

SUMMARY
MicroRNAs (miRNA) are regulatory molecules that participate in diverse biological processes in animals and plants. While thousands of mammalian genes are potentially targeted by miRNAs, the functions of miRNAs in the context of gene networks are not well understood. Specifically, it is unknown whether miRNA-containing networks have recurrent circuit motifs, as has been observed in regulatory networks of bacteria and yeast. Here we develop a computational method that utilizes gene expression data to show that two classes of circuits—corresponding to positive and negative transcriptional co-regulation of a miRNA and its targets—are prevalent in the human and mouse genomes. Additionally, we find that neuronal-enriched miRNAs tend to be coexpressed with their target genes, suggesting that these miRNAs could be involved in neuronal homeostasis. Our results strongly suggest that coordinated transcriptional and miRNA-mediated regulation is a recurrent motif to enhance the robustness of gene regulation in mammalian genomes.
INTRODUCTION
MicroRNAs (miRNA) are post-transcriptional regulatory molecules recently discovered in animals and plants (review in (Bartel, 2004)). They have been shown to regulate diverse biological processes ranging from embryonic development to the regulation of synaptic plasticity (Carthew, 2006; Kloosterman and Plasterk, 2006). Primary miRNA transcripts are predominantly transcribed by RNA Polymerase II. After multiple steps of transcript processing, the mature miRNA (∼22 bps) is incorporated into the RISC complex in the cytoplasm. Mature miRNAs suppress gene expression via imperfect base pairing to the 3′ untranslated region (3′UTR) of target mRNAs, leading to repression of protein production, and in some cases, mRNA degradation (Bartel, 2004; Carthew, 2006; Valencia-Sanchez et al., 2006). Hundreds of miRNA genes have been identified in mammalian genomes (Griffiths-Jones et al., 2006), and computational predictions indicate that thousands of genes could be targeted by miRNAs in mammals (John et al., 2004; Krek et al., 2005; Lewis et al., 2005; Rajewsky, 2006). These findings suggest that miRNAs play an integral role in genome-wide regulation of gene expression.
Similar to electronic circuits, gene regulatory networks (GRN) are made up of basic subcircuits, such as feedback and feedforward loops. Pioneering work in E. coli has shown that certain subcircuits are favored by evolution and hence are significantly more abundant than others (Shen-Orr et al., 2002). The identification of these recurring subcircuits, called network motifs (Milo et al., 2002), has offered key insights into gene regulation. For instance, ∼35% of E. coli transcription factors repress their own transcription and such negative auto-regulatory circuits can significantly accelerate transcriptional response time (Rosenfeld et al., 2002) and dampen protein expression fluctuations (Becskei and Serrano, 2000). Like transcriptional repressors, miRNAs are likely embedded in a large number of GRNs, in which certain miRNA-containing circuits may be recurrent. While all miRNAs operate through a repressive mechanism, their functions in networks need not be simply repressive; they could have diverse functions depending on the unique GRN context of individual miRNA-target interactions. Hence, the identification of recurring miRNA-containing motifs in GRNs would greatly increase our understanding of the functional roles of miRNAs in gene regulation.
Only a few studies have experimentally explored miRNA function in the context of a GRN. They suggest that a key recurring function of miRNAs in networks is to reinforce the gene expression program of differentiated cellular states. For instance, the secondary vulva cell fate in C. elegans is promoted by Notch signaling, which also activates miR-61; miR-61 in turn post-transcriptionally represses an inhibitory factor of Notch signaling, thereby stabilizing the secondary vulva fate (Yoo and Greenwald, 2005). Networks of similar architecture can also be found in the asymmetric differentiation of left-right neurons in C. elegans (Johnston et al., 2005), eye and sensory organ precursor development in Drosophila (Li and Carthew, 2005; Li et al., 2006), and granulocytic differentiation in human (Fazi et al., 2005).
The repressive effect of miRNAs on target expression is modest and is often limited to the level of translation with little effects on transcript abundance (Bartel, 2004). Thus, an important question is whether miRNAs act in concert with other regulatory processes, such as transcriptional control, to regulate target gene expression at multiple levels and with greater strength. One possibility is that the transcription of the miRNAs and their targets is oppositely regulated by common upstream factor(s) (Type II circuits, Figure 1). For instance, an upstream factor could repress the transcription of a target gene and simultaneously activate the transcription of a miRNA that inhibits target-gene translation. Type II circuits may be prevalent as genome-scale studies have shown that predicted target transcripts of several tissue-specific miRNAs tend to be expressed at a lower level in tissues where the miRNAs are expressed (Farh et al., 2005; Sood et al., 2006; Stark et al., 2005). In contrast, there is little evidence for circuits in which the transcription of the miRNAs and their targets are positively co-regulated (Type I circuits, Figure 1); only one such example has been confirmed experimentally, where miR-17-5p represses E2F1, and both are transcriptionally activated by c-Myc in human cells (O'Donnell et al., 2005). While Type I circuits may seem counterintuitive and its functional significance has not been fully characterized, Type I circuits have the potential to provide a host of regulatory and signal processing functions (Hornstein and Shomron, 2006), such as the fine-tuning and maintenance of protein steady-states (see Discussion).
Figure 1. Two Classes of miRNA-containing Circuits.
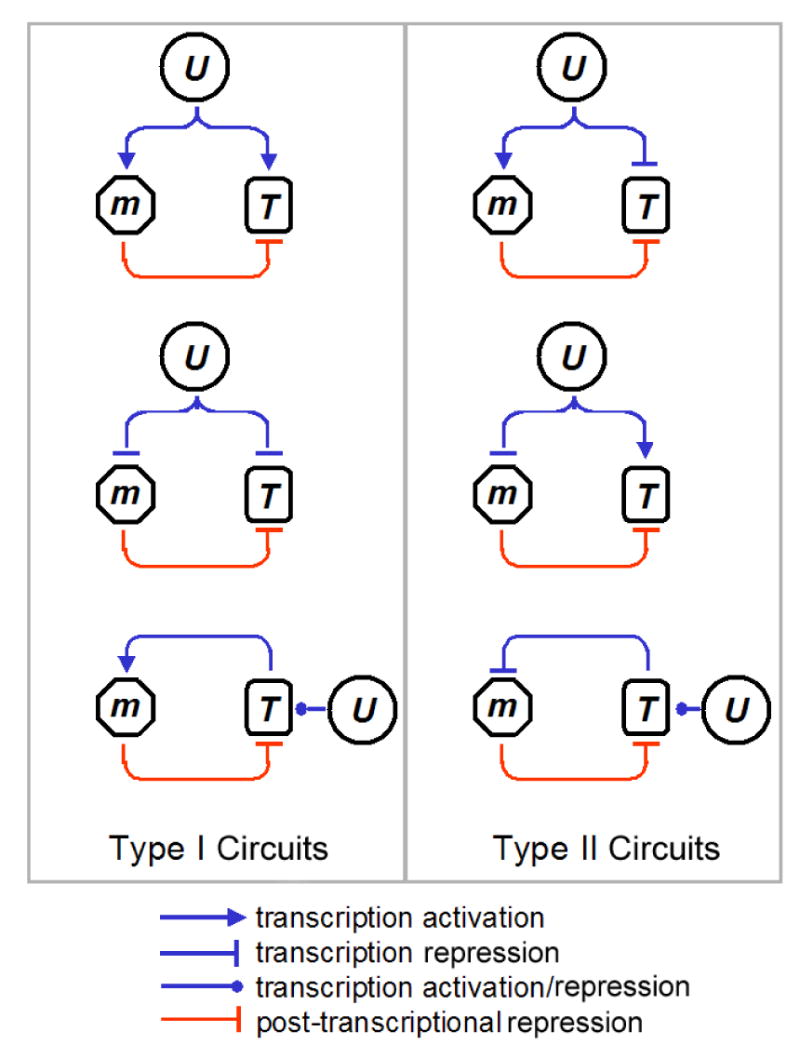
U denotes an upstream factor or a process that regulates the transcription rate of a miRNA (m) and one of its targets (T). In Type I (II) circuits, the transcription rate of m and T are positively (negatively) co-regulated by U. Both Type I and II circuits are topologically characterized by miRNA-mediated feedback or feedforward loops acting on T. Note that in the case of the positive (Type II) and negative (Type I) feedback circuits, U regulates the transcription rate of m indirectly through T.
