

Abstract
Most Gluconobacter species produce and accumulate 2-keto-d-gluconate (2KGA) and 5KGA simultaneously from d-glucose via GA in culture medium. 2KGA is produced by membrane-bound flavin adenine dinucleotide-containing GA 2-dehydrogenase (FAD-GADH). FAD-GADH was purified from “Gluconobacter dioxyacetonicus” IFO 3271, and N-terminal sequences of the three subunits were analyzed. PCR primers were designed from the N-terminal sequences, and part of the FAD-GADH genes was cloned as a PCR product. Using this PCR product, gene fragments containing whole FAD-GADH genes were obtained, and finally the nucleotide sequence of 9,696 bp was determined. The cloned sequence had three open reading frames (ORFs), gndS, gndL, and gndC, corresponding to small, large, and cytochrome c subunits of FAD-GADH, respectively. Seven other ORFs were also found, one of which showed identity to glucono-δ-lactonase, which might be involved directly in 2KGA production. Three mutant strains defective in either gndL or sldA (the gene responsible for 5KGA production) or both were constructed. Ferricyanide-reductase activity with GA in the membrane fraction of the gndL-defective strain decreased by about 60% of that of the wild-type strain, while in the sldA-defective strain, activity with GA did not decrease and activities with glycerol, d-arabitol, and d-sorbitol disappeared. Unexpectedly, the strain defective in both gndL and sldA (double mutant) still showed activity with GA. Moreover, 2KGA production was still observed in gndL and double mutant strains. 5KGA production was not observed at all in sldA and double mutant strains. Thus, it seems that “G. dioxyacetonicus” IFO 3271 has another membrane-bound enzyme that reacts with GA, producing 2KGA.
Gluconobacter species carry out incomplete oxidation of a variety of sugars, sugar alcohols, and sugar acids to accumulate the corresponding oxidized products in large amounts in culture medium. These oxidation reactions are carried out by membrane-bound dehydrogenases linked to the respiratory chain located in the cytoplasmic membrane facing the periplasm (12). Most Gluconobacter species produce both 2-keto-d-gluconate (2KGA) and 5KGA from d-glucose via GA and accumulate them to high levels in culture medium simultaneously. The production ratio of 2KGA to 5KGA depends on individual strains and also culture conditions (22). 2KGA is produced not only by Gluconobacter but also by Pseudomonas, Serratia, and Klebsiella spp. (11). On the other hand, 5KGA production has been found exclusively in Gluconobacter so far, except that the secondary alcohol dehydrogenase of Xanthomonas campestris is reported to oxidize GA to produce 5KGA (17).
2KGA is produced by the membrane-bound flavoprotein flavin adenine dinucleotide-GA 2-dehydrogenase (FAD-GADH), which consists of three subunits and has been purified from and characterized for Pseudomonas aeruginosa (11) and “Gluconobacter dioxyacetonicus” (21). The large subunit is the dehydrogenase subunit of 62 kDa and contains FAD as the prosthetic group, which is shown to be covalently bound to the histidine residue of the subunit (14). The middle subunit (45 kDa) contains heme c and is probably involved in electron transfer from FAD in the dehydrogenase subunit to ubiquinone in the cytoplasmic membrane, linking to the respiratory chain (12). The small subunit is 21 kDa, and its function is unknown. The substrate specificity of the purified enzyme was very strict for GA, only 2KGA was produced, and the Michaelis constant for GA was measured as 2.2 mM (21).
On the other hand, the membrane-bound enzyme involved in 5KGA production remained unidentified for a long time, but recently, the enzyme was shown to be identical to the membrane-bound quinoprotein glycerol dehydrogenase having pyrroloquinoline quinone as the prosthetic group (PQQ-GLDH) (10). PQQ-GLDH was shown to be a versatile enzyme in the oxidation of various sugar alcohols, such as glycerol, d-sorbitol, d-arabitol, and d-mannitol, to their corresponding oxidation products; however, oxidation proceeds stereo- and regioselectively, obeying the so-called Bertrand-Hudson rule. The Km values of PQQ-GLDH are rather high (20, 40, and 420 mM for d-arabitol, d-sorbitol, and GA, respectively [10]). 5KGA is a useful compound as a precursor of tartaric acid, xylaric acid, or a number of flavor compounds (18); thus, several attempts to produce 5KGA have been made with Gluconobacter strains, and recently, exclusive production of 5KGA was accomplished with an FAD-GADH-defective mutant of Gluconobacter oxydans 621H (5). On the other hand, 2KGA itself is also a useful chemical and is applicable as a building block in the chemical synthesis of heterocyclic compounds and for regioselective and stereoselective chemical reactions (23).
