

Abstract
Akt is a serine/threonine protein kinase that plays a vital role in promoting cellular survival. Predominantly cytosolic, upon stimulation with growth-factors or stress, active Akt translocates into mitochondria, but the functions of Akt in mitochondria are not yet fully understood. Mitochondria play a central role in apoptotic pathways and given Akt’s functions in the cytoplasm, Akt in mitochondria may help preserve mitochondrial integrity during cellular stress. To test if the translocation of Akt into mitochondria is neuroprotective, adenoviral vectors expressing a constitutively active Akt, Ad-HA-Akt (DD), and a constitutively active Akt with a mitochondrial targeting signal, Ad-Mito-HA-Akt (DD), were generated. Human SH-SY5Y neuroblastoma cells expressing the adenoviral constructs were treated with staurosporine to initiate intrinsic apoptotic cell death and several aspects of the mitochondrial apoptotic pathway were evaluated. Expression of active Akt targeted to mitochondria was found to be sufficient to significantly reduce staurosporine-induced activation of caspase-3 and caspase-9, the release of cytochrome c from mitochondria, and Bax oligomerization at mitochondria. These findings demonstrate that intramitochondrial active Akt results in efficient protection against apoptotic signaling.
Keywords: Akt, mitochondria, apoptosis, caspase, Bax, Δψm
Akt (also known as protein kinase B) is a serine/threonine protein kinase that belongs to the AGC protein kinase subfamily that includes protein kinase A (PKA) and protein kinase C (PKC) [Peterson and Schreiber, 1999]. Akt is a predominant pro-survival protein that is regulated by phosphorylation. The phosphorylation of Akt is increased in response to activation of phosphoinositol 3-kinase (PI3K)-dependent pathways [Datta et al., 1996; Bellacosa et al., 1998]. Recruitment and activation of PI3K in response to specific ligands results in the generation of 3′-phosphoinositides at the plasma membrane and recruitment of Akt to the plasma membrane via its pleckstrin homology domain [Franke et al., 1997b]. Akt undergoes a conformational change and is subsequently activated by phosphorylation at either Thr308 or Ser473, with maximal activity occurring when both sites are phosphorylated [Kohn et al., 1995; Alessi et al., 1996; Scheid et al., 2002a,b].
There is unequivocal evidence that Akt is neuroprotective. For example, in experiments where cerebellar granule neurons were deprived of either serum or growth-factors, expression of wild-type Akt enhanced survival while the expression of a dominant negative Akt mutant increased apoptosis [Dudek et al., 1997]. Similar studies conducted in sympathetic neurons also showed that Akt was necessary and sufficient for promoting cellular survival [Crowder and Freeman, 1998]. These and other studies demonstrate that activation of Akt protects against neuronal cell death in response to many different stressors [Goswami et al., 1999; Salinas et al., 2001; Yamaguchi et al., 2001; Humbert et al., 2002; Chen et al., 2003; Dhandapani et al., 2005].
Numerous studies have focused on the mechanisms by which the activation of Akt protects against cell death. Active Akt has been shown to be anti-apoptotic by phosphorylating and inhibiting numerous cytosolic pro-apoptotic proteins [Brunet et al., 2001]. For example, Akt phosphorylates the constitutively active glycogen synthase kinase-3 (GSK3) (Ser21 in GSK3α and Ser9 in GSK3β), which inhibits the activity of GSK3 and attenuates its pro-apoptotic function [Cross et al., 1995; Pap and Cooper, 1998]. Akt has also been shown to function at the transcriptional level, inhibiting the transcription of genes that function in cell cycle arrest and apoptosis [Medema et al., 2000; Nakamura et al., 2000]. Akt can suppress intrinsic apoptotic processes at both pre-mitochondrial and post-mitochondrial levels [Franke et al., 1997a; Tang et al., 2001; Zhou et al., 2001]. Mitochondria play a central role in the intrinsic apoptotic pathway which can be initiated in response to various stimuli, such as growth-factor withdrawal, heat-shock, hypoxia, DNA-damaging agents, UV-irradiation, and cytotoxic drugs [Debatin et al., 2002]. When a cell is exposed to an apoptotic stimulus, the proapoptotic Bcl-2 family members, such as Bad and Bax, translocate to the mitochondrial outer membrane and form heterodimers with anti-apoptotic Bcl-2 proteins, Bcl-2, and Bcl-xL [Zha et al., 1996a, 1997]. Bax/Bcl-2 heterodimerization inhibits Bax from oligomerizing with itself in the outer membrane forming pores through which intermembrane space proteins can be released into the cytoplasm where these proteins can act in initiating apoptotic pathways [Mikhailov et al., 2001; Danial and Korsmeyer, 2004]. The heterodimerization of Bad to Bcl-2 results in displacement of Bcl-2 away from Bax, enabling Bax to oligomerize and form pores [Yang et al., 1995]. Akt phosphorylates Bad at Ser136 producing a docking site for 14-3-3 protein and this interaction causes Bad to be sequestered in the cytosol away from mitochondria [Zha et al., 1996b; Datta et al., 1997]. Akt has also been shown to affect Bax translocation to mitochondria, possibly by phosphorylation at Ser184 but the exact mechanism of this regulation has not been defined [Gardai et al., 2004].
The localization of Akt also may play a key role in determining its anti-apoptotic effects. Active Akt is present in the cytosol and nucleus, and recently it was shown to be localized within mitochondria [Andjelkovic et al., 1997; Meier et al., 1997; Bijur and Jope, 2003]. In addition, Akt levels in mitochondria increased upon stimulation with growth-factors or an apoptotic stimulus [Bijur and Jope, 2003]. Although it is clear that active Akt is in mitochondria, the function of Akt in mitochondria is not known. Therefore, the purpose of this study was to determine if active Akt localized in mitochondria affects intrinsic apoptotic cell death processes. To accomplish this goal constitutively active Akt targeted to mitochondria was expressed in human neuroblastoma SH-SY5Y cells followed by initiation of apoptosis and examination of the mitochondrial apoptotic pathway to test if mitochondrial Akt is capable of regulating apoptotic signaling. Constitutively active Akt was created by mutating Thr308 and Ser473 to aspartic acid residues which mimics phosphorylation and has been shown to result in a highly active Akt construct [Alessi et al., 1996]. The results of this study show that in response to staurosporine, cells expressing mitochondrial-targeted active Akt display decreased poly (ADP-ribose) polymerase (PARP) proteolysis, significantly decreased caspase-3 and caspase-9 activities, and a decrease in the level of pro-apoptotic Bcl-2 protein Bax oligomerization at mitochondria. These findings demonstrate that Akt in mitochondria likely plays a key role in attenuating apoptotic cell death in response to specific stressors that activate mitochondrial-mediated cell death pathways.
MATERIALS AND METHODS
Construction of Akt Constructs
The HA-Akt (wt) was a generous gift from Dr. Thomas Franke. The hemagglutinin (HA) epitope-tag peptide sequence is MYPYDVPDYASR. The constitutively active double mutant HA-Akt Thr308Asp/Ser473Asp, designated HA-Akt (DD), was created by sequential site-directed mutagenesis using the following PCR primers: forward 5′-G GAT GGT GCC ACT ATG AAG GAC TTC TGC GGA ACG-3′, reverse 5′-CGT TCC GCA GAA GTC CTT CAT AGT GGC ACC ATC C-3′ for Thr308Asp; forward 5′-C TTC CCC CAG TTC GAC TAC TCA GCC AGT GGC ACA G-3′, reverse 5′-C TGT GCC ACT GGC TGA GTA GTC GAA CTG GGG GAA G-3′ for Ser473Asp.
For generating the mitochondrial-targeted HA-Akt constructs, HA-Akt (wt) and HA-Akt (DD) were each PCR amplified out of pcDNA3.1 expression vector (Invitrogen, Carlsbad, CA) using Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA) and cloned into the SalI and NotI sites of pCMV/myc/MTS expression vector (Invitrogen) resulting in Mito-HA-Akt (wt) and Mito-HA-Akt (DD), respectively. The mitochondrial targeting signal peptide sequence is MSVLTPLLLRGLTGSARRLPVPRAKIHSL.