While individual examples of Type I and II circuits exist in mammalian GRNs, our goal is to determine whether these circuits are recurrent (i.e. more prevalent than would be expected by chance). Although existing experimental data suggests that Type II circuits are prevalent and Type I circuits are not, the number of examples is far too few to be conclusive. It is possible that the apparent lack of evidence for the prevalence of Type I circuits is due to the bias in the choice of experimental systems, i.e., most existing studies used cellular differentiation systems where Type II circuits function to reinforce differentiation decisions.
Given the dearth of known miRNA-containing networks, it is infeasible to directly determine whether Type I/II circuits are recurrent. However, if a miRNA is involved in a larger number of Type I (Type II) circuits than expected by chance, one would expect the transcription of the miRNA and a significant number of its targets to be positively (negatively) correlated across diverse conditions. There are three challenges that complicate the identification of such correlation signatures. The first challenge is that large-scale expression data sets containing both miRNAs and protein-coding genes are lacking. We address this challenge by taking advantage of the large number of miRNAs that are embedded in the introns of protein-coding genes in human and mouse (Rodriguez et al., 2004). With few exceptions (e.g. miR-7 during Drosophila embryogenesis (Aboobaker et al., 2005)), the expression profiles of embedded miRNAs examined thus far are highly correlated to their host genes at both the tissue and individual cell levels (Aboobaker et al., 2005; Baskerville and Bartel, 2005; Li and Carthew, 2005), suggesting that they tend to be co-transcribed at identical rates from the same promoter(s) (Kim and Kim, 2007). Hence, the relative level of host-gene transcription across conditions can accurately serve as a proxy for that of the embedded miRNA(s), even though the steady-state levels of host-gene mRNA and that of the embedded miRNA(s) may be different.
The second challenge is that only a few miRNA targets have been verified in vivo and computational target predictions can be noisy (Rajewsky, 2006). We address this challenge by developing a robust method that does not rely on target prediction to detect significant over-abundance of Type I and/or II circuits.
The third challenge is that most existing mammalian expression data sets tend to study tissues, not individual cell types. While expression correlation over tissue conditions is likely due to transcriptional co-regulation by common upstream factors, cell-type heterogeneity in tissues can complicate the analysis. For example, some miRNAs and their targets could be expressed in distinct cell types within a tissue even though their averaged expression at the tissue level may suggest that their expression is correlated. To address this challenge, we analyze expression data from homogeneous neuronal cell populations (Arlotta et al., 2005; Sugino et al., 2006).
We consistently observe that Type I and/or II circuits are prevalent for a significant fraction of the embedded miRNAs we analyzed, independent of the gene expression data sets used in the analysis, suggesting that these two circuit types are recurrent motifs in mammalian genomes. Strikingly, brain-enriched miRNAs tend to target brain-enriched genes and Type I circuits are especially prevalent in mature neurons. Our findings not only confirm that Type II circuits are abundant, but reveal the surprising genome-wide prevalence of Type I circuits, suggesting that miRNAs are employed in recurrent gene regulatory circuits to perform important biological functions in mammals.
RESULTS
Genes Highly Correlated in Expression with a miRNA are More likely to be Predicted as Targets
We first sought to determine whether embedded miRNAs tend to correlate in expression with their putative targets, a phenomenon we term “targeting bias”, by analyzing the Novartis human expression atlas (Su et al., 2004) that comprises 79 distinct tissues/cell types (Figure 2A). Hierarchical clustering indicates that a large number of embedded miRNA host genes are characterized by upregulation in immune/cancer (Group II) and brain tissues (Groups I and IV), suggesting that these miRNAs are important in cell proliferation and brain functions, respectively. For embedded miRNAs (e.g. miR-9/9*, -153, -128) that are known to be expressed in the adult brain (Cao et al., 2006), their host genes belonged to Groups I and IV; while host genes of cancer-related miRNAs (Esquela-Kerscher and Slack, 2006), such as miR-15a/16, are clustered in Group II (Figure 2A). These observations further assure us that host-gene transcription patterns can be used as a proxy for that of the embedded miRNA.
Figure 2. Targeting Bias Analysis of Human Embedded miRNAs.
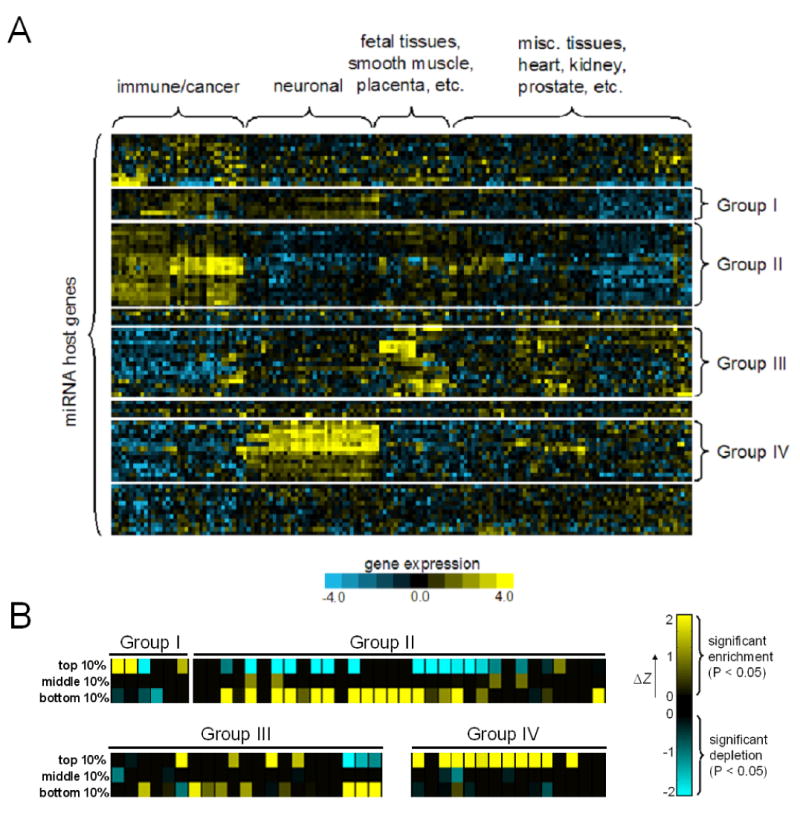
(A) The expression profiles of human embedded miRNA host genes in the Novartis atlas. Four prominent clusters were identified by hierarchical clustering.
(B) Targeting bias enrichment scores of miRNAs from major expression clusters. The color scale reflects the degree of deviation from the 5% significance level (ΔZ), while black denotes insignificant enrichment/depletion (i.e. P>0.05).
To assess targeting bias, we first made a ranked list of genes for each embedded miRNA based on the extent of their expression correlation and compiled a list of targets for each miRNA using the TargetScanS algorithm (Lewis et al., 2005). To examine if a miRNA's predicted target set is enriched in genes that are highly correlated or anti-correlated in expression with the miRNA, we devised a statistical test based on the hypergeometric distribution (see Methods). For the 60 miRNAs we analyzed, 75% have a significantly higher number of predicted targets (P < 0.05) in the top- or bottom-10 percentile of the ranked expression-correlation list. For instance, the number of predicted targets of miR-153 that fall in the top-10 percentile of its ranked list is twice more than expected (P<10−30). In contrast, the predicted target set of only 8% of the miRNA we analyzed show significant enrichment for genes in the middle-10 percentile of the ranked list (Figure 2B). Interestingly, most brain-enriched miRNAs (Group IV) exhibit targeting bias for positively correlated genes (Figure 2B, all with P<0.01). Similar targeting bias trends were observed for mouse embedded miRNAs using the Novartis mouse expression atlas (Su et al., 2004) (data not shown). From these results, we conclude that many embedded miRNAs indeed exhibit targeting bias, in which their expression is often highly correlated or anticorrelated with putative targets.
