

Abstract
Recent studies have highlighted the surprising richness of soil bacterial communities; however, bacteria are not the only microorganisms found in soil. To our knowledge, no study has compared the diversities of the four major microbial taxa, i.e., bacteria, archaea, fungi, and viruses, from an individual soil sample. We used metagenomic and small-subunit RNA-based sequence analysis techniques to compare the estimated richness and evenness of these groups in prairie, desert, and rainforest soils. By grouping sequences at the 97% sequence similarity level (an operational taxonomic unit [OTU]), we found that the archaeal and fungal communities were consistently less even than the bacterial communities. Although total richness levels are difficult to estimate with a high degree of certainty, the estimated number of unique archaeal or fungal OTUs appears to rival or exceed the number of unique bacterial OTUs in each of the collected soils. In this first study to comprehensively survey viral communities using a metagenomic approach, we found that soil viruses are taxonomically diverse and distinct from the communities of viruses found in other environments that have been surveyed using a similar approach. Within each of the four microbial groups, we observed minimal taxonomic overlap between sites, suggesting that soil archaea, bacteria, fungi, and viruses are globally as well as locally diverse.
Soil microorganisms represent a considerable fraction of the living biomass on Earth (63), with surface soils containing 103 to 104 kg of microbial biomass per hectare (7). Despite this abundance and the importance of soil microorganisms for key ecosystem functions (35, 37, 62), the diversity and structure of soil microbial communities remain poorly studied. With the advent of molecular techniques, we can now begin to survey the full extent of microbial diversity, including the vast majority of microorganisms which cannot be identified using traditional taxonomic approaches (47).
Of the microbial groups that are abundant in soil, the bacteria have been the most extensively studied. With an estimated 103 to 107 bacterial “species” per individual soil sample (15, 23, 59, 60), they are often considered to be the most diverse group of soil microorganisms (13). However, bacteria are not the only microorganisms found in soil; archaea, fungi, and viruses are also numerically abundant (58). To our knowledge, no previous studies have examined the sequence diversity of soil viruses, and no studies have compared the levels of genetic diversity found in the different taxonomic groups of soil microorganisms (bacteria, archaea, fungi, and viruses) inhabiting a given soil sample.
We propose that soil fungal, archaeal, and viral communities are likely to be as taxonomically diverse as soil bacterial communities. Although soil fungi have been studied for centuries, recent DNA-based surveys suggest that fruiting body and cultivation-based surveys have underestimated the total richness of soil fungal communities (33, 43, 54). Recent research also indicates that soil archaea are phylogenetically diverse (44, 46, 61) and are undersurveyed despite their apparent importance in soil processes (37). Soil viruses are known to be abundant, to be morphologically diverse, and to span a wide range of genome sizes (48, 64), but there are currently no published reports describing the genomic diversity of soil viral communities.
For this study, our goal was not to identify every individual microorganism found in soil. To do so would be prohibitively difficult given the magnitude of the required sequencing effort (17, 55). Rather, our goal was to compare the phylogenetic diversities of the four dominant taxonomic groups of soil microorganisms in soils collected from a tallgrass prairie, an arid desert, and a tropical rainforest. These sites were chosen because they represent globally dominant ecosystem types and span a broad gradient in aridity and productivity. We analyzed partial sequences of amplified 16S and 18S rRNA genes to characterize the phylogenetic diversity of archaeal, fungal, and bacterial communities in each soil. Because viruses lack ubiquitously conserved genetic elements, we assessed viral diversity by sequencing randomly chosen clones from viral DNA metagenomic libraries.
MATERIALS AND METHODS
Soil collection and DNA extraction.
Soil was collected from three sites: Manu National Park in Peru (Amazonian terra firme forest; 12.65oS, 71.23oW), Mojave Desert in California (desert shrubland; 33.97oN, 116.07oW), and the long-term ecological research site at Konza Prairie in Kansas (tallgrass prairie; 39.10oN, 96.60oW). Additional soil and site information is given in Table 1. At each site, mineral soil (the upper 5 cm) was collected from 10 locations within a single 100-m2 plot using a stratified random sampling approach. The individual soil samples from each plot were homogenized together, and the composited sample was sieved to 2 mm and stored either at 4°C for extraction of viral DNA or at −80°C for extraction of fungal, bacterial, and archaeal DNA.
TABLE 1.
Site information and general properties of the three soils studieda
| Soil type | Sampling location | Latitude, longitude | Elevation (m) | Dominant vegetation | Mean annual precipitation (mm) | Mean annual temp (oC) | Soil texture | Soil pH | % Organic C |
|---|---|---|---|---|---|---|---|---|---|
| Desert | Joshua Tree National Park, CA | 33.97oN, 116.07oW | 1,360 | Larrea tridentata, Ambrosia dumosa | 90 | 16 | Loamy sand | 7.6 | 0.7 |
| Prairie | Konza Prairie long-term ecological research site, KS | 39.10oN, 96.60oW | 300 | Andropogon gerardii, Sorghastrum nutans | 835 | 13 | Silt loam | 7.1 | 4.0 |
| Rainforest | Manu National Park, Peru | 12.65oS, 71.23oW | 420 | Lomariopsis sp., Piper sp., Cecropia sp., Ficus sp., | 4,000 | 25 | Clay | 4.6 | 3.4 |
Soil physiochemical properties were characterized using the methods described by Fierer and Jackson (21). The dominant plant species at each site were determined in a qualitative manner at the time of sample collection. Dominant plants are described by genera if species identification was unclear.
For the bacterial, fungal, and archaeal clone libraries, DNA was extracted from each of the three soil samples using the MoBio PowerSoil DNA kit (MoBio Laboratories, Carlsbad, CA). DNA was extracted from 10 replicate subsamples (of 1.0 g soil) from each of the three composited soil samples (one from each plot). These replicate DNA extractions provided the templates for the construction of the bacterial, archaeal, and fungal clone libraries.