In this study, we cloned the gene for FAD-GADH from “Gluconobacter dioxyacetonicus” IFO 3271, an FAD-GADH-disrupted mutant was constructed, and then 2KGA and 5KGA productions were examined. Unexpectedly, 2KGA was still produced by the FAD-GADH-disrupted mutant; therefore, it seems that “G. dioxyacetonicus” IFO 3271, which is known as a 2KGA producer (21), has another GADH yielding 2KGA.
MATERIALS AND METHODS
Materials.
All chemicals were obtained from commercial sources. Yeast extract was a kind gift from the Oriental Co.
Bacterial strains and growth conditions.
Strains used in this study are listed in Table 1. “G. dioxyacetonicus” IFO 3271 (now Gluconobacter frateurii NBRC 3271) was obtained from the Institute for Fermentation, Osaka, Japan (IFO), and the National Institute of Technology and Evaluation Biological Resource Center (NBRC). The strain was kept on an agar slant containing 0.5% CaCO3 in potato medium (24). Escherichia coli DH5α was used for cloning. The concentrations of ampicillin, kanamycin, and tetracycline used for transformants were as follows: 50, 50, and 25 μg/ml for E. coli and 500, 50, and 50 μg/ml for “G. dioxyacetonicus.” “G. dioxyacetonicus” and its mutant strains were precultured in potato medium and then inoculated (1%, vol/vol) into glucose-GA (G-GA) medium containing 1% d-glucose, 1% sodium GA (NaGA), 0.3% yeast extract, and 0.3% polypeptone. Bacterial growth was measured using a Klett-Summerson photoelectric colorimeter with a red filter.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant trait(s) | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | F− φ80dlacZΔM15 Δ(lacZYA-argF) recA1 (rK− mK+) | TOYOBO |
| “G. dioxyacetonicus” IFO 3271 (G. frateurii NBRC 3271) strains | ||
| Wild type | IFO or NBRC | |
| gndL::Km mutant | Kmr, gndL disruptant | This study |
| sldA::Km mutant | Kmr, sldA disruptant | This study |
| gndL::Tc mutant | Tcr, gndL disruptant | This study |
| gndL::Tc sldA::Km mutant | Tcr Kmr, gndL and sldA double disruptant | This study |
| Plasmids | ||
| pUC4K | Contains nonpolar Kmr cassette | Pharmacia |
| pKRP12 | Contains Tcr cassette | 16 |
| pSUP202sldA::Km | Kmr, mutation for sldA disruption | 10 |
| pUC119-BP | 4.3-kb BamHI-PstI fragment in pUC119 | This study |
| pUC119-BPgndL::Km | Kmr, for gndL disruption; Kmr cassette inserted at SalI site | This study |
| pUC119-BPgndL::Tc | Tcr, for gndL disruption; Tcr cassette inserted at SalI site | This study |
Preparation of the membrane fraction.
The membrane fraction was prepared as described previously (24) and suspended in 10 mM potassium phosphate buffer (pH 5.7).
Purification of FAD-GADH.
FAD-GADH was solubilized and purified according to the method described previously (21). Since we failed to remove alcohol dehydrogenase activity, additional CM-Toyopearl column chromatography (the third) was performed, with the column being equilibrated with 10 mM potassium phosphate buffer (pH 5.7) containing 0.1% Triton X-100 (buffer A). The column was first washed with buffer A containing 0.04 M NaCl, and then the active fraction that eluted with 0.2 M NaCl was pooled.
Enzyme assay.