A Computational Method for Predicting whether a miRNA is biased for Type I/II Circuits given an Expression Data Set
Although our observation of targeting bias is encouraging, one potential concern is that the analysis requires miRNA target prediction, which can be noisy (Lewis et al., 2005; Rajewsky, 2006). Furthermore, genes with different expression patterns might have different 3′UTR length distributions (Stark et al., 2005). In principle, these problems might have contributed to the targeting bias we observed. Therefore, we developed an alternative method that avoids target prediction and uses a measure that is independent of 3′UTR lengths. The new measure stems from the observation that putative miRNA binding sites have a higher probability of being evolutionarily conserved across mammalian genomes (Lewis et al., 2005; Xie et al., 2005). We reasoned that if a miRNA (m) is enriched in Type I (II) networks, a higher than expected proportion of putative binding sites in the 3′UTR of genes positively (negatively) correlated in expression with m should be functional in vivo, and hence evolutionarily conserved. In essence, given a group of genes (G) and a miRNA seed, our method counts the number of seed matches (S) in the 3′UTRs of G and the number of those matches that are conserved (C) (gray box in Figure 3), then it computes a conservation enrichment (CE) score to indicate whether the observed conservation rate (CR=C/S) is significantly higher than that of randomly drawn gene sets (Figure 3 step 3; CE>1.65 corresponds to P<0.05). Because the CR measure is normalized by the number of binding site occurrences, it is independent of biases in 3′UTR length and base composition.
Figure 3. Computing the CE Score Profile of a miRNA given an Expression Data Set.
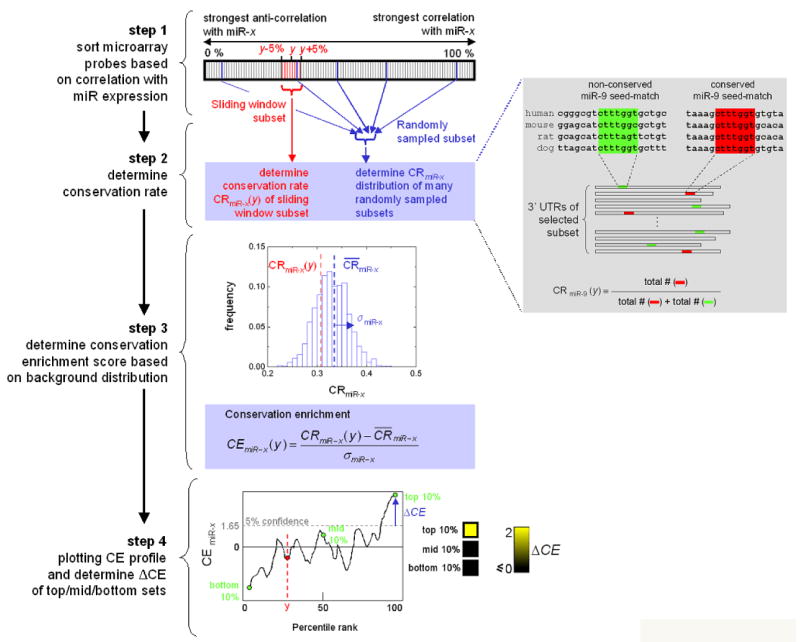
Step 1: sort all genes on the microarray based on their expression correlation to the miRNA host gene. Step 2: for each sliding-window subset, the conservation rate (CR) of the miRNA seed-matches is computed by counting the number of occurrences of conserved and non-conserved matches (gray box). Step 3: The CR is used to compute the CE score based on the background CE score distribution obtained from randomly drawn gene sets (σ denotes the standard deviation of the background CR distribution). The CE score is the number of standard deviations (σ) away from the expected conservation rate ( ) of gene sets drawn at random. Step 4: the centers of each sliding window subset (e.g. the top-10 percentile set centers at 95) are plotted against the corresponding CE scores. We summarize the data by showing the scores of the top-10, middle-10, and bottom-10 percentile sets as a heatmap. The yellow scale reflects the amount of deviation (ΔCE) from the 5% significance level (i.e. CE>1.65). Insignificant scores (CE <= 1.65) are shown in black.
To determine whether a miRNA m exhibits bias for Type I and/or II circuits given an expression data set, we first rank genes by their expression correlation with m and slide a fixed-size window across the ranked list to generate a series of gene groups (G) with decreasing degrees of expression correlation to m (Figure 3 step 1). For each group in G we compute the CE score of the two 7-mer seeds (m1-7, m2-8) of m (Figure 3 steps 2-3) (Lewis et al., 2005). If m is enriched in Type I (II) networks, the CE score of at least one of the seeds in highly (lowly) ranked groups is likely to be significant. We designate a miRNA to exhibit Type I (II) bias if the CE score of its top (bottom) 10 percentile gene set is significant (i.e. CE > 1.65; Figure 3 step 4).
Many Human and Mouse Embedded miRNAs Exhibit Bias for Type I/II Circuits
We applied CE analysis to human embedded miRNAs, again using the Novartis atlas. Of the 60 miRNAs we analyzed, 67% have a significant CE score in their top-10 or bottom-10 percentile sets (P<0.05) (Figure 4A), compared to only 8% having a significant CE score in their middle-10 percentile sets (i.e. group of genes having insignificant expression correlation). Overall, the CE scores of the top-10 and bottom-10 percentile sets are significantly higher than that of the middle-10 percentile sets (Kolmogorov-Smirnov Test, P=2×10−11 and 3.6×10−5 respectively). Representative CE score profiles are shown in Figures 4B-D. Interestingly, more miRNAs show Type I bias (48% vs 27%), partially due to the relative abundance of brain-enriched miRNAs we analyzed. As was observed in the targeting bias analysis, a large number of miRNAs (8 out of 12) in the brain cluster (Group IV) show Type I bias, but none show bias for Type II (Figure 4A), suggesting that these miRNAs are not primarily involved in reinforcing the suppression of genes expressed outside the brain (except miR-9, which shows bias for both). The biases shown by non-brain-enriched miRNAs (i.e. Groups I and IV) are more evenly distributed among the two types (e.g. Group II, Figure 4A). Several miRNAs have significant CE scores in both the top-10 and bottom-10 percentile sets, such as miR-9 and miR-15a/15b/16 (both copies: chromosomes 3 and 13), indicating that their involvement in both circuit types is prevalent.
Figure 4. CE Analysis of the Novartis Expression Atlas for Human and Mouse.
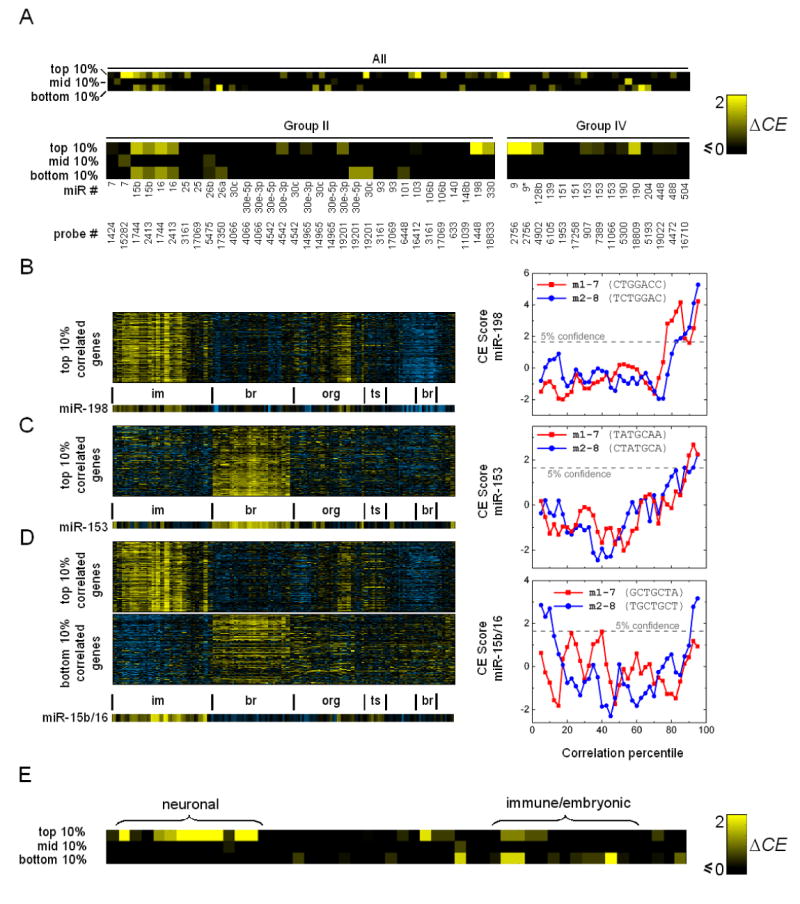
(A) ΔCE scores of the top-10, middle-10, and bottom-10 percentile sets of human embedded miRNAs we analyzed. The scores of miRNAs in Group II (immune/cancer expression signature) and IV (brain-enriched) are shown. Since some host genes have multiple microarray probes, the probe IDs are also shown.