The adenoviral constructs were generated using a protocol from Q-BIOgene’s Ad-EASY vector system (Carlsbad, CA) with the following modifications. The constitutively active Akt constructs were amplified by PCR and cloned into pShuttle-CMV adenovirus transfer vector (BD Clontech Laboratories, Inc., Palo Alto, CA). HA-Akt (DD) was cloned into the HindIII and EcoRV sites and Mito-HA-Akt was cloned into the BglII and NotI sites of pShuttle-CMV and each were transformed into BJ5183 bacteria, resulting in Ad-HA-Akt (DD) and Ad-Mito-HA-Akt (DD). All constructs were purified using Qiagen Plasmid Maxiprep kits according to the manufacturer’s protocol. The constructs were confirmed by restriction enzyme digestion and sequencing analysis by the University of Alabama at Birmingham DNA sequencing core facility. Adenoviral constructs expressing either green-fluorescent protein (Ad-GFP; a generous gift from Dr. Chandra Raman, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL) or beta-galactosidase (Ad-LacZ; BD Clontech Laboratories, Inc.; a generous gift from Dr. Bradley Yoder, Department of Cell Biology, University of Alabama at Birmingham, Birmingham, AL) were used as controls for adenoviral infection.
The adenoviruses were amplified by infecting human embryonic kidney 293A (HEK293A) cells. Infected cells were harvested and centrifuged at 1,250g at 4°C for 10 min and the resulting cellular lysate pellet was resuspended in DMEM media without supplements. The cells were disrupted using three freeze/thaw cycles and the suspension was subsequently spun at 1,250g at 4°C for 15 min to release the virus particles. The supernatant containing the viral particles was spun at 100,000g (Beckman SW28 rotor) for 16 h at 4°C through an isopycnic CsCl gradient. The viral band was isolated and dialyzed against 10% glycerol in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 1.47 mM KH2PO4, and 8.1 mM Na2HPO4, pH 7.6) for 16–24 h at 4°C. The viral particle concentration (titer) was determined by absorbance at 260 nm. Viruses were aliquoted and stored at −80°C until used.
Cell Culture, Transient Transfections, Adenoviral Infections, and Treatments
All cells were maintained in a 37°C incubator in 5% CO2. HEK293A cells were grown in DMEM media (Cellgro, Herndon, VA) containing 10% heat-inactivated fetal bovine serum (Gibco BRL, Gaithersburg, MD), 2 mM l-glutamine (Gibco BRL), and 100 U/ml penicillin G-streptomycin (Gibco BRL). Chinese hamster ovary (CHO) cells were grown in F-12 media (Cellgro) containing 5% heat-inactivated fetal bovine serum, 2 mM l-glutamine, and 100 U/ml penicillin G-streptomycin. Human SH-SY5Y neuroblastoma cells were grown in RPMI 1640 media (Cellgro) containing 10% heat-inactivated horse serum (Gibco BRL), 5% heat-inactivated Fetal Clone II (Hyclone, Logan, UT), 2 mM l-glutamine, and 100 U/ml penicillin G-streptomycin.
CHO cells were transiently transfected using FuGENE 6 transfection reagent (Roche Molecular Biochemicals, Indianapolis, IN), according to the manufacturer’s protocol.
For adenovirus infection, SH-SY5Y cells were rinsed twice with DMEM media without supplements, infected with the designated adenovirus for 30 min and the media changed to RPMI without supplements for overnight. After 16–24 h, the cells were used for experiments. All infected cultures were examined for adequate infection efficiency was approximately 80% as assessed by GFP fluorescence.
Staurosporine was prepared as a 1 mM stock solution dissolved in dimethylsulfoxide (DMSO) and stored at −80°C until use. For treatments, the 1 mM stock solution was diluted to 0.5 μM staurosporine in RPMI without supplements and added to the cells.
Immunoblotting
Cells were washed twice with ice-cold PBS and the cell lysates were collected in NP-40 lysis buffer (10 mM Tris, pH 7.5, 0.2% Nonidet P-40, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA) supplemented with protease and phosphatase inhibitors (10 μg/ml leupeptin, 10 μg/ml aprotinin, 10 μg/ml pepstatin, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μM sodium orthovanadate, 50 mM NaF, and 0.1 μM okadaic acid). The samples were sonicated on ice for 10 s and centrifuged at 16,000g for 10 min. The protein concentrations were determined by the bicinchoninic acid (BCA) assay (Pierce Chemical Co., Rockford, IL). Samples were diluted and boiled in 2×-SDS buffer (0.25 M Tris-Cl, pH 7.5, 2% SDS, 25 mM dithiothreitol (DTT), 5 mM EGTA, 5 mM EDTA, 10% glycerol, and 0.01% bromophenol blue). Equal amounts of protein for each sample were electrophoresed on 7.5, 10, 12.5, 15%, or gradient (4–15%) SDS–polyacrylamide gels and transferred onto nitrocellulose membranes. The nitrocellulose membranes were then blocked for 1 h with TBST (20 mM Tris-Cl, pH 7.6, 137 mM NaCl, 0.05% Tween 20) and 5% milk and incubated with the indicated antibodies overnight. The blots were then rinsed with TBST, incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (Jackson ImmunoResearch Labs, Bar Harbor, ME) for 2 h at room temperature. The blots were then rinsed with TBST several times and developed using enhanced chemiluminescence (ECL) (Amersham Pharmacia Biotech, Arlington Heights, IL). Antibodies used for probing the membranes were as follows: Akt, phospho-(Ser/Thr) Akt substrate (PAS), phospho-S9-GSK3β, cleaved caspase-3 (Cell Signaling, Beverly, MA); α-tubulin (Sigma, St. Louis, MO); anti-HA (HA.11, Covance Research Products, Inc., Berkeley, CA); ATP synthase β-subunit (Molecular Probes, Eugene, OR); GSK3β, poly (ADP-ribose) polymerase (PARP), cytochrome c (BD-PharMingen/Transduction Laboratories, San Diego, CA); Bax (Upstate Biotechnology, Inc., Lake Placid, NY).
Cellular Fractionation
Cells were separated into cytosolic and mitochondrial fractions according to previously published procedures [Bijur and Jope, 2003] with the following modifications. All steps were carried out at 0–4°C. Cells were washed twice with ice-cold PBS and collected in 0.5–1 ml cavitation buffer (5 mM HEPES, pH 7.5, 250 mM sucrose, 3 mM MgCl2, 1 mM EGTA) supplemented with protease and phosphatase inhibitors (10 μg/ml leupeptin, 10 μg/ml aprotinin, 10 μg/ml pepstatin, 1 μM PMSF, 1 μM sodium orthovanadate, 50 mM NaF, and 0.1 μM okadaic acid). Cells were disrupted by nitrogen cavitation at 200 p.s.i. for 5 min on ice using a cell disruption bomb (Parr Instrument Co., Moline, IL). The homogenate was centrifuged once at 500g for 5 min to remove unbroken cells and nuclei and the supernatant was further fractionated by ultracentrifugation at 100,000g (Beckman SW50.1 rotor) for 1 h at 4°C through a 1 M/1.5 M discontinuous sucrose gradient containing protease and phosphatase inhibitors. A 200 μl-aliquot of the uppermost layer of the supernatant was collected (cytosolic fraction). The diffuse white band between the sucrose layers was transferred to a microcentrifuge tube, diluted 1:1 (v/v) with cavitation buffer with protease and phosphatase inhibitors, gently mixed and centrifuged at 20,800g for 20 min at 4°C. The resulting pellet (mitochondrial fraction) was gently washed three times (20,800g for 20 min) in cavitation buffer with protease and phosphatase inhibitors. The pellet was resuspended in NP-40 lysis buffer supplemented with protease and phosphatase inhibitors. Protein concentrations were determined by the BCA assay.
In Vitro Akt Kinase Assay
To determine the Akt kinase activity of the HA-Akt constructs, CHO cells were transiently transfected as described above. Cells were washed twice with ice-cold PBS and cell lysates were collected in NP-40 lysis buffer supplemented with protease and phosphatase inhibitors. The samples were sonicated on ice for 10 s and centrifuged at 16,000g for 10 min. The protein concentrations of the cell lysates were determined by BCA assay and diluted to a concentration of 0.5 μg/μl in a final volume of 100 μl using NP-40 lysis buffer containing protease and phosphatase inhibitors. The samples were then immunoprecipitated using 4.9 μg of the monoclonal anti-HA 12CA5 antibody recognizing the HA-epitope (Sigma) (to immunoprecipitate HA-Akt) pre-coupled to protein G-Sepharose beads (Amersham Pharmacia Biotech). The samples were incubated for 3 h at 4°C on a rotational shaker with antibody-protein G complexes. After washing the protein G-Sepharose beads containing the immuno-immobilized HA-Akt twice with NP-40 lysis buffer, the beads were washed two additional times with kinase buffer (20 mM Tris-Cl, pH 7.5, 5 mM MgCl2, and 1 mM DTT).