Enzyme activities were measured at 25°C with a 0.1 M concentration of the substrate and potassium ferricyanide as the electron acceptor (24) in McIlvaine buffer (pH 5.0). One unit of enzyme activity was defined as the amount of enzyme catalyzing the oxidation of 1 μmol of the substrate per min.
Determination of protein concentration.
Protein concentration was measured by a modified Lowry method (4). Bovine serum albumin was used as the standard protein.
Determination of 2KGA and 5KGA.
2KGA and 5KGA were enzymatically quantified by 2KGA reductase and 5KGA reductase, respectively (1, 2). Qualitative analysis of 2KGA and 5KGA was performed using thin-layer chromatography analysis. Samples were spotted on a Silica Gel 60 plate (Merck) and developed with a solvent reagent containing ethyl acetate-acetic acid-methanol-deionized water (6:1.5:1.5:1). After the plate was dried, a color-developing reagent (a freshly prepared mixture of 2 g of diphenylamine, 2 ml of aniline, 100 ml of acetone, and 15 ml of phosphoric acid) was spread and heated at 120°C for 10 to 20 min to allow us to see the color development. 5KGA and 2KGA appeared as dark-purple and red-brown spots, respectively.
SDS-PAGE.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was done on a 12.5% (wt/vol) acrylamide slab gel by the methods described by Laemmli (6). Proteins were stained with Coomassie brilliant blue R-250. Before being stained, the gel was exposed to UV light to allow us to see whether the cofactor was covalently bound to the peptide. For analysis of N-terminal amino acid sequences, proteins in the gel were transferred electrophoretically onto a polyvinylidene difluoride membrane and stained with Coomassie brilliant blue G-250. The corresponding protein band was cut off and analyzed using a model PPSQ-2 protein sequencer (Shimadzu).
DNA techniques.
Restriction enzyme digestion, DNA ligation, and other DNA modifications were performed according to the manufacturer's recommendations. Plasmid DNA was prepared from E. coli strains, and other general molecular biology techniques were performed as described by Sambrook et al. (19). Genomic DNA was isolated by a modified method of Marmur (8). PCR was performed using a Ready-To-Go PCR bead kit (Amersham Biosciences). Primers GADH1-1, 5′-GTS ATY GGY TTC GGY TG-3′, and GADH2-1, 5′-TG RCA SGC YTC GCA RTC RCC-3′, which were designed from N-terminal amino acid sequences of the large and small subunits, respectively (Fig. 1), were used for PCR amplification with genomic DNA of “G. dioxyacetonicus” IFO 3271 as a template. The obtained 1.9-kb DNA fragment with an expected size was isolated by agarose gel electrophoresis and purified with a QIAquick gel extraction kit (QIAGEN) and then cloned into the pGEM-T Easy vector (Promega). Using the 1.5-kb EcoRI fragment as a probe, 4.3 kb of the BamHI-PstI fragment was obtained. Next, using the 1.7-kb BamHI-EcoRI and 2.3-kb SalI-PstI fragments from the 4.3-kb fragment, the 4.6-kb EcoRI and 2.3-kb SalI-PstI fragments, respectively, were obtained by colony hybridization (Fig. 1).
FIG. 1.

Schematic representation of the gene fragment obtained in this study. Primers for PCR are indicated by arrows. Thick lines are DNA fragments obtained in this study. Hatched boxes represent gene fragments used for hybridization. The open triangle shows the position where the kanamycin or tetracycline resistance gene was inserted for mutation. ORF-5 is not complete.
Southern hybridization.
Genomic DNA was digested with suitable restriction enzymes, electrophoresed in agarose gel, and then transferred to a Hybond N+ membrane (Amersham Biosciences). Hybridization and detection were carried out using the ECL direct nucleotide labeling system (Amersham Biosciences) according to the protocol provided by the supplier.
Colony hybridization.
Colonies of the gene library constructed from the genomic DNA of “G. dioxyacetonicus” IFO 3271 in pUC119 were grown on an LB agar plate containing 50 μg/ml of ampicillin, transferred to a Hybond-N+ membrane, and lysed. Hybridization and detection were performed using the digoxigenin system (Roche Diagnostics) according to the protocol provided by the supplier.