(B-D) Representative CE profiles of biased miRNAs along with their expression patterns. Expression conditions: im=immune/cancer; br=brain; org= organs; ts=testis-related.
(B) CE score profile of miR-198. Expression patterns of genes in the top-10 percentile set are also shown.
(C) miR-153 is brain-enriched and its Type I CE score profile is typical of miRNAs in Group IV.
(D) miR-15b and miR-16 (embedded in the same host gene) exhibit both Type I and II biases.
(E) ΔCE scores of the top-10, middle-10, and bottom-10 percentile sets of mouse embedded miRNAs we analyzed. Brain-enriched miRNAs only exhibit significant CE scores in their top-10 percentile sets.
To assess whether the abundance of Type I/II biases is a general feature of mammalian gene regulation, we conducted CE analysis on embedded miRNAs in mouse using the Novartis mouse expression atlas comprising 61 tissues/cell types (Su et al., 2004). The overall trends are consistent with those of human: of the 45 miRNAs analyzed, 69% exhibit bias for either type, with 42% and 36% displaying bias for Type I and II (9% showing both), respectively (Figure 4E). In comparison, less than 7% have a significant CE score in their middle-10 percentile set. As is the case for human, mouse brain-enriched miRNAs only exhibit Type I bias, whereas other biased miRNAs are more evenly distributed among the two types. Since Type I/II signatures are prevalent in human and mouse, these signatures are unlikely a result of data irregularity or noise particular to one expression data set, and we conclude that both circuit classes are recurrent in mammalian miRNA-containing networks. Our results independently confirm that host-gene transcription patterns largely reflect those of embedded miRNAs. If the opposite is true, one would expect a much smaller fraction of miRNAs to have significant CE scores in the top-10 and/or bottom-10 percentile sets, likely similar to the fraction of miRNAs with significant CE scores in the middle-10 percentile set (i.e. random noise).
The fact that brain-enriched miRNAs tend to target brain-enriched genes suggests that some of them may be positively co-regulated by transcription factors that specify neuronal fates (e.g. NRSF/REST (Ballas et al., 2005); see Discussion). However, due to the heterogeneity of cell types in brain tissues, it is possible that the expression of some of these miRNAs and their targets does not overlap at the single-cell level. This suggests that some brain-enriched miRNAs may also be enriched in Type II circuits, even though tissue-level analysis only shows Type I bias due to the lack of coverage of individual cell types. Therefore, expression data on homogeneous neuronal cell populations are needed to confirm whether Type I circuits are indeed prevalent in the adult brain (see below). Non-brain-enriched miRNAs with Type I bias are less likely to be confounded by cell type heterogeneity, because unlike brain-enriched miRNAs, they tend to be differentially regulated across diverse tissue and homogeneous cultured-cell conditions (e.g. miR-198; Figure 4B); hence positive expression correlation between a miRNA and its targets across diverse conditions suggests that they are transcriptionally co-regulated by common upstream factors.
The Prevalence of Type I/II Biases Persists in Homogeneous Neuronal Cell Population Expression Profiles
To address the concern that the human and mouse data sets consist mostly of tissues but are lacking in homogeneous cell-types, we conducted CE analysis on embedded miRNAs using two additional mouse expression data sets: the developmental time-course of three types of motor neurons (MDEV) (Arlotta et al., 2005) and profiles of 12 homogeneous neuronal cell types (NCELL) from five different brain regions (Sugino et al., 2006). Both data sets were obtained by careful isolation of homogeneous neuronal cell populations.
The MDEV profiles comprise three motor neuron types over four developmental stages (E18, P3, P6, P14). The samples were isolated from the mouse neocortex using a combination of anatomical and cell sorting techniques. The expression data are highly reproducible across biological replicates isolated from different mice (Arlotta et al., 2005), indicating that noise resulting from the cross-contamination of other cell types is minimal. Of the 42 embedded miRNAs analyzed, 45% exhibit bias, with 29% and 24% having significant CE scores in their top-10 and bottom-10 percentile sets, respectively; while only 12% show significant CE scores in their middle-10 percentile sets (Figure 5). It is not surprising that a smaller percentage of miRNAs exhibit bias compared to the Novartis atlas, as this data set only covers the development of a few neuronal subtypes and fewer miRNAs are expected to be regulated under these conditions. In fact, the prevalence of Type I/II signatures is quite remarkable considering the limited coverage of conditions, suggesting that miRNAs have diverse functions and are deployed in a myriad of pathways. The biased miRNAs include ones known to be neuronal (e.g. miR-7) and those whose host gene has brain-enriched expression patterns (e.g. miR-326). However, several have host genes that are broadly down-regulated in brain tissues of the adult brain according to the Novartis atlas (e.g. miR-106b), indicating that they may be transiently expressed during brain development. For instance, the host of miR-106b has a Type II signature, and its expression is elevated at E18 but continues to fall as development proceeds (data not shown), suggesting that it may function in a large number of Type II networks to reinforce the suppression of genes specific to later developmental stages.
Figure 5. CE Analysis of the Mouse Motor Neuron Development (MDEV) and Homogeneous Neuronal Cell (NCELL) Data Sets.

(A) ΔCE scores of the top/middle/bottom percentile sets of mouse embedded miRNAs in the MDEV data set.
(B-C) CE score profiles of miR-103 and miR-342, representative of miRNAs showing Type I and II bias, respectively.
(C) ΔCE scores of the top-10, middle-10, and bottom-10 percentile sets of embedded miRNAs in the NCELL data set.
Similar trends were observed in the NCELL data set, though a higher proportion (61%) of the miRNAs analyzed exhibit bias, consistent with the fact that this data set consists of more diverse conditions than the MDEV profiles (Figure 5D). Interestingly, significantly more miRNAs exhibit Type I bias than Type II (44% vs 17%). Since this data set consists of mature neurons, our results suggest that Type I circuits may be more prevalent in networks that carry out homeostatic neuronal functions (see Discussion). The large proportion of miRNAs that exhibit Type I and/or II bias at the individual cell-type level strongly suggests that cell type heterogeneity alone is unlikely to explain the similar results we observed in the human and mouse atlas data sets. We conclude that Type I and II circuits are indeed prevalent in the human and mouse genomes.
While some miRNAs consistently exhibit bias across all data sets we analyzed, others only show bias in some data sets (see Supplemental Data for examples). This is to be expected as the bias detected by our method largely depends on the conditions profiled in each data set. As with protein-coding genes, the transcription of a miRNA is likely regulated by multiple cis regulatory modules (Howard and Davidson, 2004); thus a miRNA can be involved in a large number of Type I or II circuits via different cis modules (see Discussion). A particular set of profiled conditions in a given data set may only reveal transcription patterns due to the regulation of a subset of these modules. In essence, two key factors determine whether a miRNA would exhibit bias as detected by CE analysis: first, whether the expression of the miRNA is differentially regulated across the profiled conditions; and second, whether a higher than expected number of functional targets (functional implies the seed-matches of the miRNA have a higher probability of being conserved) are positively or negatively co-regulated with the miRNA. As more expression data containing both miRNAs and protein-coding genes become available, CE analysis can readily be applied to further dissect the prevalence of Type I and II circuits in diverse biological contexts.
