the brain region and cell type. The overaction and redundancy principle ensures transmitter homeostasis under pathophysiological conditions in a special time window. The function of adenosine receptors in the regulation of the synaptic transmission is complex.
The key receptor in regulation of the neuronal transmission may be the A2AR, whereas the interaction of the A1R with metabotropic and ionotropic receptors as well as with KATP serves as fine-tuning to inhibit synaptic transmission, as mentioned by Sebastiao and Ribeiro [80]. A1Rs may play a nonessential role in normal physiology as demonstrated in mice lacking the A1Rs [134]. However, they play an important protective role under pathophysiological conditions especially during hypoxia. The activation initiates a fast inhibition of the glutamatergic neurotransmission and the receptor interactions may contribute to its maintenance or can support the A1R-mediated effects.
Most of our knowledge on receptor-receptor interactions involving the A1R results from experiments on cell cultures, slice preparation or, to a lesser extent, from in vivo experiments where regulation can be studied in principle or new drug targets can be characterized. These findings may contribute to a better understanding of disturbances in transmitter homeostasis. As our understanding of the complexity of receptor signaling and interaction develops, we may well gain new perspectives in new drug development. The clinical relevance of the testing models has often been questioned, however. Discordance between studies on cells and animal and human studies may be due to bias or failure of models to mimic clinical disease to an adequate degree. There are new techniques such as neuroimaging, nanotechnology, siPCR, and new selective receptor ligands that will help to overcome some of these aspects in the near future.
Acknowledgement
The authors thank Dr. Annette G. Beck-Sickinger, University of Leipzig for the generous gift of the Y1 receptor agonist [F7, P34]pNPY.
References
- 1.Takeda S, Kadowaki S, Haga T, Takaesu H, Mitaku S (2002) Identification of G protein-coupled receptor genes from the human genome sequence. FEBS Lett 520:97–101 [DOI] [PubMed]
- 2.Brink CB, Harvey BH, Bodenstein J, Venter DP, Oliver DW (2004) Recent advances in drug action and therapeutics: relevance of novel concepts in G-protein-coupled receptor and signal transduction pharmacology. Br J Clinical Pharmacol 57:373–387 [DOI] [PMC free article] [PubMed]
- 3.Franco R, Casado V, Mallol J, Ferrada C, Ferre S, Fuxe K, Cortes A, Ciruela F, Lluis C, Canela EI (2006) The two-state dimer receptor model: a general model for receptor dimers. Mol Pharmacol 69:1905–1912 [DOI] [PubMed]
- 4.Maudsley S, Martin B, Luttrell LM (2005) The origins of diversity and specificity in G protein-coupled receptor signaling. J Pharmacol Exp Ther 314:485–494 [DOI] [PMC free article] [PubMed]
- 5.Agnati LF, Fuxe K, Benfenati F, Zini I, Hokfelt T (1983) On the functional role of coexistence of 5-HT and substance P in bulbospinal 5-HT neurons. Substance P reduces affinity and increases density of 3H-5-HT binding sites. Acta Physiol Scand 117:299–301 [DOI] [PubMed]
- 6.Agnati LF, Ferre S, Lluis C, Franco R, Fuxe K (2003) Molecular mechanisms and therapeutical implications of intramembrane receptor/receptor interactions among heptahelical receptors with examples from the striatopallidal GABA neurons. Pharmacol Rev 55:509–550 [DOI] [PubMed]
- 7.Latini S, Pedata F (2001) Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem 79:463–484 [DOI] [PubMed]
- 8.Ribeiro JA, Sebastiao AM, de Mendonca A (2003) Participation of adenosine receptors in neuroprotection. Drug News Perspect 16:80–86 [DOI] [PubMed]