Viral community DNA was extracted from the soils using methods similar to those described elsewhere (8, 10). Soil samples (∼200 g [wet weight]) were resuspended in 0.02-μm-filtered 1× phosphate-buffered saline solution and shaken vigorously to dislodge the viruses from the soil particles. The sediments were pelleted, and the supernatant was then filtered through a 0.2-μm Sterivex filter to remove all nonviral organisms. Viruses in the filtrate were concentrated by polyethylene glycol precipitation with polyethylene glycol 8000 added to a final concentration of 10%, and the samples were incubated for 12 h at 4°C (11). The samples were then centrifuged at 13,000 × g for 30 min on an SW41 rotor to pellet the viral particles. The viral pellet was resuspended in 0.02-μm-filtered phosphate-buffered saline solution and loaded onto a cesium chloride step gradient consisting of 1 ml each of 1.7, 1.5, and 1.35 g ml−1. The gradient was centrifuged for 2 h at 22,000 rpm on an SW41 rotor (average of 60,000 × g), and the DNA was isolated from the 1.35 to 1.5 g ml−1 fraction (which contains most of the viral particles) using formamide and cetyltrimethylammonium bromide extraction (53).
Clone library construction.
For the analysis of small-subunit rRNA genes, individual bacterial, archaeal, and fungal clone libraries were constructed from each soil sample. For each library, three replicate PCRs were conducted per soil DNA template (for a total of 30 replicate PCRs per library) using group-specific primers. The bacterial clone library was constructed using a universal eubacterial primer set, Bac8f (5′-AGAGTTTGATCCTGGCTCAG-3′) and Univ529r (5′-ACCGCGGCKGCTGGC-3′) (5, 36, 49). The archaeal clone library was constructed using the archaeon-specific primer Arc21f (5′-TTCCGGTTGATCCTGCCGGA-3′) (5) and Univ529r. The fungal library was constructed with the EF4 (5′-GGAAGGGRTGTATTTATTAG-3′) and fung5 (5′-GTAAAAGTCCTGGTTCCCC-3′) primer set (57), which has previously been shown to amplify 18S rRNA genes from most fungal groups (3, 24, 26, 32). Each 50-μl PCR mixture contained 1× HotStarTaq master mix (QIAGEN, Valencia, CA), 0.5 μM of each primer, and 50 ng of template DNA. The amplification protocol consisted of 15 min at 95°C, followed by 25 cycles of 60 s at 94°C, 30 s at the appropriate annealing temperature, and 60 s at 72°C and a final 10-min extension step at 72°C. The annealing temperatures for the bacterial, archaeal, and fungal amplifications were 54°C, 55°C, and 48°C, respectively.
The amplified products from the replicate PCRs were pooled together and cloned using the TOPO-TA PCR cloning kit (Invitrogen). Clones were picked and unidirectionally sequenced following standard protocols (SymBio, Menlo Park, CA). Sequences were screened for chimeras using Bellerophon (29), trimmed at conserved motifs, and aligned using either NAST (available at http://greengenes.lbl.gov) or ARB (available at http://www.arb-home.de). Figure 1 and Table 2 indicate the number of sequences included in each library.
FIG. 1.

Rarefaction curves for the bacterial, fungal, and archaeal clone libraries constructed from each of the soil samples. Rarefaction curves were generated using EstimateS (version 7.5; R. K. Colwell, http://purl.oclc.org/estimates). In all nine libraries, there is no apparent asymptote in the rarefaction curves, suggesting that the libraries do not encompass the full extent of OTU richness in each of the communities with an OTU defined at the ≥97% sequence similarity level.
TABLE 2.
Number of OTUs observed per library versus number predicted by the power law model
| Microbial group | Soil type | No. of sequences analyzed | No. of OTUs observed | Mean (SD) no. of OTUs predicteda |
|---|---|---|---|---|
| Bacteria | Desert | 408 | 350 | 336 (17) |
| Prairie | 358 | 333 | 331 (18) | |
| Rainforest | 363 | 327 | 322 (17) | |
| Archaea | Desert | 306 | 189 | 180 (13) |
| Prairie | 86 | 53 | 53 (7) | |
| Rainforest | 135 | 84 | 84 (9) | |
| Fungi | Desert | 304 | 207 | 215 (14) |
| Prairie | 305 | 235 | 224 (15) | |
| Rainforest | 310 | 216 | 211 (14) | |
| Viruses | Desert | 1,640 | NAb | NA |
| Prairie | 1,928 | NA | NA | |
| Rainforest | 1,009 | NA | NA |
The number of OTUs was predicted from a collection of 1,000 random samples (of size equal to the library) taken from a community representing the corresponding power law model (equation 1) of each population.
NA, not applicable. For the viral metagenomic libraries, estimates of OTU numbers are obtained by modeling the contig spectra, and therefore we cannot independently compare observed and predicted OTU numbers.
Because viruses lack ubiquitously conserved genetic elements, viral diversity was assessed by sequencing randomly chosen clones from viral DNA metagenomic libraries. The viral clone libraries were constructed using a linker-amplified shotgun library technique, as described by Breitbart et al. (11). Construction of the linker-amplified shotgun libraries was performed by Lucigen Corp. (Middleton, WI), with sequencing conducted at SymBio (Menlo Park, CA) and Agencourt (Beverly, MA). The total viral community DNA was randomly sheared using a HydroShear and end repaired, and double-stranded DNA linkers were ligated to the ends. The fragments were then amplified using the high-fidelity Vent DNA polymerase, ligated into the pSMART vector, and electroporated into MC12 cells. This method circumvents problems associated with modified nucleotides and deadly genes in viral genomes, as well as the low DNA concentrations in environmental samples.
Analysis of archaeal, bacterial, and fungal libraries.
We confirmed that the sequences from each library matched the targeted taxonomic group by comparing the sequences to those in the GenBank database using the BLAST algorithm (1). The archaeal, fungal, and bacterial libraries were dereplicated into operational taxonomic units (OTUs) using Fastgroup II (65). An OTU was defined as a group with ≥97% identity in their small-subunit rRNA gene sequences following the conventional definition of a microbial “species” (52). Due to the computational challenges associated with estimating diversity indices and the associated error around these estimates, we only used a single OTU definition for this study. After grouping sequences into OTUs at the ≥97% sequence similarity level, we used EstimateS (version 7; R. K. Colwell, http://purl.oclc.org/estimates) to produce rarefaction curves (Fig. 1). Because none of the rarefaction curves approached an asymptote, we know that we have undersampled the total diversity of each microbial group, and therefore the rarefaction curves cannot be used to compare the diversities of the microbial communities.