Identification of miRNAs with Neuronal-enriched Expression Patterns
Motivated by the observation that brain-enriched miRNAs tend to target brain-enriched genes, we reasoned that brain-enriched miRNAs can be identified by searching for miRNAs with significant CE scores in a group of genes with a brain-enriched expression signature. To test this idea, we applied CE analysis to a group of mouse genes whose expression is upregulated across all neuronal tissues profiled in the Novartis atlas (Figure S1). Table 1 lists all miRNA seed-matches with a significant CE score (CE > 2.33, P<0.01). Strikingly, 9 out of 10 seeds at the top of the list correspond to miRNAs known to be brain-specific or -enriched. For instance, miR-124a is one of the most abundant and ubiquitously expressed miRNAs in the brain (Lagos-Quintana et al., 2002). Other notable brain-enriched miRNAs in the list include miR-9,-125,-153, and 218 (Cao et al., 2006). This result further supports our conclusion that brain-enriched miRNAs tend to target brain genes, and provides direct evidence that the CE score is a biologically sensitive measure of whether a miRNA functionally interacts with a statistically significant number of targets in a gene group. High-scoring miRNAs in Table 1 not previously shown to be neuronal warrant experimental confirmation of their brain-enriched expression pattern.
Table 1.
Mouse miRNAs with significant CE scores in a brain-enriched gene group.
| seed | CE score | matched miRNA(s) | brain/neuronal expression citation |
|---|---|---|---|
| AAGCCAU | 4.68 | mmu-miR-135a/mmu-miR-135b | (Sempere et al., 2004) |
| UGCCUUA | 4.42 | mmu-miR-124a | (Lagos-Quintana et al., 2002) |
| AAGCACA | 4.13 | mmu-miR-218 | (Sempere et al., 2004) |
| UGCAAAC | 4.10 | mmu-miR-452 | (Dostie et al., 2003) |
| CACUGCC | 3.79 | mmu-miR-34a/c mmu-miR-449 | (Kim et al., 2004) |
| AGCACAA | 3.74 | mmu-miR-218 | (Sempere et al., 2004) |
| CUGUAGA | 3.36 | mmu-miR-139 | (Sempere et al., 2004) |
| ACUGCCU | 3.28 | mmu-miR-34b/c | (Kim et al., 2004) |
| ACUGCCA | 3.22 | mmu-miR-34a mmu-miR-449 | (Kim et al., 2004) |
| CCUCUGC | 3.21 | mmu-miR-298 | |
| AUUCUUU | 2.87 | mmu-miR-186 | |
| GUAUGUA | 2.78 | mmu-miR-466 | |
| AGCAAUA | 2.76 | mmu-miR-137 | |
| UGCAGUA | 2.74 | mmu-miR-217 | (Dostie et al., 2003) |
| CCCAGAG | 2.71 | mmu-miR-326 | (Kim et al., 2004) |
| AGACGGA | 2.69 | mmu-miR-340 | (Kim et al., 2004) |
| GGAGAAG | 2.66 | mmu-miR-207 | (Dostie et al., 2003) |
| ACCAAAG | 2.66 | mmu-miR-9 | (Lagos-Quintana et al., 2002) |
| ACGCACA | 2.62 | mmu-miR-210 | |
| GCAGACA | 2.62 | mmu-miR-346 | (Kim et al., 2004) |
| CUAGGAA | 2.60 | mmu-miR-384 | |
| CCGUGUU | 2.57 | mmu-miR-411 | |
| UCUGAUC | 2.52 | mmu-miR-383 | |
| GGACCAA | 2.49 | mmu-miR-133a/b | (Mortazavi et al., 2006) |
| AGUCAUA | 2.47 | mmu-miR-468 | |
| GUUCUCA | 2.42 | mmu-miR-146 | |
| UAACCUA | 2.40 | mmu-miR-154 | |
| AUGGAGU | 2.38 | mmu-miR-136 | (Miska et al., 2004) |
| GCAAUAA | 2.37 | mmu-miR-137 |
miRNAs known to be expressed in the brain are highlighted in blue.
DISCUSSION
In this study, we found that the expression of embedded miRNA host genes and their predicted miRNA targets tend to be positively or negatively correlated (Figure 2), suggesting that the coordinated transcriptional regulation of a miRNA and its targets is an abundant motif in gene networks. This result encouraged us to develop a robust method that does not rely on target prediction to determine whether such circuits are prevalent in the human and mouse genomes (Figure 3). We found that many miRNA seed-matches are evolutionarily conserved at significantly higher rates in genes whose expression pattern is highly correlated (either positively or negatively) with that of the corresponding miRNA (Figures 4-5), indicating that putative targets that are coordinately regulated with a miRNA tend to be functional in vivo. This phenomenon is widely spread in mammalian genomes: a large fraction (44-69%) of the miRNAs analyzed show bias towards coordinated regulation with their targets, independent of the particular gene expression data sets analyzed (Figures 4-5). We speculate that the abundance of Type I/II circuits in the genome may be even higher because the profiled conditions in the expression data we analyzed are limited (e.g. miR-106 exhibits Type II bias in the MDEV data set but not in the Novartis atlas). Taken together, our results strongly argue that Type I and II circuits are recurrent network motifs in mammalian gene networks.
Negative expression correlation between a miRNA and its putative targets has been reported for several tissue-specific mammalian miRNAs (e.g. miR-133), where an increase in the miRNA level often coincides with a decrease in the levels of its target transcripts at the tissue level (Farh et al., 2005; Sood et al., 2006). However, this phenomenon can not be solely attributed to the repressive nature of miRNA-mediated gene regulation because targeting by miRNAs often lead to translational inhibition but has limited effects on target mRNA levels (Bartel, 2004; Doench et al., 2003; Doench and Sharp, 2004). A plausible explanation is that the observed negative correlation is due to Type II circuits where the suppression of target mRNA levels is mainly driven by transcriptional control and miRNAs play a modulatory role to reinforce such decisions (Bartel and Chen, 2004). This notion is further supported by the observed positive expression correlation between a miRNA and its targets in Type I circuits: elevated miRNA levels do not necessarily lead to lower target mRNA abundance at the tissue and individual cell type levels.
The prevalence of positive expression correlation in miRNA-target pairs is surprising and counterintuitive given the repressive nature of miRNAs. Although some positively correlated miRNA-target pairs may result from localized expression of a miRNA and its target in distinct cell types within a tissue (Stark et al., 2005), mutually exclusive expression cannot account for all positively correlated miRNA-target pairs. Indeed, Type I signatures remained prevalent when CE analysis was applied to expression data sets obtained from homogeneous neuronal cell populations in the adult mouse forebrain and at distinct stages of cortical motor neuron development (Figure 5), suggesting that these miRNA and their targets are coexpressed at the single-cell level. In Type I circuits, the expression of the target gene is controlled by two opposing pathways, one of which is miRNA-mediated and serves as a negative feedback/feedforward loop. This configuration implies that miRNAs modulate rather than solely establish the expression level of their target genes (Bartel and Chen, 2004). Supporting this notion, translation inhibition and mRNA degradation mediated by miRNAs are modest even for reporter genes with more than one miRNA binding sites in in vitro overexpression assays (Brennecke et al., 2005; Farh et al., 2005). The same notion also suggests that the functional importance of miRNAs is beyond simple gene repression but is determined by the underlying network structures.
The finding that brain-enriched miRNAs tend to target brain-enriched genes suggests that the primary function of these miRNAs is not to reinforce the suppression of genes specific to other tissues. However, our result does not imply that neuronal-enriched miRNAs tend to only participate in Type I circuits in the adult brain. Indeed, we found that these miRNAs could exhibit bias for either network type when data from homogeneous neuronal cell populations were used in our analysis. Interestingly, the number of miRNAs exhibiting Type I bias is still significantly higher than those showing Type II bias in the NCELL data set, even though this phenomenon was not observed in the MDEV data set. This suggests that Type I circuits may be more prevalent in networks operating in homeostasis—perhaps partly due to the need for such circuits to maintain protein steady-state and regulate local translation in neurons (see below)—as NCELL consists of mature neurons whereas MDEV only covers developing neurons.