- 9.Sebastiao AM, Stone TW, Ribeiro JA (1990) The inhibitory adenosine receptor at the neuromuscular junction and hippocampus of the rat: antagonism by 1,3,8-substituted xanthines. Br J Pharmacol 101:453–459 [DOI] [PMC free article] [PubMed]
- 10.De Mendonca A, Almeida T, Bashir ZI, Ribeiro JA (1997) Endogenous adenosine attenuates long-term depression and depotentiation in the CA1 region of the rat hippocampus. Neuropharmacology 36:161–167 [DOI] [PubMed]
- 11.De Mendonca A, Ribeiro JA (2000) Long-term potentiation observed upon blockade of adenosine A(1) receptors in rat hippocampus is N-methyl-D-aspartate receptor-dependent. Neurosci Lett 291:81–84 [DOI] [PubMed]
- 12.Jacoby E, Bouhelal R, Gerspacher M, Seuwen K (2006) The 7 TM G-protein-coupled receptor target family. Chem Med Chem 1:761–782 [DOI] [PubMed]
- 13.Kostenis E (2006) G proteins in drug screening: from analysis of receptor-G protein specificity to manipulation of GPCR-mediated signalling pathways. Curr Pharm Design 12:1703–1715 [DOI] [PubMed]
- 14.Biber K, Klotz KN, Berger M, Gebicke-Harter PJ, van Calker D (1997) Adenosine A1 receptor-mediated activation of phospholipase C in cultured astrocytes depends on the level of receptor expression. J Neurosci 17:4956–4964 [DOI] [PMC free article] [PubMed]
- 15.Basheer R, Arrigoni E, Thatte HS, Greene RW, Ambudkar IS, McCarley RW (2002) Adenosine induces inositol 1,4,5-triphosphate receptor-mediated mobilization of intracellular calcium stores in basal forebrain cholinergic neurons. J Neurosci 22:7680—7686 [DOI] [PMC free article] [PubMed]
- 16.Ciruela F, Ferre S, Casado V, Cortes A, Cunha RA, Lluis C, Franco R (2006) Heterodimeric adenosine receptors: a device to regulate neurotransmitter release. Cell Mol Life Sci 63:2427—2431 [DOI] [PMC free article] [PubMed]
- 17.Ciruela F, Casado V, Rodrigues RJ, Lujan R, Burgueno J, Canals M, Borycz J, Rebola N, Goldberg SR, Mallol J, Cortes A, Canela EI, Lopez-Gimenez JF, Milligan G, Lluis C, Cunha RA, Ferre S, Franco R (2006) Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci 26:2080–2087 [DOI] [PMC free article] [PubMed]
- 18.Ferre S, Agnati LF, Ciruela F, Lluis C, Woods AS, Fuxe K, Franco R. Neurotransmitter receptor heteromers an their integrative role in ‘local modules’: the striatal spine module. Brain Res Rev 2007 (in press) [DOI] [PMC free article] [PubMed]
- 19.Fragata IR, Ribeiro JA, Sebastiao AM (2006) Nitric oxide mediates interactions between GABA(A) receptors and adenosine A(1) receptors in the rat hippocampus. Eur J Pharmacol 543:32–39 [DOI] [PubMed]
- 20.Franco R, Ciruela F, Casado V, Cortes A, Canela EI, Mallol J, Agnati LF, Ferre S, Fuxe K, Lluis C (2005) Partners for adenosine A1 receptors. J Mol Neurosci 26:221–232 [DOI] [PubMed]
- 21.Zhou QY, Li C, Olah ME, Johnson RA, Stiles GL, Civelli O (1992) Molecular cloning and characterization of an adenosine receptor: the A3 adenosine receptor. Proc Natl Acad Sci USA 89:7432–7436 [DOI] [PMC free article] [PubMed]
- 22.Von Lubitz DK, Lin RC, Popik P, Carter MF, Jacobson KA (1994) Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol 263:59–67 [DOI] [PMC free article] [PubMed]
- 23.Von Lubitz DK, Ye W, McClellan J, Lin RC (1999) Stimulation of adenosine A3 receptors in cerebral ischemia. Neuronal death, recovery, or both? Ann N Y Acad Sci 890:93–106 [DOI] [PubMed]
- 24.Von Lubitz DKJE (1999) Adenosine and cerebral ischemia: therapeutic future or death of a brave concept? Eur J Pharmacol 365:9–25 [DOI] [PubMed]
- 25.Von Lubitz DKJE (2001) Adenosine in the treatment of stroke: yes, maybe, or absolutely not? Expert Opin Invest Drugs 10:619–632 [DOI] [PubMed]