The activity of Akt was measured as previously described [Burgering and Coffer, 1995; Cross et al., 1995] with the following modifications. The kinase assay was performed in 30 μl kinase buffer containing 250 μM ATP, 2 μCi of [γ-32P] ATP, and 100 μM Crosstide (GRPRTSSFAEG, peptide substrate for Akt; Upstate Biotechnology, Inc.). The samples were incubated at 30°C for 30 min. The reaction tubes were centrifuged for ~1 min and triplicate 9 μl aliquots of reaction supernatants were spotted on 2.5 cm P81 filter paper (Whatman, Florham Park, NJ). Filter papers were allowed to dry for ~1 min and washed four times in 0.5% o-phosphoric acid for 15 min on a bench-top rotational shaker. Filter papers were then washed in 95% ethanol for 15 min and allowed to dry. Dried filter papers were then subjected to scintillation counting. The counts/min for each sample was normalized to total Akt levels immunoprecipitated in the samples which were determined by immunoblotting. To determine the amount of Akt in the immunoprecipitates, 2×-SDS buffer was added to the beads containing the immunoprecipitated Akt and the samples were boiled and then centrifuged at 16,000g for 1 min. The supernatants were electrophoresed on 7.5% SDS–polyacrylamide gels and transferred to nitrocellulose membranes. The blots were then immunoblotted for Akt as described above. Immunoblots were scanned and quantitated using UNSCANIT software (Silk Scientific, Inc., Orem, UT). Akt activity was then normalized to Akt levels in the sample and the data were expressed as percentages of control. The efficiency of Akt immunoprecipitation was determined by immunoblotting total cell lysates for Akt.
To determine the Akt kinase activity of the adenoviral HA-Akt constructs, SH-SY5Y cells were infected with the adenoviral constructs as described above, cell lysates were collected and fractionated and protein concentrations of the fractions were determined by the BCA assay. The mitochondrial fractions were diluted in kinase buffer and 25 μg of the mitochondrial fraction was used to determine the activity of Akt as described above. The counts/min for each sample was normalized to total Akt levels in the mitochondrial fraction of the samples which were determined by immunoblotting. Akt activity was then normalized to Akt levels in the mitochondrial fraction and the data were expressed as percent of control.
Immunocytochemistry
SH-SY5Y cells seeded on 4-well Lab-Tek™ II Chamber Slide™ System (Nagle Nunc International Corporation, Naperville, IL) were infected with either Ad-HA-Akt (DD) or Ad-Mito-HA-Akt (DD) adenoviral constructs as described above. The cells were loaded for 30 min with the specific mitochondrial probe Mitotracker Red CM-H2TMRos (Molecular Probes) at 37°C. The following steps were conducted at room temperature (~25°C) in the dark unless otherwise indicated and all of the solutions were made in PBS. Cells were fixed using 4% paraformaldehyde for 10 min, washed with PBS, treated with 100 mM glycine for 15 min, permeabilized with 1% Triton X-100 for 1 min, and rinsed three times with PBS prior to incubation with 5% FBS for 1 h at 37°C to reduce the background. Cells were incubated overnight with the monoclonal anti-HA 12CA5 antibody (1:1,000) in 10% FBS. Cells were rinsed three times with PBS and incubated for 3 h with 488 Alexa anti-mouse IgG secondary antibody (1:5,000; Molecular Probes) in 5% FBS. Cells were rinsed three times with PBS and incubated for 30 min with 4′,6-diamidino-2-phenylindole (DAPI, 1:10,000; Sigma) in 5% FBS. Slides were washed extensively in PBS prior to coverslip mounting with Immun-mount (Shandon). Cells were visualized using a Nikon Diaphot 200 epifluorescence microscope and a Digital Spot CCD camera (Diagnostic Instruments) was used to capture images which were digitally stored, and displayed using Image-Pro Plus 5.1 software (Media Cybernetic, Silver Spring, MD).
Caspase Assays
After adenoviral-infected SH-SY5Y cells were treated, the cells were washed twice with ice-cold PBS and collected in ice-cold NP-40 lysis buffer supplemented with protease inhibitors (10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin, 100 μM PMSF). Cell lysates were sonicated, centrifuged, and total protein concentration of the supernatant was determined using the BCA assay. Caspase activity was measured in 25 μg of protein as described previously [Bijur et al., 2000] with the following modifications. Peptide substrates for caspase-3 (Ac-DEVD-AMC) and caspase-9 (Ac-LEHD-AMC) were purchased from Alexis Biochemicals (San Diego, CA). The fluorometric assay was conducted in 96-well clear-bottom plates with all measurements done in triplicate wells. Assay plates were incubated at 37°C for 1 h for measurement of caspase-3 and 3 h for measurement of caspase-9. Caspase activity was calculated as ((mean AMC fluorescence from triplicate wells)−(background fluorescence))/μg of protein.
Cytochrome c Release
To determine cytochrome c released from mitochondria, the cytosol was isolated from adenoviral-infected SH-SY5Y cells that had been disrupted by nitrogen cavitation. The homogenate was centrifuged once at 7,000g for 5 min at 4°C to remove unbroken cells and nuclei. The supernatant was centrifuged at 10,000g for 5 min at 4°C and the resulting supernatant (crude cytosolic fraction) was further fractionated by ultracentrifugation at 100,000g (Beckman 50 Ti rotor) for 1 h at 4°C. A 200 μl-aliquot of the uppermost portion of the supernatant was collected (cytosolic fraction). Protein concentration was determined using the BCA assay.
The amount of cytochrome c released into the cytosol was determined using a human cytochrome c ELISA kit (Quantikine; R&D Systems, Minneapolis, MN) according to the manufacturer’s protocol. Samples were run in triplicate and cytochrome c released into the cytosol was expressed as a fold of the level of cytochrome c present in the cytosol of untreated samples for each adenovirus (Ad-GFP, Ad-HA-Akt (DD), and Ad-Mito-HA-Akt (DD)).
Detection of the Mitochondrial Membrane Potential (Δψm)
To determine cell Δψm, SH-SY5Y cells were grown in 48-well tissue culture plates at 5 × 105 cells/well and infected with the adenoviral constructs as described above. After treatment, cell Δψm was determined using 5,5′,6, 6′-tetrachloro-1,1′,3,3-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1; Molecular Probes) as previously described [Ruan et al., 2004].
Bax Oligomerization
To determine Bax homo-oligomerization, mitochondria were isolated from adenoviral-infected SH-SY5Y cells as described above with the following modifications. After cells were disrupted by nitrogen cavitation (200 p.s.i. for 5 min at 4°C), the homogenate was centrifuged once at 500g for 5 min at 4°C to remove unbroken cells and nuclei and the supernatant was further fractionated by centrifugation at 10,000g for 5 min at 4°C. The resulting pellet (mitochondrial fraction) was gently washed two times (10,000g for 5 min at 4°C) in cavitation buffer with protease and phosphatase inhibitors.
Bax homo-oligomerization was determined as previously described [Mikhailov et al., 2003] with the following modifications. The pellet was resuspended with CHAPS buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 2% CHAPS supplemented with protease and phosphatase inhibitors) and extracted for 1 h on ice. At the end of the incubation, protein concentrations were determined by the BCA assay and 50 μg of mitochondrial protein was incubated with the bifunctional cross-linker EGS (ethylene glycolbis(succinic acid N-hydroxysuccinimide ester); Sigma) for 30 min at room temperature with shaking. The reaction was stopped by the addition of 4×-SDS buffer (0.25 M Tris-Cl, pH 7.5, 4% SDS, 25 mM DTT, 5 mM EGTA, 5 mM EDTA, 10% glycerol, and 0.01% bromophenol blue) and the samples were stored at −20°C until use. Samples were boiled, electrophoresed on 4–15% gradient SDS–polyacrylamide gels, transferred to nitrocellulose membranes and then immunoblotted for Bax as described above.
Statistical Analysis
Data are given as the mean±standard error of the mean (SEM). All data were analyzed using a two-tailed paired Student’s t-test or one-way ANOVA and values were considered to be significantly different when P < 0.05.