The following aspects might complicate the interpretation of our results. First, the prevalence of Type I/II circuits could be a special feature of embedded miRNAs. This is unlikely, however, because more than 80% of all known miRNAs in human and mouse reside in introns of coding or non-coding genes (Kim and Kim, 2007; Rodriguez et al., 2004). In this sense, there are no obvious features that distinguish the miRNAs we analyzed from the rest. In addition, embedded miRNAs are not homogeneous by any measure: they have diverse expression patterns and likely function in disparate biological processes. Second, post-transcriptional control of the host gene and/or the miRNA could uncouple their expression (Obernosterer et al., 2006; Thomson et al., 2006; Wulczyn et al., 2007). However, this phenomena is likely condition-specific (e.g. early development (Thomson et al., 2006)) as the steady-state expression of host genes and their embedded miRNA(s) tend to be correlated (Aboobaker et al., 2005; Baskerville and Bartel, 2005; Li and Carthew, 2005; Rodriguez et al., 2004). Importantly, the topology of Type I and II circuits does not preclude the possibility of post-transcriptional control of the miRNA. For example, the miRNA need not be immediately active after transcription in both circuit types (e.g. a delay between transcription and maturation).
Negative Auto-regulatory Feedback by Embedded miRNAs
The prevalence of Type I signatures suggests that miRNAs may be often employed in negative feedback circuits (Figure 1). An interesting instance of negative feedback circuits involves the targeting of a host gene by its own embedded miRNA(s). Based on TargetScanS predictions, we identified three auto-feedback miRNAs in human (miR-26a, -128b, -488; Table S1a). Since prior experiments have shown that non-conserved sites can confer normal levels of repression when the target transcript is coexpressed with the miRNA (Brennecke et al., 2005; Farh et al., 2005), and that high affinity sites without perfect seed matches may also be functional in vivo (e.g. good 3′ compensatory matches (Brennecke et al., 2005)), we identified 18 additional putative negative auto-feedback loops (Table S1b/c) by searching for non-conserved seed matches and high affinity sites (ΔG < −20 kcal/mol).
Type I and II Circuits in Neural Development
The positively correlated expression of miRNAs and targets in Type I circuits could be due to their sharing of cis regulatory modules regulated by common upstream transcription factors. In neuronal development, NRSF is a master transcriptional repressor that inhibits the expression of neuronal genes in non-neuronal cells and in neuronal progenitors prior to differentiation (Chong et al., 1995). Experimental and computational studies have identified hundreds of protein-coding genes and a handful of miRNAs (e.g. miR-9, -29, and -135b) under the control of NRSF (Conaco et al., 2006; Mortazavi et al., 2006; Wu and Xie, 2006). Notably, miR-29 and miR-135b have statistically significant CE scores (2.27 and 2.68, respectively) in genes with NRSF binding sites and brain-enriched expression patterns (data not shown), suggesting that a larger than expected number of functional targets of miR-29 and miR-135b are co-repressed by NRSF, and they may be co-activated as NRSF level goes down during neuronal development. Interestingly, the 3′UTR of NRSF has conserved putative binding sites for both miR-29 and miR-135b, likely forming Type II circuits (Wu and Xie, 2006). Indeed, in principle a miRNA can be involved in a large number of Type I and/or II circuits because a miRNA can target multiple genes (Figure 6A) and can be transcriptionally regulated via different cis regulatory modules (Figure 6B).
Figure 6. Circuits with miRNA-mediated Positive and Negative Feedback Loops.
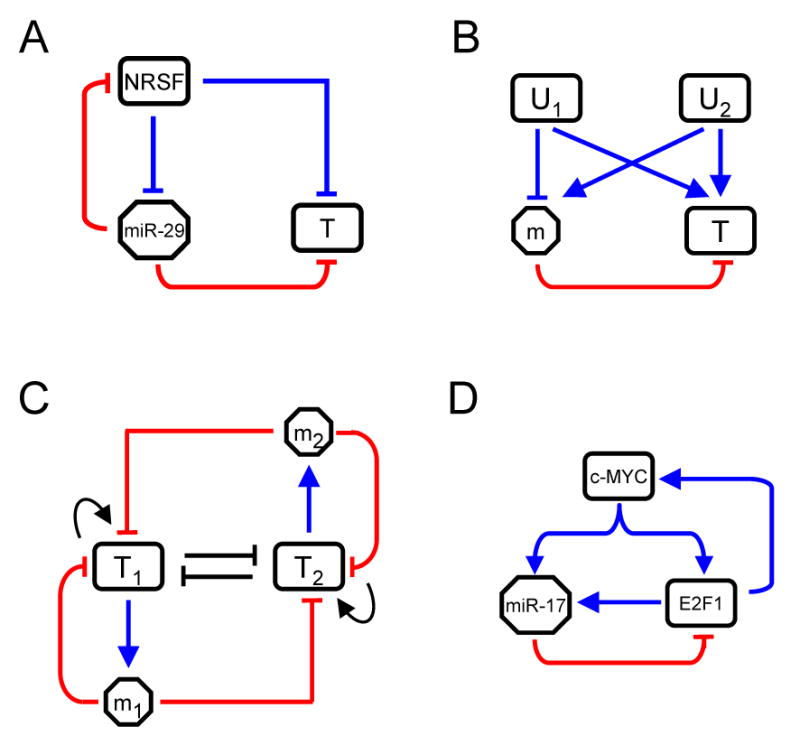
(A) The NRSF/miR-29 network. A miRNA can be embedded in both Type I and II circuits. In addition to potentially targeting NRSF, miR-29 putatively targets a significant number of neuronal genes (T) that are co-repressed by NRSF.
(B) Illustrates how a miRNA can be embedded simultaneously in both Type I and II circuits in relation to a single target. U1 and U2 regulate the miRNA and its target via different cis regulatory modules. Depending on whether U1 or U2 is active, the expression of the miRNA will positively or negatively correlate with that of the target.
(C) A hypothetical toggle-switch network with two transcription factors (T1 and T2) and two miRNAs (m1 and m2) with both Types I (T1-m1/T2-m2) and II (T1-m1-T2 and T2-m2-T1) circuits. Such networks are typically characterized by two stable states where only one of T1 or T2 is active. Each miRNA is involved in a positive and a negative feedback loop. The former functions in conjunction with other positive feedbacks to reinforce the active state (T1 or T2 active) and allow transient signals to turn the circuit on or off. The latter could buffer fluctuations and prevent random switching events. MiRNAs may be especially effective at providing feedback with short delays.
(D) The c-Myc/E2F1/miR-17-20 network in human. c-Myc and E2F1 can activate each other's transcription and both can activate the transcription of the miR-17 miRNA cluster. Two Type I circuits are present: the miR-17-mediated negative feedback to E2F1, and the c-Myc-activated feedforward loop to E2F1. These negative loops mediated by miRNAs could prevent random activation of c-Myc/E2F1 due to fluctuations in their expression.
Potential Functions of Type I Circuits
Recurrent network motifs are likely a result of convergent evolution at the network level, presumably because certain circuit topologies are particularly versatile in carrying out important functions in cells. A plausible function of the miRNA-mediated negative feedback/feedforward loop (MNFL) in Type I circuits is to define and maintain target-protein steady-states. The eukaryotic cell is a noisy environment in which transcription often occurs in a bursting manner (Blake et al., 2006; Golding et al., 2005; Raj et al., 2006), causing the number of mRNAs per cell—which can go as low as fractions of a copy when averaged over a population—to fluctuate significantly. Since other processes in gene expression, such as mRNA degradation and protein translation, are also stochastic in nature, protein levels in turn may fluctuate considerably over time (Kaern et al., 2005). Importantly, such fluctuations can propagate through the network, e.g. fluctuations in the level of an upstream transcription factor can contribute significantly to expression fluctuations of downstream genes (Pedraza and van Oudenaarden, 2005; Rosenfeld et al., 2005).