For each of the nine libraries, the rank-abundance data (where the observed OTUs are ordered from most to least abundant on the x axis and the abundance of each OTU is plotted on the y axis) were fit to four possible models: logarithmic, log-normal, exponential, and power law models. The equations for these four models are provided in reference 4. These equations describe the community structure by expressing the fraction fi of the community in the ith ranked OTU in terms of the model parameters a, b, and M. As an example, the equation describing the community structure of the power law model is
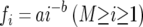 |
(1) |
where M is the total predicted richness of the population, a is the proportional abundance of the most abundant genotype, and b−1 is a measure of the evenness of the population. The other models also express the fi in terms of model parameters a, b, and M, although the functional dependence is different. Note that since the sum of the fis is equal to 1, any two of the three parameters a, b and M determine the third.
The parameters for all the models for all of the libraries were estimated using maximum-likelihood methods. The estimates for the viral communities followed the procedure described by Breitbart et al. (11) and are further described in “Viral sequence analysis” below. The maximum-likelihood estimates of M and a for the other communities proceeded by minimizing the variance-weighted sum of squared deviations Y between the observed and the predicted number of OTUs sampled exactly k times in a sample of size n:
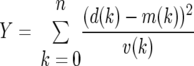 |
(2) |
In this formula, d(k) is the number of OTUs that were actually observed k times, m(k) is the expected number of such OTUs and v(k) is the variance of the number of such OTUs based on the model. The corresponding likelihood (the probability that the hypothesis is true given the data) is proportional to exp(−Y/2), which is maximized by minimizing Y. The minimum values of Y are shown in Table 3 (model error).
TABLE 3.
Estimation of error for the parametric models used to describe the OTU abundance distribution in each community and estimates of OTU richnessa
| Model | Microbial group | Soil type | Model error | Estimated OTU richness |
|---|---|---|---|---|
| Power law | Bacteria | Desert | 15.2 | 6 × 103 |
| Prairie | 1.50 | 2 × 104 | ||
| Rainforest | 4.50 | 2 × 104 | ||
| Archaea | Desert | 66.4 | >1 × 107 | |
| Prairie | 17.3 | 2 × 103 | ||
| Rainforest | 21.3 | 1 × 103 | ||
| Fungi | Desert | 30.0 | 2 × 104 | |
| Prairie | 20.5 | 2 × 109 | ||
| Rainforest | 9.66 | 2 × 103 | ||
| Viruses | Desert | 1.08 | 1 × 103 | |
| Prairie | 0.62 | 2 × 104 | ||
| Rainforest | 7.6 | >1 × 108 | ||
| Log-normal | Bacteria | Desert | 14.8 | 6 × 104 |
| Prairie | 1.90 | 4 × 105 | ||
| Rainforest | 5.32 | >1 × 106 | ||
| Archaea | Desert | 104 | 3 × 105 | |
| Prairie | 20.6 | 3 × 103 | ||
| Rainforest | 25.2 | 3 × 103 | ||
| Fungi | Desert | 42.7 | 7 × 104 | |
| Prairie | 23.7 | >1 × 106 | ||
| Rainforest | 9.00 | 2 × 104 | ||
| Viruses | Desert | 1.08 | 1 × 103 | |
| Prairie | 0.66 | 1 × 105 | ||
| Rainforest | 10.4 | >6 × 106 | ||
| Logarithmic | Bacteria | Desert | 11.3 | 2 × 103 |
| Prairie | 5.70 | 4 × 103 | ||
| Rainforest | 13.4 | 4 × 103 | ||
| Archaea | Desert | 142 | 1 × 103 | |
| Prairie | 23.2 | 2 × 102 | ||
| Rainforest | 36.3 | 2 × 102 | ||
| Fungi | Desert | 62.0 | 2 × 103 | |
| Prairie | 29.0 | 2 × 103 | ||
| Rainforest | 28.1 | 1 × 103 | ||
| Viruses | Desert | 1.08 | 1 × 103 | |
| Prairie | 0.91 | 5 × 103 | ||
| Rainforest | 3.0 | >1 × 106 |
The best descriptive function of the community structure is the one that minimizes the calculated error (shown in boldface). The results for the exponential model are not shown because this model had the highest model error in all 12 cases.
To calculate m(k) and v(k), we make use of random variables Xi,k that take value 1 or 0, depending on whether or not the ith OTU is sampled exactly k times. Since the probability of this happening is
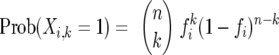 |
(3) |
it follows that this same expression also represents the expected value
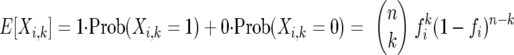 |
(4) |
The variables Xi,k are useful for us since the number of OTUs observed exactly k times is just 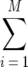 Xi,k. Thus, knowing the expected value of the Xi,k enables us to calculate
Xi,k. Thus, knowing the expected value of the Xi,k enables us to calculate
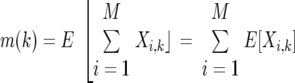 |
(5) |
and
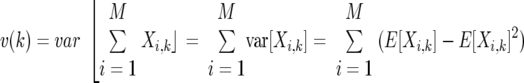 |
(6) |
where we have made use of the fact that since Xi,k takes only the value 1 or 0, Xi,k = Xi,k2. For practical evaluation, it is convenient to invoke the Poisson approximation for equation 4:
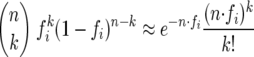 |
(7) |
Confidence intervals for the predicted number of OTUs sampled k times can be obtained using the likelihood ratio test (27). Since the likelihood of any given pair (a,M) of parameter values is proportional to exp(−Y/2), we can determine the ratio, R, of that likelihood to the maximum likelihood. The likelihood ratio test relates the distribution of log(R) to a certain chi-square distribution. In fact, 2log(R) follows a chi-square distribution with N − 1 degrees of freedom, where N is the number of parameters involved (two in this case). This fact enables us to establish a region in the (a,M) plane containing the point of maximum likelihood as well as all points with a value of 2log(R) of >1, corresponding to 68.26% confidence (standard error). The standard error intervals shown in Fig. 2a and b represent, respectively, the projections onto the M and a axes of the confidence regions so determined.
FIG. 2.