RESULTS
Characterization of Akt Constructs
To determine the role of Akt in mitochondria, constitutively active HA-tagged Akt constructs were made in which Thr308 and Ser473 were mutated to an aspartic acid (D) residue to mimic phosphorylated Akt (pseudo-phosphorylated). In addition, a mitochondrial targeting signal sequence was added to the N-terminus of the wild-type (wt) and constitutively active (DD) HA-tagged Akt constructs. Figure 1 shows the organization of these constructs. The constructs were transiently expressed in CHO cells to verify cellular expression (Fig. 2A) and subcellular localization (Fig. 2B). As expected, HA-Akt constructs with the mitochondrial targeting signal localize preferentially to the mitochondrial fraction. When mitochondria of cells transiently transfected with Mito-HA-Akt (DD) were further subfractionated to isolate outer membrane, intermembrane space, inner membrane and matrix, mitochondrial-targeted active Akt localized predominantly with the mitochondrial inner membrane fraction (data not shown). To verify that the aspartic acid mutations resulted in a constitutively active Akt, in vitro kinase assays were carried out (Fig. 3). HA-Akt (wt) and HA-Akt (DD) were transiently expressed in CHO cells, incubated overnight in serum free (SF) media and treated either with or without 10 nM IGF-1 for 15 min. Consistent with previous results [Alessi et al., 1996], IGF-1 treatment significantly activated HA-Akt (wt) (~threefold over untreated (SF) HA-Akt (wt)). HA-Akt (DD) showed kinase activity which was significantly increased over untreated (SF) HA-Akt (wt) and not affected by IGF-1 treatment, thus the double aspartic acid mutation resulted in a constitutively active Akt (Fig. 3A).
Fig. 1.
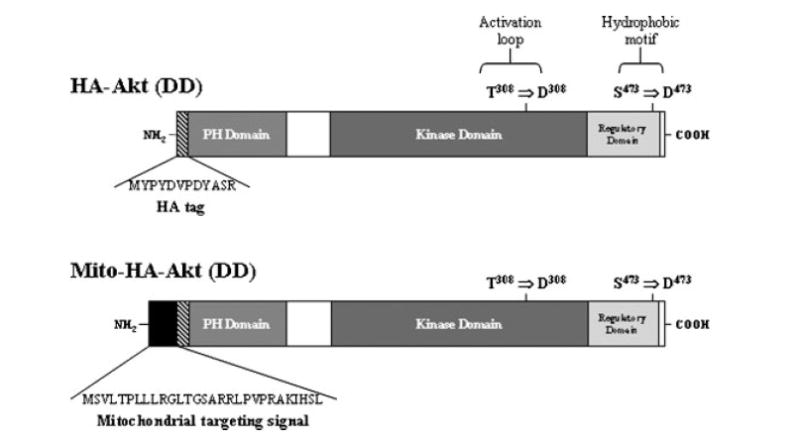
Structure of HA-Akt constructs. The constructs used have an N-terminal HA-tag (diagonal lines) and the mitochondrial-targeted constructs have an N-terminal mitochondrial targeting signal (black). Using site-directed mutagenesis, the phosphorylation sites (T308 and S473) of HA-tagged Akt were mutated to aspartic acid residues (D308 and D473) to generate the constitutively active constructs.
Fig. 2.
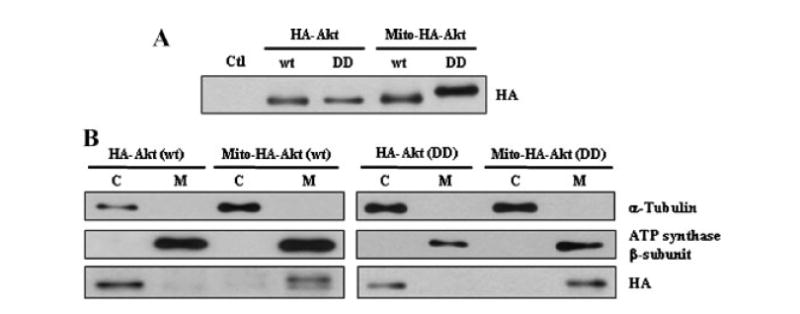
Expression and localization of HA-Akt constructs. A: Cell lysates of CHO cells transiently transfected with empty vector (pcDNA3.1; Ctl) or the HA-Akt constructs (wildtype,wt; constitutively active,DD) were collected and subjected to SDS–PAGE, followed by Western blot analysis with anti-HA antibody to determine expression level of the constructs. B: CHO cells transiently transfected with the HA-Akt constructs were separated into cytosolic (C) and mitochondrial (M) fractions and immunoblotted with the following antibodies: α-tubulin (cytosolic protein), ATP synthase β-subunit (mitochondrial protein), and anti-HA.
Fig. 3.
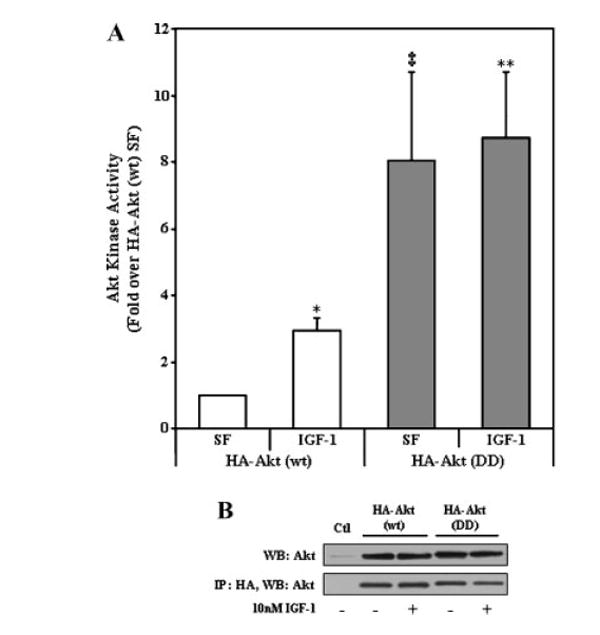
Akt kinase activity of HA-Akt (wt) and HA-Akt (DD) constructs. A: CHO cells transiently transfected with HA-Akt constructs were serum starved overnight and incubated in the absence (serum free [SF]) or presence (IGF-1) of 10 nM IGF-1 for 15 min. Cell lysates were collected, immunoprecipitated with an antibody to HA and Akt kinase activity was determined by in vitro kinase assay. The data are presented as mean ± SEM, n = 3 independent experiments; *, P < 0.05 versus HA-Akt (wt) SF; ‡, P < 0.001 versus HA-Akt (wt) SF; **, P < 0.005 versus HA-Akt (wt) SF (two-tailed, paired Student’s t-test). B: Representative immunoblot of cells transiently transfected with empty vector (pcDNA3.1; Ctl) or the HA-Akt constructs (upper panel). Representative immunoblot of HA immunoprecipitates probed with a polyclonal Akt antibody (lower panel).
Since transfection efficiency is low in SH-SY5Y neuroblastoma cells, we sub-cloned the constitutively active Akt constructs into adenoviral vectors to achieve a higher level of efficiency. The expression and subcellular localization of the adenoviral constitutively active Akt constructs were verified (Fig. 4A, B). Immunocytochemical analysis revealed that the adenoviral vectors provided high expression efficiency with ~80% of the cells expressing the respective construct (data not shown) and that the infection alone did not substantially stress the cells, as nuclear staining appeared normal (Fig. 4A, Dapi panels). Ad-HA-Akt (DD) exhibited diffuse localization throughout the cytosol and colocalized with mitochondria and nuclei (Fig. 4A, left panels). Ad-Mito-HA-Akt (DD) localized in the cytosol, predominantly colocalizing with mitochondria (Fig. 4A, right panels). Further immunoblotting of fractionated infected SH-SY5Y cells verified that Ad-Mito-HA-Akt (DD) overexpressed in SH-SY5Y neuroblastoma cells was enriched in mitochondria (Fig. 4B).
Fig. 4.