In Type I circuits (Figure 1), any deviation from the upstream factor (U)'s steady-state would drive the target (T) and miRNA (m) away from their steady-states in the same direction, thus m could tune the production rate of T opposite to the direction of U's fluctuation. Such noise buffering helps to maintain target protein homeostasis and ensures more uniform expression of T within a cell population. This may be desirable for cells that are ultra-sensitive to the level of T, where any significant drift from the desired steady-state may lead to pathological consequences. In addition, since the level of m defines T's translation rate, their co-expression may allow m to fine-tune T's steady-state (Bartel and Chen, 2004). Compared to transcriptional repressors, miRNAs may be especially effective in such circuits because they likely can tune target protein levels more rapidly at the post-transcriptional level. Thus, miRNAs could significantly shorten the response delay, leading to more effective noise buffering, as well as precise definition and maintenance of steady-states.
Noise buffering by Type I circuits may be especially common in circuits with positive feedback loops where fluctuations in any component can be amplified, driving the system to switch states (Figure 6C). Having MNFL in such networks may dampen network component fluctuations and avoid random switching events. The c-Myc/E2F1/miR-17-92 network (O'Donnell et al., 2005; Sylvestre et al., 2006) is a good example of such a circuit (Figure 6D), where the MNFLs may prevent noise-driven transitions into proliferation. An example of such random-switching phenotypes was observed in the yeast Galactose Network (GAL), which contains both positive and negative feedbacks, although the negative feedback loop is mediated by proteins instead of miRNAs. When the negative feedback in the network was removed, the GAL genes in the network would randomly switch on and off over time regardless of whether the network is induced (Acar et al., 2005). Given that positive feedback circuits are abundant in genomes (Brandman et al., 2005; Ferrell, 1999), we surmise that miRNAs are frequently coupled to such networks to provide fast feedback response.
Another potential function of Type I circuits is the regulation of local translation in neurons. It has been proposed that the translation of neuronal mRNAs is often repressed in transport granules and at local synapses, and that the inhibition can be released in response to synaptic activity (reviewed in Kiebler and Bassell, 2006). Neuronal miRNAs may be transcriptionally co-activated with their targets and constitutively lower their targets' translation rate to facilitate activity-dependent local translation. Consistent with this notion, a brain-specific microRNA, miR-134, has been shown to function directly as a repressor of LimK1 translation under basal conditions. This inhibition can be relieved by the BDNF signaling pathway, thereby allowing for local translation and spine growth (Schratt et al., 2006). Although the detailed molecular mechanisms remain to be identified, the enrichment of Type I bias in the NCELL data set supports the idea that miRNAs have important functions in local translation and synaptic plasticity.
Potential Functions of Type II Circuits
In Type II circuits, a miRNA regulates its targets coherently with transcriptional control, thereby reinforcing transcriptional logic at the post-transcriptional level. Under conditions where the target genes are transcriptionally suppressed, such circuits can serve as a surveillance mechanism to suppress “leaky” transcription of target genes (Bartel and Chen, 2004; Hornstein and Shomron, 2006; Stark et al., 2005). While the basic function of Type II circuits is intuitive, it can have sophisticated functions in networks. For instance, the miRNA-mediated repression in Type II circuits can be part of a positive feedback loop, in which the target gene encodes a transcription factor that can down-regulate the miRNA's expression. Such an example can be found in Drosophila eye development, where the reciprocal repression between miR-7 and Yan ensures their mutually exclusive expression pattern: Yan is expressed in progenitor cells and miR-7 in photoreceptor cells (Li and Carthew, 2005). This circuit can be switched by EGFR signaling, which transiently triggers Yan degradation. A decrease in Yan levels relieves miR-7 from transcriptional repression, subsequently leading to the depletion of Yan in photoreceptor cells (Li and Carthew, 2005). Positive feedback loops are often employed in such toggle-switch circuits where a transient signal can be converted into a long-lasting cellular response (Ferrell, 2002). Since most putative targets have only one binding site for a miRNA, it is likely that miRNAs would act in concert with other miRNAs and/or regulatory processes to increase the feedback strength. We speculate that miRNAs may be involved in a large number of similar positive feedback loops to enhance the robustness of irreversible cellular differentiation.
Conclusion
In conclusion, our results provide strong evidence that coordinated transcriptional and post-transcriptional regulation via miRNAs is a recurrent motif to enhance the robustness of gene regulation in mammalian genomes. If, as suggested by our findings, miRNA-mediated repression tends to play modulatory and/or reinforcing roles in networks, miRNA loss-of-function phenotypes may be subtle and quantitative experimentation at the single-cell level (Acar et al., 2005), for instance, may be necessary to reveal their functions. Further exploration of miRNA-mediated repression in the context of gene regulatory networks will provide a comprehensive view on how gene expression is regulated at the systems level.
EXPERIMENTAL PROCEDURES
Embedded microRNAs
Human and mouse RefSeq (Wheeler et al., 2006) genes and miRNA genomic coordinates were extracted from the UCSC 2004/May human and 2005/March mouse databases, respectively (http://genome.ucsc.edu/). We picked embedded miRNAs that reside on the same strand as the host, either in introns or UTRs of Refseq genes.
Gene Expression Data Processing and Host Transcript Mapping
The pre-normalized Novartis human/mouse atlas data, along with the probe annotations were downloaded from Novartis (http://wombat.gnf.org/index.html). The pre-normalized MDEV and NCELL data were downloaded from the NCBI GEO database (http://www.ncbi.nlm.nih.gov/projects/geo/). We assigned an integer id to probes on each microarray for ease of reference (Table S7-9). Probes for individual miRNA host genes were mapped and erroneous probes were removed before further analysis (see Supp. Data). To obtain relative expression levels, log-transformed expression of individual probes on an array is further normalized by subtracting the probe-median across conditions and divide by the corresponding standard deviation.
Hierarchical Clustering and Gene Groups
The hierarchical clustering module in GenePattern (Reich et al., 2006) was used. The parameters used were: average linkage, row centered with median, and distance using Pearson correlation. Spearman ranked correlation was also tested as the distance measure, but the results were essentially the same.
Target Predictions
We implemented the TargetScanS (Lewis et al., 2005) algorithm where we searched for 7-mer seed matches in the 3′UTRs of RefSeq genes. 3′UTRs were downloaded from the UCSC human/mouse database (see above), along with their multiz alignments. Given the multiple alignment of each UTR, we searched for 7-mer (no gaps) human miRNA seed-matches (m1-m7 and m2-m8) that are perfectly conserved across the human, mouse, rat, and dog genomes.
Targeting Bias Calculations
For each embedded miRNA host probe, we compiled a ranked list of genes based on their expression correlation to the probe. We then counted the number of genes (Kx) in a miRNA's predicted target set (T) that overlap with each of the X percentiles (where X can be top-10, middle-10, or bottom-10) of the ranked list. If miRNA target prediction is random, on average we expect 10% of the predicted target set to overlap each of these sets. Since target prediction corresponds to sampling without replacement, the sampling distribution is hypergeometric with parameters (N, 0.1N, |T|), where N is the total number of genes; and the sampling variance (s) can be computed exactly as:
We then computed the z-score for each of the X percentile sets to indicate the degree of enrichment:
The exact P value for enrichment (or depletion) can be obtained by computing the cumulative distribution of the above hypergeometric distribution.
Conservation Enrichment Analysis
For each miRNA host probe, we generated a ranked list of genes based on their expression correlation to the host probe. Since some genes can be represented by multiple probes on the microarray, we removed such redundancies so that each entry in the ranked list is unique. The rest of the procedure is as described in the main text (Figure 3). For the background distribution estimation, we randomly sampled 1000 times K genes from all unique genes on the microarray, where K equals 10% of the total number of unique genes on the array. For each random sample we computed the CE score for miRNA seeds of interest. The empirical CE score distributions of miRNA seeds are sufficiently normal so that less than 5% of the random samples have CE>1.65 for all seeds, so we report the CE score only. To generate the ΔCE heatmaps (e.g. Fig 4A), the maximum ΔCE score of the two seed-matches (m1-7 or m2-8) are displayed for each miRNA/probe combination.
Predicting Auto-feedback Loops
Conserved loops came directly from TargetScanS predictions. For non-conserved loops, the 3′UTRs of host genes were scanned for 7-mer seed-matches of the respective embedded miRNA. We used Miranda (Enright et al., 2003; John et al., 2004) to scan for energetically favorable binding sites. The parameters used were: score cutoff = 50, free energy cutoff = −20, scale=4 (to bias 5′ matches with a factor of 4), gap extension penalty=−4 (to allow loops towards the 3′ end).