Estimation of OTU richness (M in equation 1) (panel a) and the abundance of the most common OTU (a in equation 1) (panel b) in each of the three soils. Symbols correspond to soil type (▴, prairie; ▪, rainforest; •, desert). Parameters were estimated by fitting a power law function to OTU abundance distributions. Maximum-likelihood values are denoted with symbols, and bars indicate 68% confidence regions for the parameter estimates of the actual community (see Materials and Methods). Due to the high range of isolikelihood estimates for OTU richness in the desert archaeal, prairie fungal, and rainforest viral communities, we can conclude only that the number of OTUs in each of these communities is likely to exceed 106. The asterisks indicate that the maximum-likelihood estimates of OTU richness for the desert archaeal and rainforest viral communities exceeded 1010 OTUs.
Viral sequence analysis.
For the viral clone libraries, all sequences (average sequence length, 493 bp) were trimmed and assembled with Sequencher 4.0 (Gene Codes Corp., Ann Arbor, MI). Since the viral sequences did not originate from a single locus, standard gene alignments could not be used to differentiate viral genotypes. Instead, we used a metagenomic definition of a “species” where an OTU has ≥98% identity over a minimum of 20 bp, as per Breitbart et al. (11). The contig spectra were as follows: rainforest [980, 8, 3, 1, 0, 0, 0…], desert [1592, 24, 0, 0, 0…], and prairie [1,899, 13, 1, 0, 0, 0…]. The resulting contig spectra were mathematically modeled to predict community structure using PHACCS (4) and Monte Carlo simulations as described previously (8, 11). To determine the identities of the environmental viruses, the viral metagenomic sequences were compared against the GenBank nonredundant database using TBLASTX. Significant hits to GenBank entries (E value of <0.001) were classified into groups based on sequence annotation in the nonredundant database. To determine the types of phages found in the soils, the sequences were compared against a database containing 510 complete phage genomes (51) using TBLASTX (http://phage.sdsu.edu/oceanviruses). Hits with an E value of <10−6 against this database (approximately equivalent to an E value of 0.001 against the nonredundant database) were considered significant.
Nucleotide sequence accession numbers.
The nonredundant sequences from this study have been deposited in the GenBank nonredundant database and have accession numbers EF429664 through EF431845 (bacteria, archaea, and fungi). All viral sequences from this study have been deposited in the GenBank GSS database with accession numbers ER781257 through ER785833.
RESULTS AND DISCUSSION
The rarefaction results (Fig. 1) indicate that only a portion of the richness in the bacterial, fungal, and archaeal communities (at the ≥97% sequence similarity level) was surveyed with the clone libraries, as none of the curves reached an asymptote. However, coarse estimates of microbial diversity can be obtained without sampling every individual OTU in a given community (15, 28), and we can compare relative levels of community richness and evenness in the targeted microbial taxa. Nonparametric estimators (i.e., Chao I and ACE) (41) are frequently used to estimate the total number of OTUs in a given community (6, 30). However, in all cases, the nonparametric estimates of total OTU richness failed to stabilize or reach an asymptote (data not shown), so they cannot be used to estimate the total number of OTUs within each community (34). Instead, we used a parametric technique, based on the observed OTU abundance distribution, to predict the community-level diversity of these three groups, assuming that the form of the OTU abundance distribution is the same for both the libraries and the communities as a whole. For the viral communities, which were surveyed by constructing metagenomic libraries, the OTU abundance distribution was predicted by mathematically modeling the contig spectra.
We tested four different models that are commonly used to describe microbial community structure (23, 28) and used the most appropriate model (a power law function [Table 3]) to estimate the OTU richness and evenness of each community. While more complex parametric models have been used to estimate OTU richness (23, 55), these models were not tested because there is no a priori reason to choose one type of model over another and because less parsimonious models (those with a larger number of parameters) are likely to underestimate model error. The power law model yielded the lowest model error in 9 of the 12 cases (Table 3). Table 2 shows the close correspondence between the observed number of OTUs and the power law model prediction of OTU numbers for each library. The second-best-performing model, the log-normal model, yielded estimates of OTU richness across soils and taxonomic groups that were generally similar to the estimates obtained using the power law model (Table 3). Since the levels of diversity are estimated from the OTU abundance curve, the estimates of OTU richness should be relatively robust to changes in library size (Table 2). However, for some of the OTU richness estimates, there was a wide range in the 70% confidence regions around the maximum-likelihood values (Fig. 2). This high degree of uncertainty in richness estimates reflects the difficulties associated with reliably fitting the tail of a given distribution. This is readily apparent in Table 3 and in the extremely high richness estimates for the desert archaeal and prairie fungal communities (Fig. 2). Although our clone libraries are larger than most clone libraries published to date, they are still miniscule considering the overwhelming complexity of the soil microbial communities, making it difficult to estimate the exact number of OTUs in each taxonomic group. Due to this high degree of uncertainty, the richness estimates should be considered carefully, as they are likely to be more useful for comparing richness levels between taxonomic groups than for defining the exact number of OTUs in each of the collected soil samples. However, it is worth noting that there is far less uncertainty associated with the estimates of evenness for the individual communities (Fig. 2), as the evenness estimates are less susceptible to errors associated with predicting the specific shape of the tail end of the OTU distribution.
The model results suggest that the total OTU-level richness of bacteria, archaea, fungi, and viruses was extremely high at all sites (Fig. 2a), with the estimated richness of the last three groups equaling or exceeding the richness of soil bacteria in all habitats. The desert archaeal, prairie fungal, and rainforest viral communities were particularly OTU rich, with a minimum estimate of >106 unique OTUs each (Fig. 2a), more than an order of magnitude higher than bacterial richness at the same sites. Of course, given the caveats detailed above, it is important to recognize the high degree of uncertainty inherent in these richness estimates.
The estimated differences in evenness between taxa are likely to be more robust than our estimates of total OTU richness (Fig. 2). Of the four taxonomic groups, the archaeal communities were the least even, with a single OTU accounting for >8% of the population in a given community (Fig. 2b). The fungal and archaeal communities had lower evenness levels than bacterial communities, an observation consistent with results reported elsewhere (43, 46, 61). There was no apparent correlation between the estimated evenness and richness of the communities (r2 = 0.05; P > 0.5). Interestingly, the estimated probabilities of selecting two individuals of the same OTU from a community (Simpson's diversity index) (41) were relatively consistent within each taxonomic group regardless of soil type (Fig. 3). This consistency suggests that the overall structure of each of these communities is controlled by the type of microbe in question rather than the specific features of the soil environment.