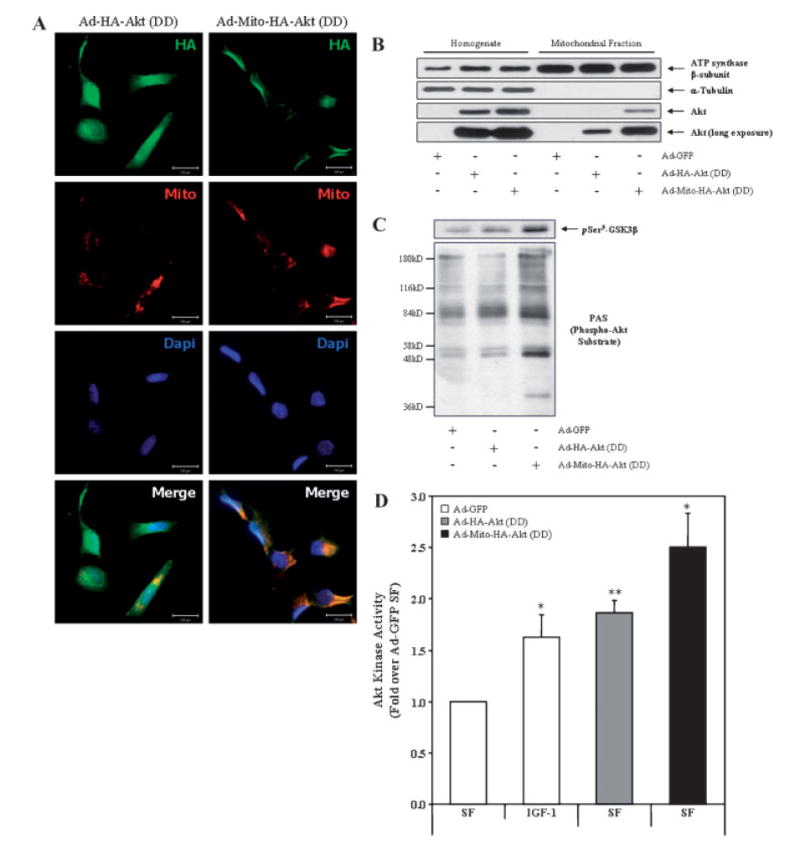
Ad-Mito-HA-Akt (DD) colocalizes with mitochondria and exhibits constitutive activation. A: SH-SY5Y cells seeded on chamber sides were infected with Ad-HA-Akt (DD) or Ad-Mito-HA-Akt (DD) and serum starved overnight. Cells were first loaded with Mitotracker Red CM-H2TMRos (Mito; red) to label mitochondria then fixed and labeled with the monoclonal anti-HA antibody (HA; green) to visualize the Akt adenoviral constructs and Dapi (Dapi; blue) to counterstain the nuclei. B: Homogenates and mitochondrial fractions from SH-SY5Y cells infected overnight with the indicated adenoviral constructs were immunoblotted for ATP synthase β-subunit (mitochondrial protein), α-tubulin (cytosolic protein), and Akt. C: SH-SY5Y cells infected with the designated adenoviral constructs were serum starved overnight, mitochondrial fractions prepared and probed for GSK3β phospho-Ser9 (pSer9-GSK3β) and phospho-Akt Substrate (PAS). D: SH-SY5Y cells infected with designated adenoviral constructs were serum starved overnight prior to incubation in the absence (serum free [SF]) or presence (IGF-1) of 10 nM IGF-1 for 15 min followed by preparation of mitochondria. Akt was immunoprecipitated from the mitochondrial fractions, and Akt kinase activity was determined by an in vitro kinase assay. The data are presented as mean ± SEM, n = 5 independent experiments; *, P < 0.05 versus Ad-GFP SF; **, P < 0.005 versus Ad-GFP SF (two-tailed, paired Student’s t-test).
It is well established that GSK3β is a substrate of Akt and phosphorylation of GSK3β on Ser9 is catalyzed by Akt [Cross et al., 1995]. Further, GSK3β is present within mitochondria and increased phosphorylation of GSK3β at Ser9 was detected in mitochondria from cells expressing Ad-Mito-HA-Akt (DD) (Fig. 4C) [Hoshi et al., 1995]. Increased immunoreactivity with the phospho-(Ser/Thr) Akt substrate (PAS) antibody was also evident in the mitochondrial fraction of cells expressing Ad-Mito-HA-Akt (DD) (Fig. 4C) [Zhang et al., 2002]. To further verify the activity of the adenoviral Akt constructs, in vitro kinase assays were carried out using mitochondrial fractions of infected SH-SY5Y cells (Fig. 4D). SH-SY5Y cells infected with the adenoviral construct expressing green fluorescent protein (Ad-GFP) was used as a control for adenoviral infection. After infection with adenovirus, cells were serum starved overnight prior to measurement of Akt activity in the mitochondrial fractions. As a positive control, SH-SY5Y cells expressing Ad-GFP were treated with 10 nM IGF-1 for 15 min before harvesting. The expression of Ad-HA-Akt (DD) significantly increased mitochondrial Akt kinase activity similar to GFP-expressing cells treated with IGF-1 (~1.75-fold over Ad-GFP untreated cells). The expression of Mito-HA-Akt also significantly increased mitochondrial Akt activity (~2.5-fold) compared with untreated cells expressing Ad-GFP.
Mitochondrial-Targeted Active Akt Significantly Attenuates Caspase-3 Activity and Decreases PARP Proteolysis in Staurosporine-Treated SH-SY5Y Cells
Caspase-3 was highly activated 2 h after treatment of SH-SY5Y cells with 0.5 μM staurosporine (Fig. 5A). Expression of active Akt significantly reduced both basal and staurosporine-induced caspase-3 activity compared with control cells expressing GFP. Remarkably, expression of active Akt targeted to mitochondria was sufficient to reduce basal and staurosporine-induced caspase-3 activity, and these reductions were greater than that achieved by expression of untargeted active Akt. These changes in caspase-3 activity were confirmed by immunoblotting cleaved, active caspase-3 and a proteolytic fragment of PARP which is generated by caspase-cleavage of intact PARP. Staurosporine treatment (0.5 μM, 2 h) caused a large increase in cleaved caspase-3 and cleaved PARP and these were reduced in cells expressing active Akt (Fig. 5B). As was observed by measuring caspase-3 activity, expression of active Akt targeted to mitochondria was sufficient to decrease the production of cleaved caspase-3 and cleaved PARP, and this protection was greater than that achieved by expression of cellular active Akt. Thus, Akt expressed in an active form targeted to mitochondria provided efficient protection of cells from staurosporine-induced caspase-3 activation.
Fig. 5.
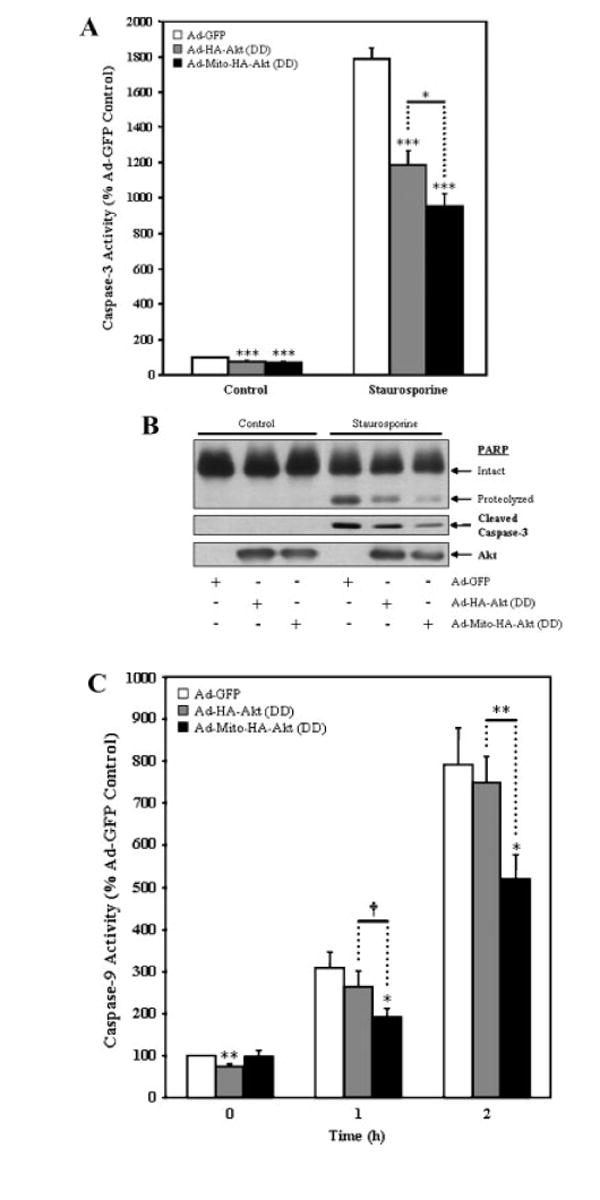
Mitochondrial-targeted active Akt protects SH-SY5Y cells from staurosporine-induced caspase activation. SH-SY5Y cells infected with the designated adenoviral constructs were serum starved overnight and incubated in the absence (control) or presence (staurosporine) of 0.5 μM staurosporine for 2 h (caspase-3 assay and PARP) or 0–2 h (caspase-9 activity). A: Caspase-3 activity. The presence of Ad-HA-Akt (DD) attenuated caspase-3 activation in response to staurosporine, with a significantly greater decrease being observed with Ad-Mito-HA-Akt (DD). The data are presented as mean ± SEM, n = 7 independent experiments; *, P < 0.05; ***, P < 0.0005 (one-way ANOVA). B: Representative blots showing the presence of cleaved PARP, cleaved caspase-3 and Akt. C: Caspase-9 activity. The presence of Ad-HA-Akt (DD) attenuated caspase-9 activation in response to staurosporine, with a significantly greater decrease being observed with Ad-Mito-HA-Akt (DD). The data are presented as mean ± SEM, n = 10 independent experiments; *, P < 0.05; †, P < 0.001; **, P <0.005 (two-tailed, paired Student’s t-test).