- 26.Rubaj A, Zgodzinski W, Sieklucka-Dziuba M (2003) The influence of adenosine A3 receptor agonist: IB-MECA, on scopolamine- and MK-801-induced memory impairment. Behav Brain Res 141:11–17 [DOI] [PubMed]
- 27.Sei Y, von Lubitz DK, AMP, Ji X, Jacobson KA (2006) Adenosine A3 receptor agonist-induced neurotoxicity in rat cerebellar granule neurons. Drug Dev Res 40:267–273
- 28.Pugliese AM, Latini S, Corradetti R, Pedata F (2003) Brief, repeated, oxygen-glucose deprivation episodes protect neurotransmission from a longer ischemic episode in the in vitro hippocampus: role of adenosine receptors. Br J Pharmacol 140:305–314 [DOI] [PMC free article] [PubMed]
- 29.Pugliese AM, Coppi E, Spalluto G, Corradetti R, Pedata F (2006) A3 adenosine receptor antagonists delay irreversible synaptic failure caused by oxygen and glucose deprivation in the rat CA1 hippocampus in vitro. Br J Pharmacol 147:524–532 [DOI] [PMC free article] [PubMed]
- 30.Ramkumar V, Stiles GL, Beaven MA, Ali H (1993) The A(3) adenosine receptor is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J Biol Chem 268:16887–16890 [PubMed]
- 31.Abbracchio MP, Brambilla R, Ceruti S, Kim HO, von Lubitz DK, Jacobson KA, Cattabeni F (1995) G protein-dependent activation of phospholipase C by adenosine A3 receptors in rat brain. Mol Pharmacol 48:1038–1045 [PubMed]
- 32.Gessi S, Varani K, Merighi S, Cattabriga E, Iannotta V, Leung E, Baraldi PG, Borea PA (2002) A(3) adenosine receptors in human neutrophils and promyelocytic HL60 cells: a pharmacological and biochemical study. Mol Pharmacol 61:415–424 [DOI] [PubMed]
- 33.Kohno Y, Ji X, Mawhorter SD, Koshiba M, Jacobson KA (1996) Activation of A3 adenosine receptors on human eosinophils elevates intracellular calcium. Blood 88:3569–3574 [PMC free article] [PubMed]
- 34.Dunwiddie TV, Diao L, Kim HO, Jiang JL, Jacobson KA (1997) Activation of hippocampal adenosine A3 receptors produces a desensitization of A1 receptor-mediated responses in rat hippocampus. J Neurosci 17:607–614 [DOI] [PMC free article] [PubMed]
- 35.Brand A, Vissiennon Z, Eschke D, Nieber K (2000) Adenosine A(1) and A(3) receptors mediate inhibition of synaptic transmission in rat cortical neurons. Neuropharmacology 40:85–95 [DOI] [PubMed]
- 36.Hentschel S, Lewerenz A, Nieber K (2003) Activation of A(3) receptors by endogenous adenosine inhibits synaptic transmission during hypoxia in rat cortical neurons. Restor Neurol Neurosci 21:55–63 [PubMed]
- 37.Lopes LV, Rebola N, Costenla AR, Halldner L, Jacobson MA, Oliveira CR, Richardson PJ, Fredholm BB, Ribeiro JA, Cunha RA (2003) Adenosine A(3) receptors in the rat hippocampus: lack of interaction with A(1) receptors. Drug Dev Res 58:428–438
- 38.Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM (2005) Adenosine and brain function. Int Rev Neurobiol 63:191–270 [DOI] [PubMed]
- 39.Rebola N, Rodrigues RJ, Lopes LV, Richardson PJ, Oliveira CR, Cunha RA (2005) Adenosine A1 and A2A receptors are co-expressed in pyramidal neurons and co-localized in glutamatergic nerve terminals of the rat hippocampus 1. Neuroscience 133:79–83 [DOI] [PubMed]
- 40.Cunha RA (2005) Neuroprotection by adenosine in the brain: from A1 receptor activation to A2A receptor blockade. Purinergic Signalling 1:111–134 [DOI] [PMC free article] [PubMed]
- 41.Popoli P, Betto P, Reggio R, Riccaiarello G (1995) Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur J Pharmacol 287:215–217 [DOI] [PubMed]
- 42.Corsi C, Melani A, Biachi L, Pedata F (2000) Striatal A2A adenosine receptor antagonism differentially modifies striatal glutamate outflow in vivo in young and aged rats. Neuroreport 11:2591–2595 [DOI] [PubMed]
- 43.Xu K, Xu YH, Chen JF, Schwarzschild MA (2002) Caffeine’s neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity shows no tolerance to chronic caffeine administration in mice. Neurosci Lett 322:13–16 [DOI] [PubMed]