FIG. 3.

Predicted values of Simpson's diversity index for each of the 12 communities. Since Simpson's index (D) is defined as the probability that two individuals taken at random from the community belong to the same species (or, in this case, OTU) (41), higher values of D−1 indicate higher overall diversity. Symbols correspond to soil type (▴, prairie; ▪, rainforest; •, desert). The mean value for D−1 (with one standard error in parentheses) for each taxonomic group is denoted above each set of symbols.
Although the slopes of the rarefaction curves were lower for archaea and fungi than for bacteria (Fig. 1), the differences in slopes reflect a lower community-level evenness in these groups (Fig. 2b), not necessarily a lower overall OTU richness. This point is worth reiterating; the slopes of rarefaction curves reflect both the richness and evenness of communities, and therefore, in most cases, rarefaction analyses alone cannot be used to compare richness levels of different microbial communities (30).
Not only are soil bacteria, archaea, fungi, and viruses locally diverse, but our results indicate that these groups are also globally diverse, as we observed little phylogenetic overlap between soils. None of the identified archaeal, fungal, or bacterial OTUs was found at more than one site, and we observed only one instance of an overlapping viral sequence (≥98% identity over 20 bp) between sites when all viral sequences (4,577 in total) were assembled together. While we have no way of estimating the global richness of these groups, the lack of overlap in observed OTUs between sites tells us that the global diversity of each of these groups must be very high. The century-old speculation that the global diversity of the smallest organisms should be relatively low (22) appears to be incorrect.
The estimated number of bacterial OTUs in the three plots (≈104 unique OTUs [Fig. 2a]) closely matches the estimates obtained in other studies (59, 60). Our estimates of fungal richness are substantially higher than estimates obtained using classical taxonomic approaches (a maximum of 3,000 fungal species identified from a single 400-ha site) (25), confirming the results of other studies showing that molecular surveys can uncover a large pool of fungal diversity that has been overlooked (2, 33, 40, 43). Soil archaea also appear to have an equivalent, if not greater, OTU richness than soil bacterial communities, consistent with the high levels of phylogenetic diversity observed in other studies of soil archaea (46, 61). To our knowledge, there are no comparable studies of phylogenetic richness in soil viral communities. However, it is important to note that because we examined only viruses with double-stranded DNA, the true richness of viral communities at each site is likely to be even higher than our estimates.
Of the three soils examined, no individual soil harbored the most diverse community of microorganisms. The estimated number of OTUs was highest in the desert soil for archaea, the prairie soil for fungi, and the rainforest soil for viruses, while the richness of bacterial OTUs was very similar across the three soils (Fig. 2a). Due to a paucity of studies comparing microbial diversity across soils from different ecosystems and the large number of possible mechanisms that may influence levels of taxonomic richness, it is unclear how to interpret these results. Fierer and Jackson (21) found the lowest levels of bacterial diversity in rainforest soils, but their study (which estimated diversity by terminal restriction fragment length polymorphism fingerprinting) was not necessarily examining diversity at the same level of taxonomic resolution as in this study. The high estimated richness of archaeal OTUs in the desert soil is surprising considering the challenging nature of this environment, but other studies have also observed high levels of archaeal diversity in soils and other environments that are likely to be suboptimal for microbial growth (50, 61). The fungal results (Fig. 2a) are consistent with a study by Jumpponen and Johnson (33) in which high fungal diversity was also observed in soils collected from Konza Prairie, KS.
To our knowledge, this is the first study to use sequencing to characterize soil viral communities. TBLASTX comparison of the soil sequences against the GenBank nonredundant database revealed that the majority of the viral sequences showed no significant similarity to previously described sequences (E value of <0.001). Among the identifiable hits, there were numerous similarities to phages (viruses that infect bacteria) (Table 4) and to herpesviruses (data not shown). While there was very little overlap in viral sequences (≥98% identity over 20 bp) between sites (see above), comparison of the sequences against a database containing the genomes of 510 completely sequenced phages demonstrated that similar types of phages were found in all three soil types (Table 4; Fig. 4). The most abundant phage types observed in the soil samples were similar to phages that infect the soil bacteria Actinoplanes, Mycobacterium, Myxococcus, and Streptomyces, as well as the halophilic archaeon Haloarcula (Table 4). The phage types observed in the soil samples were significantly different from the dominant types found in marine or fecal samples (8, 9, 11) (Table 4; Fig. 4), suggesting that distinct habitat types harbor distinct viral communities.
TABLE 4.