Identifying Brain-enriched miRNAs
Standard k-means (k=40) clustering was applied to the Novartis mouse atlas using the Cluster tool (Eisen et al., 1998). We computed the CE scores of all known miRNA seeds for one of the resulting clusters that contains 1012 Refseq genes that are expressed in the brain tissues profiled.
Supplementary Material
Supplemental Data include Figures S1-6, Tables S1-S11, additional details on probe selection, and comparison of results among the four data sets we analyzed.
Acknowledgments
This work was supported by a NSERC PGS Scholarship to J.T., a Basil O'Connor Starter Scholar award to J.Z., and grants from the NIH and NSF to A.v.O. We thank Mike Hu for introducing us to mammalian embedded miRNAs; Dale Muzzey, Margaret Ebert, Arjun Raj, Scott Rifkin, Phillip Sharp, Hunt Willard, and Han Wu for comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aboobaker AA, Tomancak P, Patel N, Rubin GM, Lai EC. Drosophila microRNAs exhibit diverse spatial expression patterns during embryonic development. Proc Natl Acad Sci U S A. 2005;102:18017–18022. doi: 10.1073/pnas.0508823102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acar M, Becskei A, van Oudenaarden A. Enhancement of cellular memory by reducing stochastic transitions. Nature. 2005;435:228–232. doi: 10.1038/nature03524. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. Rna. 2005;11:241–247. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becskei A, Serrano L. Engineering stability in gene networks by autoregulation. Nature. 2000;405:590–593. doi: 10.1038/35014651. [DOI] [PubMed] [Google Scholar]
- Blake WJ, Balazsi G, Kohanski MA, Isaacs FJ, Murphy KF, Kuang Y, Cantor CR, Walt DR, Collins JJ. Phenotypic consequences of promoter-mediated transcriptional noise. Mol Cell. 2006;24:853–865. doi: 10.1016/j.molcel.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Brandman O, Ferrell JE, Jr, Li R, Meyer T. Interlinked fast and slow positive feedback loops drive reliable cell decisions. Science. 2005;310:496–498. doi: 10.1126/science.1113834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol. 2005;3:e85. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Yeo G, Muotri AR, Kuwabara T, Gage FH. Noncoding RNAs in the Mammalian Central Nervous System. Annu Rev Neurosci. 2006 doi: 10.1146/annurev.neuro.29.051605.112839. [DOI] [PubMed] [Google Scholar]
- Carthew RW. Gene regulation by microRNAs. Curr Opin Genet Dev. 2006;16:203–208. doi: 10.1016/j.gde.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Chong JA, Tapia-Ramirez J, Kim S, Toledo-Aral JJ, Zheng Y, Boutros MC, Altshuller YM, Frohman MA, Kraner SD, Mandel G. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- Conaco C, Otto S, Han JJ, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad Sci U S A. 2006;103:2422–2427. doi: 10.1073/pnas.0511041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J, Mourelatos Z, Yang M, Sharma A, Dreyfuss G. Numerous microRNPs in neuronal cells containing novel microRNAs. Rna. 2003;9:180–186. doi: 10.1261/rna.2141503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, Bozzoni I. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell. 2005;123:819–831. doi: 10.1016/j.cell.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Ferrell JE., Jr Building a cellular switch: more lessons from a good egg. Bioessays. 1999;21:866–870. doi: 10.1002/(SICI)1521-1878(199910)21:10<866::AID-BIES9>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Ferrell JE., Jr Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr Opin Cell Biol. 2002;14:140–148. doi: 10.1016/s0955-0674(02)00314-9. [DOI] [PubMed] [Google Scholar]
- Golding I, Paulsson J, Zawilski SM, Cox EC. Real-time kinetics of gene activity in individual bacteria. Cell. 2005;123:1025–1036. doi: 10.1016/j.cell.2005.09.031. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornstein E, Shomron N. Canalization of development by microRNAs. Nat Genet. 2006;38 Suppl 1:S20–24. doi: 10.1038/ng1803. [DOI] [PubMed] [Google Scholar]
- Howard ML, Davidson EH. cis-Regulatory control circuits in development. Dev Biol. 2004;271:109–118. doi: 10.1016/j.ydbio.2004.03.031. [DOI] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Jr, Chang S, Etchberger JF, Ortiz CO, Hobert O. MicroRNAs acting in a double-negative feedback loop to control a neuronal cell fate decision. Proc Natl Acad Sci U S A. 2005;102:12449–12454. doi: 10.1073/pnas.0505530102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- Kim J, Krichevsky A, Grad Y, Hayes GD, Kosik KS, Church GM, Ruvkun G. Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc Natl Acad Sci U S A. 2004;101:360–365. doi: 10.1073/pnas.2333854100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Kim VN. Processing of intronic microRNAs. Embo J. 2007;26:775–783. doi: 10.1038/sj.emboj.7601512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Li X, Carthew RW. A microRNA mediates EGF receptor signaling and promotes photoreceptor differentiation in the Drosophila eye. Cell. 2005;123:1267–1277. doi: 10.1016/j.cell.2005.10.040. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang F, Lee JA, Gao FB. MicroRNA-9a ensures the precise specification of sensory organ precursors in Drosophila. Genes Dev. 2006;20:2793–2805. doi: 10.1101/gad.1466306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U. Network motifs: simple building blocks of complex networks. Science. 2002;298:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Townsend M, Yoshii A, Sestan N, Rakic P, Constantine-Paton M, Horvitz HR. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 2004;5:R68. doi: 10.1186/gb-2004-5-9-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Thompson EC, Garcia ST, Myers RM, Wold B. Comparative genomics modeling of the NRSF/REST repressor network: from single conserved sites to genome-wide repertoire. Genome Res. 2006;16:1208–1221. doi: 10.1101/gr.4997306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- Obernosterer G, Leuschner PJ, Alenius M, Martinez J. Post-transcriptional regulation of microRNA expression. Rna. 2006;12:1161–1167. doi: 10.1261/rna.2322506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedraza JM, van Oudenaarden A. Noise propagation in gene networks. Science. 2005;307:1965–1969. doi: 10.1126/science.1109090. [DOI] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38(Suppl):S8–13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld N, Elowitz MB, Alon U. Negative autoregulation speeds the response times of transcription networks. J Mol Biol. 2002;323:785–793. doi: 10.1016/s0022-2836(02)00994-4. [DOI] [PubMed] [Google Scholar]
- Rosenfeld N, Young JW, Alon U, Swain PS, Elowitz MB. Gene regulation at the single-cell level. Science. 2005;307:1962–1965. doi: 10.1126/science.1106914. [DOI] [PubMed] [Google Scholar]
- Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Orr SS, Milo R, Mangan S, Alon U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat Genet. 2002;31:64–68. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- Sood P, Krek A, Zavolan M, Macino G, Rajewsky N. Cell-type-specific signatures of microRNAs on target mRNA expression. Proc Natl Acad Sci U S A. 2006;103:2746–2751. doi: 10.1073/pnas.0511045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- Sylvestre Y, De Guire V, Querido E, Mukhopadhyay UK, Bourdeau V, Major F, Ferbeyre G, Chartrand P. An E2F/miR-20a auto-regulatory feed-back loop. J Biol Chem. 2006 doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–2207. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006;20:515–524. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2006;34:D173–180. doi: 10.1093/nar/gkj158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Xie X. Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol. 2006;7:R85. doi: 10.1186/gb-2006-7-9-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulczyn FG, Smirnova L, Rybak A, Brandt C, Kwidzinski E, Ninnemann O, Strehle M, Seiler A, Schumacher S, Nitsch R. Post-transcriptional regulation of the let-7 microRNA during neural cell specification. Faseb J. 2007;21:415–426. doi: 10.1096/fj.06-6130com. [DOI] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, Greenwald I. LIN-12/Notch activation leads to microRNA-mediated down-regulation of Vav in C. elegans. Science. 2005;310:1330–1333. doi: 10.1126/science.1119481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data include Figures S1-6, Tables S1-S11, additional details on probe selection, and comparison of results among the four data sets we analyzed.