- 44.Popoli P, Reggio R, Pezzola A (2000) Effects of SCH 58261, an adenosine A2A receptor antagonist, on quinpirole-induced turning in 6-hydroxydopamine-lesioned rats. Lack of tolerance after chronic caffeine intake. Neuropsychopharmacology 22:522–529 [DOI] [PubMed]
- 45.Dixon AK, Widdowson L, Richardson PJ (1997) Desensitisation of the adenosine A1 receptor by the A2A receptor in the rat striatum. J Neurochem 69:315–321 [DOI] [PubMed]
- 46.Lopes LV, Cunha RA, Ribeiro JA (1999) Cross talk between A(1) and A(2A) adenosine receptors in the hippocampus and cortex of young adult and old rats. J Neurophysiol 82:3196–3203 [DOI] [PubMed]
- 47.Lopes LV, Cunha RA, Kull B, Fredholm BB, Ribeiro JA (2002) Adenosine A(2A) receptor facilitation of hippocampal synaptic transmission is dependent on tonic A(1) receptor inhibition. Neuroscience 112:319–329 [DOI] [PubMed]
- 48.Quarta D, Borycz J, Solinas M, Patkar K, Hockemeyer J, Ciruela F, Lluis C, Franco R, Woods AS, Goldberg SR, Ferre S (2004) Adenosine receptor-mediated modulation of dopamine release in the nucleus accumbens depends on glutamate neurotransmission and N-methyl-D-aspartate receptor stimulation. J Neurochem 91:873–880 [DOI] [PubMed]
- 49.Latini S, Bordoni F, Corradetti R, Pepeu G, Pedata F (1999) Effect of A2A adenosine receptor stimulation and antagonism on synaptic depression induced by in vitro ischaemia in rat hippocampal slices. Br J Pharmacol 128:1035–1044 [DOI] [PMC free article] [PubMed]
- 50.Gines S, Hillion J, Torvinen M, Le Crom S, Casado V, Canela EI, Rondin S, Lew JY, Watson S, Zoli M, Agnati LF, Vernier P, Lluis C, Ferre S, Fuxe K, Franco R (2000) Dopamine D-1 and adenosine A(1) receptors form functionally interacting heteromeric complexes. Proc Natl Acad Sci 97:8606–8611 [DOI] [PMC free article] [PubMed]
- 51.Ferre S, Popoli P, Gimenez-Llort L, Finnman UB, Martinez E, Scotti de Carolis A, Fuxe K (1994) Postsynaptic antagonistic interaction between adenosine A1 and dopamine D1 receptors. Neuroreport 6:73–76 [DOI] [PubMed]
- 52.Mayfield RD, Jones BA, Miller HA, Simosky JK, Larson GA, Zahniser NR (1999) Modulation of endogenous GABA release by an antagonistic adenosine A1/dopamine D1 receptor interaction in rat brain limbic regions but not basal ganglia. Synapse 33:274–281 [DOI] [PubMed]
- 53.Floran B, Barajas C, Floran L, Erlij D, Aceves J (2002) Adenosine A1 receptors control dopamine D1-dependent [(3)H]GABA release in slices of substantia nigra pars reticulata and motor behavior in the rat. Neuroscience 115:743–751 [DOI] [PubMed]
- 54.Ferre S, Popoli P, Gimenez-Llort L, Rimondini R, Muller CE, Stromberg I, Ogren SO, Fuxe K (2001) Adenosine/dopamine interaction: implications for the treatment of Parkinson’s disease. Parkinsonism Relat Disord 7:235–241 [DOI] [PubMed]
- 55.Savitt JM, Dawson VL, Dawson TM (2006) Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest 116:1744–1754 [DOI] [PMC free article] [PubMed]
- 56.Migita K, Loewy AD, Ramabhadran TV, Krause JE, Waters SM (2001) Immunohistochemical localization of the neuropeptide Y Y1 receptor in rat central nervous system. Brain Res 889:23–37 [DOI] [PubMed]
- 57.Kopp J, Xu ZQ, Zhang X, Pedrazzini T, Herzog H, Kresse A, Wong H, Walsh JH, Hokfelt T (2002) Expression of the neuropeptide Y Y1 receptor in the CNS of rat and of wild-type and Y1 receptor knock-out mice. Focus on immunohistochemical localization. Neuroscience 111:443–532 [DOI] [PubMed]
- 58.Wolak ML, DeJoseph MR, Cator AD, Mokashi AS, Brownfield MS, Urban JH (2003) Comparative distribution of neuropeptide Y Y1 and Y5 receptors in the rat brain by using immunohistochemistry. J Comp Neurol 464:285–311 [DOI] [PubMed]