Mitochondrial-Targeted Active Akt Significantly Decreases Caspase-9 Activity in Staurosporine-Treated SH-SY5Y Cells
Staurosporine activates the intrinsic apoptosis pathway which causes disruption of mitochondria leading to the activation of caspase-9, an initiator caspase that activates caspase-3 [Kuida, 2000]. Therefore, we hypothesized that mitochondrial-targeted active Akt would reduce caspase-9 activation following staurosporine treatment. Measurements of caspase-9 activity demonstrated that expression of mitochondrial-targeted active Akt attenuated caspase-9 activation 1 and 2 h after staurosporine treatment (Fig. 5C).
Active Akt Significantly Decreases the Level of Cytochrome c Released from Mitochondria in SH-SY5Y Cells Treated With Staurosporine
Cytochrome c resides in the mitochondrial intermembrane space and during mitochondrial apoptotic cell death, cytochrome c is released from the intermembrane space into the cytosol where it can interact with Apaf-1 [Liu et al., 1996; Kroemer and Reed, 2000; Ott et al., 2002]. Since this is an upstream event essential for initiation of the caspase cascade, we measured cytochrome c release from mitochondria in response to staurosporine treatment. Expression of either Ad-HA-Akt (DD) or Ad-Mito-HA-Akt (DD) resulted in a significant decrease in the level of cytochrome c released from staurosporine-treated SH-SY5Y cells compared to cells expressing GFP (Fig. 6).
Fig. 6.
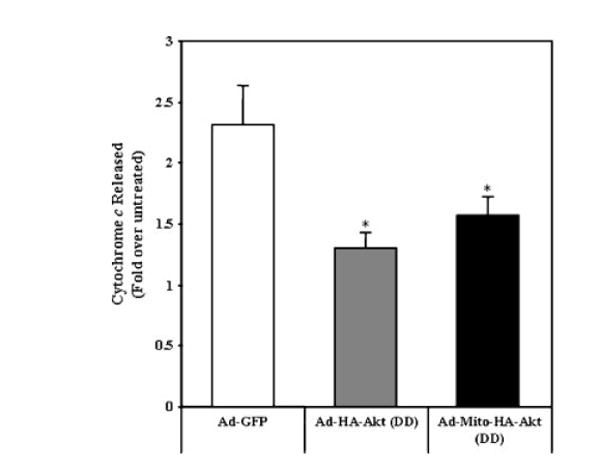
Active Akt reduces cytochrome c released from mitochondria of SH-SY5Y cells treated with staurosporine. SH-SY5Y cells infected with the designated adenoviral constructs were serum starved overnight and treated ± 0.5 μM staurosporine for 2 h. The amount of cytochrome c release from mitochondria was determined by ELISA. The data are presented as mean ± SEM, n = 6 independent experiments; *, P < 0.05 versus staurosporine-treated Ad-GFP (one-way ANOVA).
Active Akt Does not Affect the Mitochondrial Transmembrane Potential (Δψm) in Response to Staurosporine Treatment
When the outer membrane of mitochondria becomes permeabilized, cytochrome c and other proapoptotic proteins are released from the intermembrane space into the cytosol. The mechanism by which mitochondrial outer membrane permeabilization (MOMP) occurs during apoptotic paradigms has not been fully elucidated. One model suggests mitochondrial swelling is a primary mechanism by which cytochrome c is released [Skulachev, 1996; Green and Reed, 1998]. A mechanism for mitochondrial swelling involves mitochondrial permeability transition (MPT) [Susin et al., 1998]. MPT refers to an abrupt transition in permeability of the mitochondrial inner membrane which causes two major changes in mitochondria: (1) depolarization of mitochondria and (2) entry of ions and water into the matrix. Depolarization of mitochondria refers to a decrease in the mitochondrial transmembrane potential (Δψm), which is a loss in the H+ gradient across the inner membrane, causing interference in electron transport and respiration. To determine if this could be the mechanism which resulted in the cytochrome c release observed in staurosporine-treated SH-SY5Y cells we analyzed Δψm using JC-1 as previously described [Ruan et al., 2004]. As expected, staurosporine treatment resulted in hyperpolarization of mitochondria [Vander Heiden et al., 1997; Kennedy et al., 1999; Scarlett et al., 2000; Poppe et al., 2001] with a peak at approximately 1 h and subsequently decreased with no differences being observed between control (Ad-LacZ) infected cells and cells expressing either of the active Akt constructs (Fig. 7).
Fig. 7.
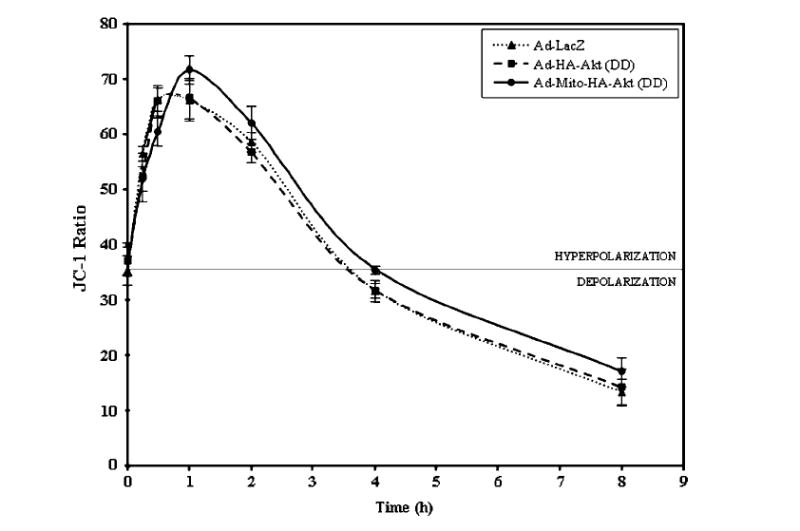
Mitochondrial membrane potential is not altered by the presence of active Akt in SH-SY5Y cells treated with staurosporine. SH-SY5Y cells infected with the designated adenoviral constructs were serum starved overnight and incubated with 0.5 μM staurosporine for the times indicated. The ratio was obtained by dividing JC-1 aggregate fluorescence by monomer fluorescence. The data are presented as mean ± SEM, n = 8 independent experiments.
Mitochondrial-Targeted Active Akt Decreases Bax Oligomerization in Staurosporine-Treated SH-SY5Y Cells
Another model for MOMP suggests that the formation of a protein-permeable pore in the mitochondrial outer membrane is a key factor [Liu et al., 1996; Pavlov et al., 2001; Wei et al., 2001; De Giorgi et al., 2002; Guo et al., 2004; Ruan et al., 2004]. The Bcl-2 proapoptotic family member Bax which has been shown to oligomerize in the outer membrane in response to mitochondrial apoptotic stimuli has been postulated to form the pores [McGinnis et al., 1999; Degli Esposti and Dive, 2003; Scorrano and Korsmeyer, 2003]. Bax translocation and oligomerization in mitochondria in response to staurosporine treatment was evaluated. Staurosporine treatment induced an increase in the level of monomeric Bax at mitochondria in all cells and resulted in the presence of a slower migrating band which corresponded to the migration rate of a dimerized form of Bax (Fig. 8). Staurosporine induced the formation of Bax dimers in Ad-GFP cells which was decreased in cells expressing Ad-HA-Akt (DD) or mitochondrial-targeted active Akt (Fig. 8).
Fig. 8.