Comparison of viral communities from soil and other environments
| Phage | Phage type | Habitata | % of total phage hitsb
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Soil
|
Fecal samples | Marine sediment | Seawater
|
||||||
| Desert | Prairie | Rainforest | MB | SP | |||||
| Actinoplanes φAsp2 | Unclassified | Soil | 9.6 | 11.3 | 6.5 | 0.0 | 0.9 | 1.6 | 0.5 |
| Mycobacterium φBxz1 | Myoviridae | Soil | 7.0 | 6.7 | 5.8 | 0.0 | 0.0 | 0.0 | 0.5 |
| Streptomyces venezuelae φVWB | λ-like siphophage | Soil | 5.3 | 6.5 | 1.4 | 0.0 | 0.0 | 0.8 | 0.0 |
| Haloarcula hispanica φSH1 | Unclassified | Lake | 4.5 | 5.3 | 6.5 | 0.0 | 0.5 | 0.0 | 0.0 |
| Mycobacterium φRosebush | Corndog-like siphophage | Soil | 3.7 | 4.5 | 5.8 | 0.0 | 0.0 | 0.8 | 0.5 |
| Myxococcus xanthus φMx8 | Corndog-like siphophage | Soil | 1.8 | 2.8 | 4.3 | 1.1 | 1.4 | 1.6 | 0.9 |
| Bordetella φBMP-1 | Podoviridae | Path | 0.6 | 1.0 | 0.0 | 9.6 | 1.9 | 1.6 | 0.9 |
| Bordetella φBPP-1 | Podoviridae | Path | 1.2 | 1.4 | 1.4 | 4.3 | 0.0 | 0.8 | 0.0 |
| Salmonella φepsilon15 | λ-like siphophage | Path | 0.2 | 0.6 | 1.4 | 4.3 | 2.3 | 7.3 | 1.4 |
| Listeria innocua φList-I6 | Prophage | Path | 0.2 | 0.0 | 0.0 | 3.2 | 0.0 | 0.0 | 0.5 |
| Vibrio harveyi φVHML | P2-like myophage | Inv | 0.2 | 0.0 | 0.0 | 3.2 | 0.0 | 0.0 | 0.5 |
| Pseudomonas φPaP3 | T7-like podophage | Unk | 2.3 | 1.0 | 0.0 | 0.0 | 6.6 | 4.0 | 2.7 |
| Alphaproteobacterium φJL001 | Siphoviridae | Sea | 0.6 | 1.4 | 0.7 | 0.0 | 4.2 | 0.8 | 2.7 |
| Prochlorococcus phage P-SSM4 | T4-like myophage | Sea | 0.2 | 0.4 | 2.0 | 0.0 | 4.2 | 2.4 | 16.7 |
| Prochlorococcus phage P-SSM2 | T4-like myophage | Sea | 0.2 | 1.0 | 1.4 | 0.0 | 2.8 | 2.4 | 10.8 |
| Burkholderia cepacia φBcepC6B | Unclassified | Soil | 1.8 | 1.2 | 2.2 | 1.1 | 2.8 | 4.0 | 0.9 |
| Pseudomonas PP03 | Prophage | Soil | 0.4 | 0.4 | 0.0 | 1.0 | 0.9 | 4.8 | 5.9 |
| Synechococcus phage S-PM2 | T4-like myophage | Sea | 0.4 | 0.2 | 0.0 | 0.0 | 1.4 | 0.0 | 5.4 |
| Roseophage SIO1 | T7-like podophage | Sea | 0.2 | 0.4 | 0.7 | 0.0 | 2.3 | 7.3 | 7.2 |
| Xylella fastidiosa φXfP2 | Prophage | Path | 0.0 | 0.4 | 0.0 | 1.1 | 1.4 | 4.0 | 0.0 |
Environment from which the phage was first isolated. Lake, hypersaline lake; Path, plant or animal (pathogen); Inv, marine invertebrate intestine; Unk, unknown; Sea, seawater.
The top significant TBLASTX hit for each sequence against a database of completely sequenced phage genomes was recorded. The five phage genomes with the most top TBLASTX hits from each sample are indicated by boldface, while the numbers indicate the percentage of total phage hits from the sample that each sequence comprised. The corresponding abundance of each phage in the other samples is shown as a percentage but not in boldface. The percentages do not add up to 100% because we show only the five most abundant phages in each environment. The two seawater samples were collected from Mission Bay (MB), San Diego, CA, and Scripps Pier (SP), La Jolla, CA. For further information on the seawater, sediment, and fecal viral communities, see references 8 to 10.
FIG. 4.

Hierarchical clustering showing the phylogenetic distance between viral communities from soil (this study), marine sediment (8), human fecal samples (9), and seawater environments (11). Distances were estimated with the weighted Unifrac algorithm (38, 39) using only those sequences from the metagenomic libraries with significant hits to the Phage Proteomic Tree (http://phage.sdsu.edu/oceanviruses) to generate the input phylogenetic trees. A sequence jackknifing technique was applied to each cluster to determine the sensitivity of the relationships to sample size. Asterisks indicate that the nodes are well supported, having been observed in >95% of the jackknifing runs. The soil viral communities were significantly different from the viral communities in the other environments (P < 0.02 in all cases with the UniFrac significance test) (39).
A number of mechanisms may contribute to the surprising local richness of soil microbial communities (Fig. 2a). Such factors may include a high degree of microscale variability in soil properties, rapid rates of speciation, high immigration rates, and low rates of extinction (14, 18, 21, 22, 31, 66). In addition, it is important to recognize that small body size alone may partially account for the high diversity of soil microorganisms at individual sites. Since richness is often correlated with the abundance of a taxon in a given area (16, 56), which is largely a function of body size (42, 45), surveying microbial diversity in individual soils may be similar in magnitude to surveying the diversity of “macro-organisms” at continental scales. For example, estimating microbial richness in our 100-m2 plots is likely to be analogous in terms of scale to estimating bird species richness (assume a body size of 10−3 m3) in a 108-km2 area. While body size alone is not likely to account for the high diversity of soil microorganisms, once we reconcile differences in spatial scale, the local richness of soil microorganisms may be more comparable to the observed levels of plant and animal richness.
Together our results confirm that we have only begun to explore the diversity of soil microorganisms. In an individual sample, our data suggest that the actual number of archaeal, fungal, bacterial, and viral “species” (or OTUs) exceeds the total number of microbial species that have been named to date (≈7,500 named archaea and bacteria combined, ≈80,000 fungi, and ≈2,000 viruses) (12, 19, 20). Clearly, the majority of the microbial diversity on Earth remains undiscovered.
Acknowledgments
We are grateful to J. Blair and M. Silman for their help with soil collection and to P. Holden, W. Cook, and M. Wallenstein for their valuable assistance on this project. We thank P. Adler, A. Martin, and two anonymous reviewers for comments on previous drafts of the manuscript.
This work was supported by grants from the Mellon Foundation and NSF to N.F.; grants from the Mellon Foundation, NIGEC/NICCR/DOE, IAI, and NSF to R.B.J.; and grants from the Gordon and Betty Moore Foundation and NSF to F.R.
Footnotes
Published ahead of print on 7 September 2007.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson, I. C., and J. W. G. Cairney. 2004. Diversity and ecology of soil fungal communities: increased understanding through the application of molecular techniques. Environ. Microbiol. 6:769-779. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, I. C., C. D. Campbell, and J. I. Prosser. 2003. Potential bias of fungal 18S rDNA and internal transcribed spacer polymerase chain reaction primers for estimating fungal biodiversity in soil. Environ. Microbiol. 5:36-47. [DOI] [PubMed] [Google Scholar]
- 4.Angly, F., B. Rodrigues-Brito, D. Bangor, P. McNairnie, M. Breitbart, P. Salamon, B. Felts, J. Nulton, J. Mahaffy, and F. Rohwer. 2005. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinformatics 6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker, G. C., J. J. Smith, and D. A. Cowan. 2003. Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods 55:541-555. [DOI] [PubMed] [Google Scholar]
- 6.Bohannan, B. J. M., and J. Hughes. 2003. New approaches to analyzing microbial biodiversity data. Curr. Opin. Microbiol. 6:282-287. [DOI] [PubMed] [Google Scholar]
- 7.Brady, N., and R. Weil. 2002. The nature and properties of soils. Pearson Education, Inc., Upper Saddle River, NJ.