- 59.Bijak M (2000) Neuropeptide Y reduces epileptiform discharges and excitatory synaptic transmission in rat frontal cortex in vitro. Neuroscience 96:487–494 [DOI] [PubMed]
- 60.Cabrele C, Beck-Sickinger AG (2000) Molecular characterization of the ligand-receptor interaction of the neuropeptide Y family. J Pept Sci 6:97–122 [DOI] [PubMed]
- 61.Parrado C, Diaz-Cabiale Z, Garcia-Coronel M, Agnati LF, Covenas R, Fuxe K, Narvaez JA (2007) Region specific galanin receptor/neuropeptide YY1 receptor interactions in the tel- and diencephalon of the rat. Relevance for food consumption. Neuropharmacology 52:684–692 [DOI] [PubMed]
- 62.Sichardt K, Beck-Sickinger AG, Nieber K (2006) Modulation of synaptic transmission by adenosine A1 and NPY receptors on rat cortical neurons. Purinergic Signalling 2:258–259
- 63.Burnstock G (2007) Purine and pyrimidine receptors. Cell Mol Life Sci 64:1471–1483 [DOI] [PMC free article] [PubMed]
- 64.Yoshioka K, Hosoda R, Kuroda Y, Nakata H (2002) Hetero-oligomerization of adenosine A1 receptors with P2Y1 receptors in rat brains. FEBS Lett 531:299–303 [DOI] [PubMed]
- 65.Ikeuchi Y, Nishizaki T, Mori M, Okada Y (1996) Adenosine activates the K+ channel and enhances cytosolic Ca2+ release via a P2Y purinoceptor in hippocampal neurons. Eur J Pharmacol 304:191–199 [DOI] [PubMed]
- 66.Yoshioka K, Saitoh O, Nakata H (2001) Heteromeric association creates a P2Y-like adenosine receptor. Proc Natl Acad Sci USA 98:7617–7622 [DOI] [PMC free article] [PubMed]
- 67.Yoshioka K, Saitoh O, Nakata H (2002) Agonist-promoted heteromeric oligomerization between adenosine A(1) and P2Y(1) receptors in living cells. FEBS Lett 523:147–151 [DOI] [PubMed]
- 68.Nakata H, Yoshioka K, Saitoh S (2003) Hetero-oligomerization between adenosine A(1) and P2Y(1) receptors in living cells: formation of ATP-sensitive adenosine receptors. Drug Dev Res 58:340–349
- 69.Nakata H, Yoshioka K, Kamiya T (2004) Purinergic-receptor oligomerization: implications for neural functions in the central nervous system. Neurotox Res 6:291–297 [DOI] [PubMed]
- 70.Nakata H, Yoshioka K, Kamiya T, Tsuga H, Oyanagi K (2005) Functions of heteromeric association between adenosine and P2Y receptors. J Mol Neurosci 26:233–238 [DOI] [PubMed]
- 71.Mori M, Heuss C, Gahwiler BH, Gerber U (2001) Fast synaptic transmission mediated by P2X receptors in CA3 pyramidal cells of rat hippocampal slice cultures. J Physiol 535:115–123 [DOI] [PMC free article] [PubMed]
- 72.Nieber K, Poelchen W, Illes P (1997) Role of ATP in fast excitatory synaptic potentials in locus coeruleus neurones of the rat. Br J Pharmacol 122:423–430 [DOI] [PMC free article] [PubMed]
- 73.Poelchen W, Sieler D, Wirkner K, Illes P (2001) Co-transmitter function of ATP in central catecholaminergic neurons of the rat. Neuroscience 102:593–602 [DOI] [PubMed]
- 74.Halliwell RF (2007) A short history of the rise of the molecular pharmacology of ionotropic drug receptors. Trends Pharmacol Sci 28:214–219 [DOI] [PubMed]
- 75.Romanelli MN, Gratteri P, Guandalini L, Martini E, Bonaccini C, Gualtieri F (2007) Central nicotinic receptors: structure, function, ligands, and therapeutic potential. Chem Med Chem 2:746–767 [DOI] [PubMed]
- 76.Boehm S, Kubista H (2002) Fine tuning of sympathetic transmitter release via ionotropic and metabotropic presynaptic receptors. Pharmacol Rev 54:43–99 [DOI] [PubMed]
- 77.Gundlfinger A, Bischofberger J, Johenning FW, Torvinen M, Schmitz D, Breustedt J (2007) Adenosine modulates transmission at the hippocampal mossy fibre synapse via direct inhibition of presynaptic calcium channels. J Physiol 582(Pt 1):263–277 [DOI] [PMC free article] [PubMed]
- 78.Dunwiddie TV, Masino SA (2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24:31–55 [DOI] [PubMed]