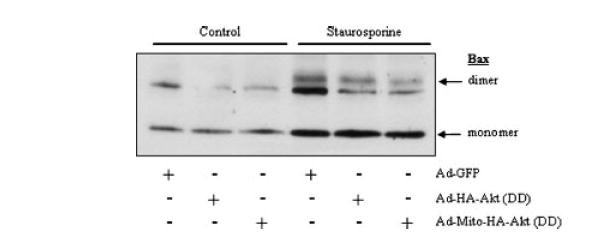
Mitochondrial-targeted active Akt reduces Bax oligomerization in response to staurosporine treatment. SH-SY5Y cells infected with the designated adenoviral constructs were serum starved overnight and incubated in the absence (control) or presence (staurosporine) of 0.5 μM staurosporine for 2 h. The oligomerization of Bax was determined by crosslinking the mitochondrial followed by immunoblotting with a Bax antibody.
DISCUSSION
The results of this study demonstrate for the first time that active Akt within mitochondria attenuates intrinsic apoptotic cell death pathways. Specifically, this capacity of mitochondrial Akt to provide protection was demonstrated using a very powerful inducer of apoptosis, staurosporine, to activate apoptotic signaling in SH-SY5Y neuroblastoma cells. Untargeted Akt, which can be found in the cytosol, nucleus, and mitochondria of cells, is well-known to provide substantial protection from intrinsic-mitochondrial apoptotic pathways [Franke et al., 1997a,b; Zhou et al., 2000; Tang et al., 2001]. Although actions of Akt in the cytosol have been shown to contribute to its anti-apoptotic effects, such as its phosphorylation of cytosolic GSK3β, Bad, and other proteins, no studies have investigated the possibility that Akt in mitochondria may also provide an important component of its anti-apoptotic capacity, primarily because Akt’s import into mitochondria was only recently discovered [Bijur and Jope, 2003]. Considering the central role that mitochondrial events play in intrinsic apoptotic signaling and the well-documented protective effects of Akt in many apoptotic conditions, the presence of Akt in mitochondria places it in a critical position, providing Akt the potential to attenuate mitochondrial apoptotic events. Indeed, this study found that introducing constitutively active Akt into mitochondria attenuated staurosporine-induced disruption of mitochondria that resulted in cytochrome c release, as well as attenuating the subsequent consecutive activation of caspase-9 and caspase-3. Interestingly, expression of mitochondrial-targeted active Akt was more effective than expression of untargeted active Akt in reducing apoptotic signaling, suggesting that both cytosolic and mitochondrial pools of Akt contribute to its anti-apoptotic capacity.
The release of cytochrome c and other intermembrane space proteins from mitochondria occurs as a result of MOMP which has been proposed to occur via a mechanism involving either mitochondrial swelling or the formation of protein-permeable pores [Skulachev, 1996; Pavlov et al., 2001; De Giorgi et al., 2002; Danial and Korsmeyer, 2004]. Since mitochondrial swelling is dependent on MPT with mitochondrial depolarization an indication of MPT the Δψm of staurosporine-treated SH-SY5Y cells was evaluated. Staurosporine has been shown to induce acute hyperpolarization in Jurkat, Rat1a, and SH-SY5Y cells and it is postulated that cytochrome c release and subsequent caspase activation may be preceded by mitochondrial hyperpolarization [Vander Heiden et al., 1997; Kennedy et al., 1999; Scarlett et al., 2000; Poppe et al., 2001]. This suggests that mitochondrial swelling is not the mechanism of MOMP due to staurosporine. In our studies, staurosporine induced transient hyperpolarization prior to cytochrome c release and caspase activation (caspase-3 and caspase-9) in SH-SY5Y cells expressing adenoviral constructs. This is in accordance with the previous studies and the finding that mitochondrial depolarization is not required for cytochrome c release to occur [Krohn et al., 1999].
MOMP is also postulated to result from the formation of protein-permeable pores in the mitochondrial outer membrane, specifically by the Bcl-2 pro-apoptotic family member Bax [Liu et al., 1996; McGinnis et al., 1999; Pavlov et al., 2001; Wei et al., 2001; De Giorgi et al., 2002; Degli Esposti and Dive, 2003; Scorrano and Korsmeyer, 2003; Guo et al., 2004]. Cells treated with staurosporine displayed an increase in monomeric Bax and produced Bax dimers at mitochondria, suggesting that the mechanism for MOMP induced by staurosporine in SH-SY5Y cells is via protein-permeable pores. Expression of mitochondrial-targeted active Akt did not change the level of monomeric Bax; however the level of Bax dimerization was reduced. The exact mechanism through which Bax forms pores in the mitochondrial outer membrane is unclear. A recent paper presented evidence that Bax initially inserts into the mitochondrial outer membrane as a monomer and then undergoes a concerted conformational change and homo-oligomerization to form pores [Annis et al., 2005]. A previous study proposed active Akt to affect the conformation of Bax and thus inhibit Bax oligomerization in the mitochondrial outer membrane [Yamaguchi et al., 2001]. Our results further support this hypothesis and suggest that Akt in mitochondria functions in altering the mechanism through which Bax oligomerizes in the mitochondrial outer membrane.
In summary, it is known that Akt functions to promote cellular survival during growth-factor withdrawal and after cytotoxic insults that initiate the intrinsic apoptotic pathway. Published evidence has shown that Akt can function at both pre-mitochondrial and post-mitochondrial points in the apoptotic cascade. In its regulation of apoptosis, phosphorylation of substrates by cytosolic Akt results in direct inhibition (e.g., GSK3) or the redistribution of pro-apoptotic factors away from their point of action (e.g., Bad) [Cross et al., 1997; Datta et al., 1997; Pap and Cooper, 1998]. This study shows for the first time that Akt in the mitochondrion also suppresses apoptotic signaling. Further studies need to be done to elucidate the precise mechanism by which mitochondrial Akt attenuated apoptotic signaling, which could include directly impeding Bax oligomerization or other actions that reduce mitochondrial dysfunction caused by apoptotic signaling.
Acknowledgments
The authors thank Dr. Thomas Franke for the gift of the HA-Akt (wt) construct, Dr. Chandra Raman for the gift of Ad-GFP, and Dr. Bradley Yoder for the gift of Ad-LacZ.
Grant sponsor: NIH; Grant numbers: NS051279, NS037768.