- 8.Breitbart, M., B. Felts, S. Kelley, J. M. Mahaffy, J. Nulton, P. Salamon, and F. Rohwer. 2004. Diversity and population structure of a near-shore marine-sediment viral community. Proc. R. Soc. London Ser. B 271:565-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breitbart, M., I. Hewson, B. Felts, J. Mahaffy, J. Nulton, P. Salamon, and F. Rohwer. 2003. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 85:6220-6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breitbart, M., J. H. Miyake, and F. Rohwer. 2004. Global distribution of nearly identical phage-encoded DNA sequences. FEMS Microbiol. Lett. 236:249-256. [DOI] [PubMed] [Google Scholar]
- 11.Breitbart, M., P. Salamon, B. Andresen, J. M. Mahaffy, A. M. Segall, D. Mead, F. Azam, and F. Rohwer. 2002. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. USA 99:14250-14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bridge, P., and B. Spooner. 2001. Soil fungi: diversity and detection. Plant Soil 232:147-154. [Google Scholar]
- 13.Buckley, D., and T. Schmidt. 2002. Exploring the biodiversity of soil—a microbial rain forest, p. 183-208. In J. Staley and A. Reysenbach (ed.), Biodiversity of microbial life. John Wiley & Sons, New York, NY.
- 14.Crawford, J., J. Harris, K. Ritz, and I. Young. 2005. Towards an evolutionary ecology of life in soil. Trends Ecol. Evol. 20:81-87. [DOI] [PubMed] [Google Scholar]
- 15.Curtis, T. P., W. T. Sloan, and J. W. Scannell. 2002. Estimating prokaryotic diversity and its limits. Proc. Natl. Acad. Sci. USA 99:10494-10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diamond, J. 1988. Factors controlling species diversity: overview and synthesis. Ann. Missouri Bot. Garden 75:117-129. [Google Scholar]
- 17.Dunbar, J., S. M. Barns, L. O. Ticknor, and C. R. Kuske. 2002. Empirical and theoretical bacterial diversity in four Arizona soils. Appl. Environ. Microbiol. 68:3035-3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dykhuizen, D. E. 1998. Santa Rosalia revisited: why are there so many species of bacteria? Antonie Leeuwenhoek 73:25-33. [DOI] [PubMed] [Google Scholar]
- 19.Euzeby, J. 1997. List of bacterial names with standing in nomenclature: a folder available on the internet. Int. J. Syst. Bacteriol. 47:590-592. [DOI] [PubMed] [Google Scholar]
- 20.Fauquet, C., M. Mayo, J. Maniloff, U. Desselberger, and L. Ball (ed.). 2005. Virus taxonomy. Eighth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, CA.
- 21.Fierer, N., and R. Jackson. 2006. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 103:626-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finlay, B. J. 2002. Global dispersal of free-living microbial eukaryote species. Science 296:1061-1063. [DOI] [PubMed] [Google Scholar]
- 23.Gans, J., M. Wolinsky, and J. Dunbar. 2005. Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309:1387-1390. [DOI] [PubMed] [Google Scholar]
- 24.Hagn, A., K. Pritsch, W. Ludwig, and M. Schloter. 2003. Theoretical and practical approaches to evaluate suitable primer sets for the analysis of soil fungal communities. Acta Biotechnol. 4:373-381. [Google Scholar]
- 25.Hawksworth, D. L. 2001. The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol. Res. 105:1422-1432. [Google Scholar]
- 26.He, J., Z. Xu, and J. Hughes. 2005. Analyses of soil fungal communities in adjacent natural forest and hoop pine plantation ecosystems of subtropical Australia using molecular approaches based on 18S rRNA genes. FEMS Microbiol. Lett. 247:91-100. [DOI] [PubMed] [Google Scholar]
- 27.Hilborn, R., and M. Mangel. 1997. The ecological detective. Princeton University Press, Princeton, NJ.
- 28.Hong, S. H., J. Bunge, S. O. Jeon, and S. S. Epstein. 2006. Predicting microbial species richness. Proc. Natl. Acad. Sci. USA 103:117-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huber, T., G. Faulkner, and P. Hugenholtz. 2004. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20:2317-2319. [DOI] [PubMed] [Google Scholar]
- 30.Hughes, J. B., J. J. Hellmann, T. H. Ricketts, and B. J. M. Bohannan. 2001. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl. Environ. Microbiol. 67:4399-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hughes-Martiny, J. B., B. J. M. Bohannan, J. Brown, R. Colwell, J. Fuhrman, J. Green, M. Horner-Devine, M. Kane, J. Krumins, C. Kuske, P. Morin, S. Naeem, L. Ovreas, A. Reysenbach, V. Smith, and J. Staley. 2006. Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4:102-112. [DOI] [PubMed] [Google Scholar]
- 32.Hunt, J., L. Boddy, P. F. Randerson, and H. J. Rogers. 2004. An evaluation of 18S rDNA approaches for the study of fungal diversity in grassland soils. Microb. Ecol. 47:385-395. [DOI] [PubMed] [Google Scholar]
- 33.Jumpponen, A., and L. C. Johnson. 2005. Can rDNA analyses of diverse fungal communities in soil and roots detect effects of environmental manipulations—a case study from tallgrass prairie. Mycologia 97:1177-1194. [DOI] [PubMed] [Google Scholar]
- 34.Kemp, P., and J. Aller. 2004. Estimating prokaryotic diversity: when are 16S rDNA libraries large enough? Limnol. Oceanogr. Methods 2:114-125. [Google Scholar]
- 35.Kent, A. D., and E. W. Triplett. 2002. Microbial communities and their interactions in soil and rhizosphere ecosystems. Annu. Rev. Microbiol. 56:211-236. [DOI] [PubMed] [Google Scholar]
- 36.Lane, D. 1991. 16S/23S rRNA sequencing, p. 115-175. In E. Stackebrandt and M. Goodfellow (ed.), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, West Sussex, United Kingdom.