- 79.Abbracchio MP, Cattabeni F (1999) Brain adenosine receptors as targets for therapeutic intervention in neurodegenerative diseases. Ann N Y Acad Sci 890:79–92 [DOI] [PubMed]
- 80.Sebastiao AM, Ribeiro JA (2000) Fine-tuning neuromodulation by adenosine. Trends Pharmacol Sci 21:341–346 [DOI] [PubMed]
- 81.Li H, Henry JL (2000) Adenosine receptor blockade reveals N-methyl-D-aspartate receptor- and voltage-sensitive dendritic spikes in rat hippocampal CA1 pyramidal cells in vitro. Neuroscience 100:21–31 [DOI] [PubMed]
- 82.Nikbakht MR, Stone TW (2001) Suppression of presynaptic responses to adenosine by activation of NMDA receptors. Eur J Pharmacol 427:13–25 [DOI] [PubMed]
- 83.Boeck CR, Kroth EH, Bronzatto MJ, Vendite D (2005) Adenosine receptors co-operate with NMDA preconditioning to protect cerebellar granule cells against glutamate neurotoxicity. Neuropharmacology 49:17–24 [DOI] [PubMed]
- 84.Schotanus SM, Fredholm BB, Chergui K (2006) NMDA depresses glutamatergic synaptic transmission in the striatum through the activation of adenosine A(1) receptors: evidence from knockout mice. Neuropharmacology 51:272–282 [DOI] [PubMed]
- 85.Heinbockel T, Pape HC (1999) Modulatory effects of adenosine on inhibitory postsynaptic potentials in the lateral amygdala of the rat. Br J Pharmacol 128:190–196 [DOI] [PMC free article] [PubMed]
- 86.Shen KZ, Johnson SW (1997) Presynaptic GABAB and adenosine A1 receptors regulate synaptic transmission to rat substantia nigra reticulata neurones. J Physiol 505(Pt 1):153–163 [DOI] [PMC free article] [PubMed]
- 87.Ulrich D, Huguenard JR (1995) Purinergic inhibition of GABA and glutamate release in the thalamus: implications for thalamic network activity. Neuron 15:909–918 [DOI] [PubMed]
- 88.Chen G, van den Pol AN (1997) Adenosine modulation of calcium currents and presynaptic inhibition of GABA release in suprachiasmatic and arcuate nucleus neurons. J Neurophysiol 77:3035–3047 [DOI] [PubMed]
- 89.Neary JT, Zhu Q, Kang Y, Dash PK (1996) Extracellular ATP induces formation of AP-1 complexes in astrocytes via P2 purinoceptors. Neuroreport 7:2893–2896 [DOI] [PubMed]
- 90.Cunha RA (2001) Regulation of the ecto-nucleotidase pathway in rat hippocampal nerve terminals. Neurochem Res 26:979–991 [DOI] [PubMed]
- 91.Illes P, Ribeiro JA (2001) Molecular physiology of P2 receptors in the central nervous system. Eur J Pharmacol 483:5–17 [DOI] [PubMed]
- 92.Hebb MO, White TD (1998) Co-administration of adenosine kinase and deaminase inhibitors produces supra-additive potentiation of N-methyl-D-aspartate-evoked adenosine formation in cortex. Eur J Pharmacol 344:121–125 [DOI] [PubMed]
- 93.Cunha RA, Correia-de-Sa P, Sebastiao AM, Ribeiro JA (1996) Preferential activation of excitatory adenosine receptors at rat hippocampal and neuromuscular synapses by adenosine formed from released adenine nucleotides. Br J Pharmacol 119:253–260 [DOI] [PMC free article] [PubMed]
- 94.Masino SA, Diao L, Illes P, Zahniser NR, Larson GA, Johansson B, Fredholm BB, Dunwiddie TV (2002) Modulation of hippocampal glutamatergic transmission by ATP is dependent on adenosine a(1) receptors. J Pharmacol Exp Ther 303:356–363 [DOI] [PubMed]
- 95.De Mendonca A, Sebastiao AM, Ribeiro JA (1995) Inhibition of NMDA receptor-mediated currents in isolated rat hippocampal neurones by adenosine A1 receptor activation. Neuroreport 6:1097–1100 [DOI] [PubMed]
- 96.Costenla AR, de Mendonca A, Sebastiao A, Ribeiro JA (1999) An adenosine analogue inhibits NMDA receptor-mediated responses in bipolar cells of the rat retina. Exp Eye Res 68:367–370 [DOI] [PubMed]
- 97.Dunwiddie TV, Hoffer BJ, Fredholm BB (1981) Alkylxanthines elevate hippocampal excitability. Evidence for a role of endogenous adenosine. Naunyn Schmiedebergs Arch Pharmacol 316:326–330 [DOI] [PubMed]