References
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- Annis MG, Soucie EL, Dlugosz PJ, Cruz-Aguado JA, Penn LZ, Leber B, Andrews DW. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 2005;24:2096–2103. doi: 10.1038/sj.emboj.7600675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P. Akt activation by growth factors is a multiple-step process: The role of the PH domain. Oncogene. 1998;17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem. 2003;87:1427–1435. doi: 10.1046/j.1471-4159.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, De Sarno P, Jope RS. Glycogen synthase kinase-3beta facilitates staurosporine- and heat shock-induced apoptosis. Protection by lithium. J Biol Chem. 2000;275:7583–7590. doi: 10.1074/jbc.275.11.7583. [DOI] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EM, Orr HT, Botas J, Zoghbi HY. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3mediates neurodegeneration in spinocerebellar ataxia type 1. Cell. 2003;113:457–468. doi: 10.1016/s0092-8674(03)00349-0. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Cross DA, Watt PW, Shaw M, van der Kaay J, Downes CP, Holder JC, Cohen P. Insulin activates protein kinase B, inhibits glycogen synthase kinase-3 and activates glycogen synthase by rapamycin-insensitive pathways in skeletal muscle and adipose tissue. FEBS Lett. 1997;406:211–215. doi: 10.1016/s0014-5793(97)00240-8. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: Critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Datta K, Bellacosa A, Chan TO, Tsichlis PN. Akt is a direct target of the phosphatidylinositol 3-kinase. Activation by growth factors, v-src and v-Ha-ras, in Sf9 and mammalian cells. J Biol Chem. 1996;271:30835–30839. doi: 10.1074/jbc.271.48.30835. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- De Giorgi F, Lartigue L, Bauer MK, Schubert A, Grimm S, Hanson GT, Remington SJ, Youle RJ, Ichas F. The permeability transition pore signals apoptosis by directing Bax translocation and multimerization. Faseb J. 2002;16:607–609. doi: 10.1096/fj.01-0269fje. [DOI] [PubMed] [Google Scholar]
- Debatin KM, Poncet D, Kroemer G. Chemotherapy: Targeting the mitochondrial cell death pathway. Oncogene. 2002;21:8786–8803. doi: 10.1038/sj.onc.1206039. [DOI] [PubMed] [Google Scholar]
- Degli Esposti M, Dive C. Mitochondrial membrane permeabilisation by Bax/Bak. Biochem Biophys Res Commun. 2003;304:455–461. doi: 10.1016/s0006-291x(03)00617-x. [DOI] [PubMed] [Google Scholar]
- Dhandapani KM, Wade FM, Wakade C, Mahesh VB, Brann DW. Neuroprotection by stem cell factor in rat cortical neurons involves AKT and NFkappaB. J Neurochem. 2005;95:9–19. doi: 10.1111/j.1471-4159.2005.03319.x. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC. PI3K: Downstream AKTion blocks apoptosis. Cell. 1997a;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997b;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- Gardai SJ, Hildeman DA, Frankel SK, Whitlock BB, Frasch SC, Borregaard N, Marrack P, Bratton DL, Henson PM. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem. 2004;279:21085–21095. doi: 10.1074/jbc.M400063200. [DOI] [PubMed] [Google Scholar]
- Goswami R, Kilkus J, Dawson SA, Dawson G. Overexpression of Akt (protein kinase B) confers protection against apoptosis and prevents formation of ceramide in response to pro-apoptotic stimuli. J Neurosci Res. 1999;57:884–893. [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Guo L, Pietkiewicz D, Pavlov EV, Grigoriev SM, Kasianowicz JJ, Dejean LM, Korsmeyer SJ, Antonsson B, Kinnally KW. Effects of cytochrome c on the mitochondrial apoptosis-induced channel MAC. Am J Physiol Cell Physiol. 2004;286:C1109–C1117. doi: 10.1152/ajpcell.00183.2003. [DOI] [PubMed] [Google Scholar]
- Hoshi M, Sato M, Kondo S, Takashima A, Noguchi K, Takahashi M, Ishiguro K, Imahori K. Different localization of tau protein kinase I/glycogen synthase kinase-3 beta from glycogen synthase kinase-3 alpha in cerebellum mitochondria. J Biochem (Tokyo) 1995;118:683–685. doi: 10.1093/oxfordjournals.jbchem.a124965. [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19:5800–5810. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn AD, Kovacina KS, Roth RA. Insulin stimulates the kinase activity of RAC-PK, a pleckstrin homology domain containing ser/thr kinase. EMBO J. 1995;14:4288–4295. doi: 10.1002/j.1460-2075.1995.tb00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Krohn AJ, Wahlbrink T, Prehn JH. Mitochondrial depolarization is not required for neuronal apoptosis. J Neurosci. 1999;19:7394–7404. doi: 10.1523/JNEUROSCI.19-17-07394.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K. Caspase-9. Int J Biochem Cell Biol. 2000;32:121–124. doi: 10.1016/s1357-2725(99)00024-2. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- McGinnis KM, Gnegy ME, Wang KK. Endogenous bax translocation in SH-SY5Y human neuroblastoma cells and cerebellar granule neurons undergoing apoptosis. J Neurochem. 1999;72:1899–1906. doi: 10.1046/j.1471-4159.1999.0721899.x. [DOI] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- Meier R, Alessi DR, Cron P, Andjelkovic M, Hemmings BA. Mitogenic activation, phosphorylation, and nuclear translocation of protein kinase Bbeta. J Biol Chem. 1997;272:30491–30497. doi: 10.1074/jbc.272.48.30491. [DOI] [PubMed] [Google Scholar]
- Mikhailov V, Mikhailova M, Pulkrabek DJ, Dong Z, Venkatachalam MA, Saikumar P. Bcl-2 prevents Bax oligomerization in the mitochondrial outer membrane. J Biol Chem. 2001;276:18361–18374. doi: 10.1074/jbc.M100655200. [DOI] [PubMed] [Google Scholar]
- Mikhailov V, Mikhailova M, Degenhardt K, Venkatachalam MA, White E, Saikumar P. Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J Biol Chem. 2003;278:5367–5376. doi: 10.1074/jbc.M203392200. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969–8982. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- Pavlov EV, Priault M, Pietkiewicz D, Cheng EH, Antonsson B, Manon S, Korsmeyer SJ, Mannella CA, Kinnally KW. A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J Cell Biol. 2001;155:725–731. doi: 10.1083/jcb.200107057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson RT, Schreiber SL. Kinase phosphorylation: Keeping it all in the family. Curr Biol. 1999;9:R521–R524. doi: 10.1016/s0960-9822(99)80326-1. [DOI] [PubMed] [Google Scholar]
- Poppe M, Reimertz C, Dussmann H, Krohn AJ, Luetjens CM, Bockelmann D, Nieminen AL, Kogel D, Prehn JH. Dissipation of potassium and proton gradients inhibits mitochondrial hyperpolarization and cytochrome c release during neural apoptosis. J Neurosci. 2001;21:4551–4563. doi: 10.1523/JNEUROSCI.21-13-04551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Q, Lesort M, MacDonald ME, Johnson GV. Striatal cells from mutant huntingtin knock-in mice are selectively vulnerable to mitochondrial complex II inhibitor-induced cell death through a non-apoptotic pathway. Hum Mol Genet. 2004;13:669–681. doi: 10.1093/hmg/ddh082. [DOI] [PubMed] [Google Scholar]
- Salinas M, Martin D, Alvarez A, Cuadrado A. Akt1/PKBalpha protects PC12 cells against the parkinsonism-inducing neurotoxin 1-methyl-4-phenylpyridinium and reduces the levels of oxygen-free radicals. Mol Cell Neurosci. 2001;17:67–77. doi: 10.1006/mcne.2000.0921. [DOI] [PubMed] [Google Scholar]
- Scarlett JL, Sheard PW, Hughes G, Ledgerwood EC, Ku HH, Murphy MP. Changes in mitochondrial membrane potential during staurosporine-induced apoptosis in Jurkat cells. FEBS Lett. 2000;475:267–272. doi: 10.1016/s0014-5793(00)01681-1. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Huber M, Damen JE, Hughes M, Kang V, Neilsen P, Prestwich GD, Krystal G, Duronio V. Phosphatidylinositol (3,4,5)P3 is essential but not sufficient for protein kinase B (PKB) activation; phosphatidylinositol (3,4)P2 is required for PKB phosphorylation at Ser-473: Studies using cells from SH2-containing inositol-5-phosphatase knockout mice. J Biol Chem. 2002a;277:9027–9035. doi: 10.1074/jbc.M106755200. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Marignani PA, Woodgett JR. Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol Cell Biol. 2002b;22:6247–6260. doi: 10.1128/MCB.22.17.6247-6260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun. 2003;304:437–444. doi: 10.1016/s0006-291x(03)00615-6. [DOI] [PubMed] [Google Scholar]
- Skulachev VP. Why are mitochondria involved in apoptosis? Permeability transition pores and apoptosis as selective mechanisms to eliminate superoxide-producing mitochondria and cell. FEBS Lett. 1996;397:7–10. doi: 10.1016/0014-5793(96)00989-1. [DOI] [PubMed] [Google Scholar]
- Susin SA, Zamzami N, Kroemer G. Mitochondria as regulators of apoptosis: Doubt no more. Biochim Biophys Acta. 1998;1366:151–165. doi: 10.1016/s0005-2728(98)00110-8. [DOI] [PubMed] [Google Scholar]
- Tang D, Okada H, Ruland J, Liu L, Stambolic V, Mak TW, Ingram AJ. Akt is activated in response to an apoptotic signal. J Biol Chem. 2001;276:30461–30466. doi: 10.1074/jbc.M102045200. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem. 2001;276:5256–5264. doi: 10.1074/jbc.M008552200. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Zha H, Aime-Sempe C, Sato T, Reed JC. Proapoptotic protein Bax heterodimerizes with Bcl-2 and homodimerizes with Bax via a novel domain (BH3) distinct from BH1 and BH2. J Biol Chem. 1996a;271:7440–7444. doi: 10.1074/jbc.271.13.7440. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996b;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Osipov K, Jockel J, Waksman G, Korsmeyer SJ. BH3 domain of BAD is required for heterodimerization with BCL-XL and pro-apoptotic activity. J Biol Chem. 1997;272:24101–24104. doi: 10.1074/jbc.272.39.24101. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zha X, Tan Y, Hornbeck PV, Mastrangelo AJ, Alessi DR, Polakiewicz RD, Comb MJ. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J Biol Chem. 2002;277:39379–39387. doi: 10.1074/jbc.M206399200. [DOI] [PubMed] [Google Scholar]
- Zhou H, Li XM, Meinkoth J, Pittman RN. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol. 2000;151:483–494. doi: 10.1083/jcb.151.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]