- 37.Leininger, S., T. Urich, M. Schloter, L. Schwark, J. Qi, G. Nicol, J. Prosser, S. Schuster, and C. Schleper. 2006. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442:806-809. [DOI] [PubMed] [Google Scholar]
- 38.Ley, R. E., F. Backhed, P. Turnbaugh, C. A. Lozupone, R. D. Knight, and J. I. Gordon. 2005. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 102:11070-11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lozupone, C., and R. Knight. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71:8228-8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lynch, M. D. J., and R. G. Thorn. 2006. Diversity of basidiomycetes in Michigan agricultural soils. Appl. Environ. Microbiol. 72:7050-7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magurran, A. 2004. Measuring biological diversity. Blackwell Publishing, Oxford, United Kingdom.
- 42.May, R. 1988. How many species are there on Earth? Science 247:1441-1449. [DOI] [PubMed] [Google Scholar]
- 43.O'Brien, H., J. Parrent, J. Jackson, J. Moncalvo, and R. Vilgalys. 2005. Fungal community analysis by large-scale sequencing of environmental samples. Appl. Environ. Microbiol. 71:5544-5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ochsenreiter, T., D. Selesi, A. Quaiser, L. Bonch-Osomolovskaya, and C. Schleper. 2003. Diversity and abundance of Crenarchaeota in terrestrial habitats studied by 16S RNA surveys and real time PCR. Environ. Microbiol. 5:787-797. [DOI] [PubMed] [Google Scholar]
- 45.Oindo, B., A. Skidmore, and H. Prins. 2001. Body size and abundance relationship: an index of diversity for herbivores. Biodiv. Conserv. 10:1923-1931. [Google Scholar]
- 46.Oline, D., S. Schmidt, and M. Grant. 2006. Biogeography and landscape-scale diversity of the dominant Crenarchaeota of soil. Microbial Ecol. 52:480-490. [DOI] [PubMed] [Google Scholar]
- 47.Pace, N. R. 1997. A molecular view of microbial diversity and the biosphere. Science 276:734-739. [DOI] [PubMed] [Google Scholar]
- 48.Prigent, M., M. Leroy, F. Confalonieri, M. Dutertre, and M. Dubow. 2005. A diversity of bacteriophage forms and genomes can be isolated from the surface sands of the Sahara Desert. Extremophiles 9:289-296. [DOI] [PubMed] [Google Scholar]
- 49.Reysenbach, A., and N. Pace. 1995. Reliable amplification of hyperthermophilic archaeal 16S rRNA genes by the polymerase chain reaction, p. 101-107. In F. Robb (ed.), Archaea: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 50.Robertson, C., J. Harris, J. Spear, and N. Pace. 2005. Phylogenetic diversity and ecology of environmental Archaea. Curr. Opin. Microbiol. 8:638-642. [DOI] [PubMed] [Google Scholar]
- 51.Rohwer, F., and R. Edwards. 2002. The Phage Proteomic Tree: a genome-based taxonomy for phage. J. Bacteriol. 184:4529-4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rossello-Mora, R., and R. Amann. 2001. The species concept for prokaryotes. FEMS Microbiol. Rev. 25:39-67. [DOI] [PubMed] [Google Scholar]
- 53.Sambrook, J., and D. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed., vol. 3. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 54.Schadt, C. W., A. P. Martin, D. A. Lipson, and S. K. Schmidt. 2003. Seasonal dynamics of previously unknown fungal lineages in tundra soils. Science 301:1359-1361. [DOI] [PubMed] [Google Scholar]
- 55.Schloss, P. D., and J. Handelsman. 2006. Toward a census of bacteria in soil. PLoS Comp. Biol. 2:e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siemann, E., D. Tilman, and J. Haarstad. 1996. Insect species diversity, abundance and body size relationships. Nature 380:704-706. [Google Scholar]
- 57.Smit, E., P. Leeflang, B. Glandorf, J. D. van Elsas, and K. Wernars. 1999. Analysis of fungal diversity in the wheat rhizosphere by sequencing of cloned PCR-amplified genes encoding 18S rRNA and temperature gradient gel electrophoresis. Appl. Environ. Microbiol. 65:2614-2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sylvia, D., J. Fuhrman, P. Hartel, and D. Zuberer (ed.). 1998. Principles and applications of soil microbiology. Prentice-Hall, Saddle River, NJ.
- 59.Torsvik, V., L. Ovreas, and T. F. Thingstad. 2002. Prokaryotic diversity: magnitude, dynamics, and controlling factors. Science 296:1064-1066. [DOI] [PubMed] [Google Scholar]
- 60.Tringe, S., C. von Mering, A. Kobayashi, A. Salamov, K. Chen, H. Chang, M. Podar, J. Short, E. Mathur, J. Detter, P. Bork, P. Hugenholtz, and E. Rubin. 2005. Comparative metagenomics of microbial communities. Science 308:554-557. [DOI] [PubMed] [Google Scholar]
- 61.Walsh, D. A., R. T. Papke, and W. F. Doolittle. 2005. Archaeal diversity along a soil salinity gradient prone to disturbance. Environ. Microbiol. 7:1655-1666. [DOI] [PubMed] [Google Scholar]
- 62.Wardle, D. A., R. D. Bardgett, J. N. Klironomos, H. Setala, W. H. van der Putten, and D. H. Wall. 2004. Ecological linkages between aboveground and belowground biota. Science 304:1629-1633. [DOI] [PubMed] [Google Scholar]
- 63.Whitman, W., D. Coleman, and W. Wiebe. 1998. Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. USA 95:6578-6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Williamson, K. E., M. Radosevich, and K. E. Wommack. 2005. Abundance and diversity of viruses in six Delaware soils. Appl. Environ. Microbiol. 71:3119-3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu, Y., M. Breitbart, P. McNairnie, and F. Rohwer. 2006. FastGroupII: a web-based bioinformatics platform for analyses of large 16S rDNA libraries. BMC Bioinformatics 7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou, J., B. Xia, D. S. Treves, L. Y. Wu, T. L. Marsh, R. V. O'Neill, A. V. Palumbo, and J. M. Tiedje. 2002. Spatial and resource factors influencing high microbial diversity in soil. Appl. Environ. Microbiol. 68:326-334. [DOI] [PMC free article] [PubMed] [Google Scholar]