- 98.Mori A, Shindou T, Ichimura M, Nonaka H, Kase H (1996) The role of adenosine A2A receptors in regulating GABAergic synaptic transmission in striatal medium spiny neurons. J Neurosci 16:605–611 [DOI] [PMC free article] [PubMed]
- 99.Huang CC, Liang YC, Hsu KS (2001) Characterization of the mechanism underlying the reversal of long term potentiation by low frequency stimulation at hippocampal CA1 synapses. J Biol Chem 276:48108–48117 [DOI] [PubMed]
- 100.Scammell TE, Arrigoni E, Thompson MA, Ronan PJ, Saper CB, Greene RW (2003) Focal deletion of the adenosine A1 receptor in adult mice using an adeno-associated viral vector. J Neurosci 23:5762–5770 [DOI] [PMC free article] [PubMed]
- 101.Wardas J (2002) Neuroprotective role of adenosine in the CNS. Polh J Pharmacol 54:313–326 [PubMed]
- 102.Kittler JT, Moss SJ (2003) Modulation of GABA(A) receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr Opin Neurobiol 13:341–347 [DOI] [PubMed]
- 103.Li H, Wu L, Li YQ (2004) Adenosine suppresses GABAA receptor–mediated responses in rat sacral dorsal commissural neurons. Auton Neurosci 111:71–79 [DOI] [PubMed]
- 104.Akhondzadeh S, Stone TW (1994) Interaction between adenosine and GABAA receptors on hippocampal neurones. Brain Res 665:229–236 [DOI] [PubMed]
- 105.Madden KP (1994) Effect of gamma-aminobutyric acid modulation on neuronal ischemia in rabbits. Stroke 25:2271–2274 [DOI] [PubMed]
- 106.O’Regan MH, Simpson RE, Perkins LM, Phillis JW (1992) Adenosine receptor agonists inhibit the release of gamma-aminobutyric acid (GABA) from the ischemic rat cerebral cortex. Brain Res 582:22–26 [DOI] [PubMed]
- 107.Heron A, Lekieffre D, Le Peillet E, Lasbennes F, Seylaz J, Plotkine M, Boulu RG (1994) Effects of an A1 adenosine receptor agonist on the neurochemical, behavioral and histological consequences of ischemia. Brain Res 641:217–224 [DOI] [PubMed]
- 108.Lucchi R, Latini S, deMendonca A, Sebastiao AM, Ribeiro JA (1996) Adenosine by activating A(1) receptors prevents GABA(A)-mediated actions during hypoxia in the rat hippocampus. Brain Res 732:261–266 [DOI] [PubMed]
- 109.Rufke C, Scheibner T, Klotz KN, Nieber K (2006) In vitro desensitization of adenosine A1 receptors after exposure to adenosine analogues. Arch Pharm 372:117/41
- 110.Wetherington JP, Lambert NA (2002) Differential desensitization of responses mediated by presynaptic and postsynaptic A1 adenosine receptors. J Neurosci 22:1248–1255 [DOI] [PMC free article] [PubMed]
- 111.Dar MS (2002) Mouse cerebellar adenosine-glutamate interactions and modulation of ethanol-induced motor incoordination. Alcohol Clin Exp Res 26:1395–1403 [DOI] [PubMed]
- 112.North RA (2002) Molecular physiology of P2X receptors. Physiol Rev 82:1013–1067 [DOI] [PubMed]
- 113.Burnstock G (2007) Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev 87:659–797 [DOI] [PubMed]
- 114.Sneddon P, Westfall TD, Todorov LD, Mihaylova-Todorova S, Westfall DP, Kennedy C (1999) Modulation of purinergic neurotransmission. Prog Brain Res 120:11–20 [DOI] [PubMed]
- 115.Proctor WR, Dunwiddie TV (1987) Pre- and postsynaptic actions of adenosine in the in vitro rat hippocampus. Brain Res 426:187–190 [DOI] [PubMed]
- 116.Cunha RA, Vizi ES, Ribeiro JA, Sebastiao AM (1996) Preferential release of ATP and its extracellular catabolism as a source of adenosine upon high- but not low-frequency stimulation of rat hippocampal slices. J Neurochem 67:2180–2187 [DOI] [PubMed]
- 117.Zimmermann H (2000) Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol 362:299–309 [DOI] [PubMed]
- 118.Cunha RA, Sebastiao AM, Ribeiro JA (1998) Inhibition by ATP of hippocampal synaptic transmission requires localized extracellular catabolism by ecto-nucleotidases into adenosine and channeling to adenosine A1 receptors. J Neurosci 18:1987–1995 [DOI] [PMC free article] [PubMed]
- 119.Sebastiao AM, Cunha RA, Cascalheira JF, Ribeiro JA (1999) Adenine nucleotides as inhibitors of synaptic transmission: role of localised ectonucleotidases. Prog Brain Res 120:183–192 [DOI] [PubMed]
- 120.Coppi E, Pugliese AM, Stephan H, Mueller CE, Pedata F (2007) Role of P2 purinergic receptors in synaptic transmission under normoxic and ischaemic conditions in the CA1 region of rat hippocampal slices. Purinergic Signalling 3:203–219 [DOI] [PMC free article] [PubMed]
- 121.Lazdunski M (1994) ATP-sensitive potassium channels: an overview. J Cardiovasc Pharmacol 24(Suppl 4):S1–S5 [PubMed]
- 122.Pissarek M, Garcia de Arriba S, Schäfer M, Sieler D, Nieber K, Illes P (1998) Changes by short-term hypoxia in the membrane properties of pyramidal cells and the levels of purine and pyrimidine nucleotides in slices of rat neocortex; effects of agonists and antagonists of ATP-dependent potassium channels. Naunyn Schmiedebergs Arch Pharmacol 358:430–439 [DOI] [PubMed]
- 123.Mironov SL, Langohr K, Richter DW (1999) A1 adenosine receptors modulate respiratory activity of the neonatal mouse via the cAMP-mediated signaling pathway. J Neurophysiol 81:247–255 [DOI] [PubMed]
- 124.Andoh T, Ishiwa D, Kamiya Y, Echigo N, Goto T, Yamada Y (2006) A1 adenosine receptor-mediated modulation of neuronal ATP-sensitive K channels in rat substantia nigra. Brain Res 1124:55–61 [DOI] [PubMed]
- 125.Shan HQ, Cheng JS (2000) Effect of adenosine on adenosine triphosphate-sensitive potassium channel during hypoxia in rat hippocampal neurons. Neurosci Lett 286:45–48 [DOI] [PubMed]
- 126.Hu K, Huang CS, Jan YN, Jan LY (2003) ATP-sensitive potassium channel traffic regulation by adenosine and protein kinase C. Neuron 38:417–432 [DOI] [PubMed]
- 127.Svenningsson P, Le Moine C, Fisone G, Fredholm BB (1999) Distribution, biochemistry and function of striatal adenosine A2A receptors. Prog Neurobiol 59:355–396 [DOI] [PubMed]
- 128.Boison D (2005) Adenosine and epilepsy: from therapeutic rationale to new therapeutic strategies. Neuroscientist 11:25–36 [DOI] [PubMed]
- 129.Pagonopoulou O, Efthimiadou A, Asimakopoulos B, Nikolettos NK (2006) Modulatory role of adenosine and its receptors in epilepsy: possible therapeutic approaches. Neurosci Res 56:14–20 [DOI] [PubMed]
- 130.Sadigh-Lindell B, Sylven C, Berglund M, Eriksson BE (2003) Role of adenosine and opioid-receptor mechanisms for pain in patients with silent myocardial ischemia or angina pectoris: a double-blind, placebo-controlled study. J Cardiovasc Pharmacol 42:757–763 [DOI] [PubMed]
- 131.Porkka-Heiskanen T, Kalinchuk A, Alanko L, Urrila A, Stenberg D (2003) Adenosine, energy metabolism, and sleep. Sci World J 3:790–798 [DOI] [PMC free article] [PubMed]
- 132.Franco R, Lluis C, Canela EI, Mallol J, Agnati L, Casado V, Ciruela F, Ferre S, Fuxe K (2007) Receptor-receptor interactions involving adenosine A(1) or dopamine D (1) receptors and accessory proteins. J Neural Transm 114:93–104 [DOI] [PubMed]
- 133.Guttinger M, Padrun V, Pralong WF, Boison D (2005) Seizure suppression and lack of adenosine A(1) receptor desensitization after focal long-term delivery of adenosine by encapsulated myoblasts. Exp Neurol 193:53–64 [DOI] [PubMed]
- 134.Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Gimenez-Llort L, Escorihuela RM, Fernandesz-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hardemark A, Betshotz C, Herlenius E, Fredholm BB (2001) Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci USA 98:9407–9412 [DOI] [PMC free article] [PubMed]