Nucleotide sequence analysis.
Plasmids for sequencing were prepared using the QIAprep Spin miniprep kit (QIAGEN). Sequencing was performed using an ABI PRISM 310 sequencer (Applied Biosystems). Homology search analysis and alignment were performed with BLAST (http://www.ncbi.nlm.nih.gov/BLAST/).
Construction of mutants.
Plasmids for the disruption of gndL were constructed by inserting the Kmr or Tcr cassette from pUC4K or pKRP12 (16), respectively, at the SalI site in the 4.3-kb BamHI-PstI fragment (Fig. 1). For sldA disruption, pSUP202sldA::Km was used (10). Plasmids for mutation were introduced into “G. dioxyacetonicus” IFO 3271 by electroporation as described previously (24), and the cells were incubated in potato medium for 8 hours at 30°C for gene replacement. Colonies obtained after 2 to 3 days on agar plates containing 1% sorbitol, 1% glycerol, 0.3% polypeptone, 0.3% yeast extract, and 0.5% CaCO3 with kanamycin and/or tetracycline were selected by patching them on two new agar plates containing kanamycin and/or tetracycline with or without ampicillin. Double-crossover mutants were obtained as ampicillin-sensitive mutants, and gene disruption was confirmed by PCR with a set of specific primers characterized in Fig. 1: GADH1-4, 5′-AGA TGG AAA AGG GAA GCC TC-3′, and GADH1-5, 5′-TTG AAC GAT CCG CAT CGA AC-3′ for gndL disruption and primers SldA-1 and SldA-2 used previously (10) for sldA disruption.
Nucleotide sequence accession number.
The sequence described in this work has been deposited with the DNA Data Bank of Japan (DDBJ) under accession number AB292729.
RESULTS
Purification of membrane-bound FAD-GADH from “G. dioxyacetonicus” IFO 3271.
FAD-GADH was purified from “G. dioxyacetonicus” IFO 3271 by following the method previously reported (21). The enzyme showed slightly different properties upon column chromatography: for example, in the previous report, enzyme activity was eluted from the CM-Toyopearl column (second chromatography) with buffer containing 0.2 M NaCl, but in this study, major activity had already been eluted with 0.05 M NaCl containing the quinohemoprotein alcohol dehydrogenase; therefore, a third CM-Toyopearl column chromatography procedure was performed to remove alcohol dehydrogenase (0.04 M NaCl), and FAD-GADH was eluted with 0.2 M NaCl. Purified FAD-GADH had a specific activity of 137 U/mg (Table 2), which is much lower than reported previously (237 U/mg [21]). SDS-PAGE analysis showed three major bands (65, 44, and 23 kDa); however, several minor bands were still seen in this purified fraction (Fig. 2). Under UV light, the 65-kDa subunit showed fluorescence, indicating covalently bound FAD. N-terminal amino acid sequences of these proteins determined by Edman degradation were as follows: the 65-kDa subunit was VSRNEKKTDVVIVGFGWV, the 44-kDa subunit was ADADLIKRGAYVAVLGDXEAXHTAHDGK, and the 23-kDa subunit was ADREYAYRPVFFSDDE. The first two sequences were similar to those of FAD-GADH reported from Erwinia cypripedii (25), but the sequence of the 23-kDa subunit showed no similarity to other sequences.
TABLE 2.
Purification summary of FAD-GADH from “G. dioxyacetonicus” IFO 3271
| Step | Total protein (mg) | Total activity (U) | Sp act (U/mg) | Recovery (%) | Purification (fold) |
|---|---|---|---|---|---|
| Membrane | 1,220 | 2,470 | 2.03 | 100 | 1 |
| Solubilization | 242 | 2,180 | 9.03 | 88 | 4.4 |
| DEAE-cellulose | 79 | 1,920 | 24.5 | 78 | 12 |
| CM-Toyopearl (first) | 16 | 1,500 | 96.0 | 61 | 47 |
| CM-Toyopearl (second) | 3.7 | 556 | 149 | 22 | 73 |
| CM-Toyopearl (third) | 2.0 | 271 | 137 | 11 | 67 |
FIG. 2.
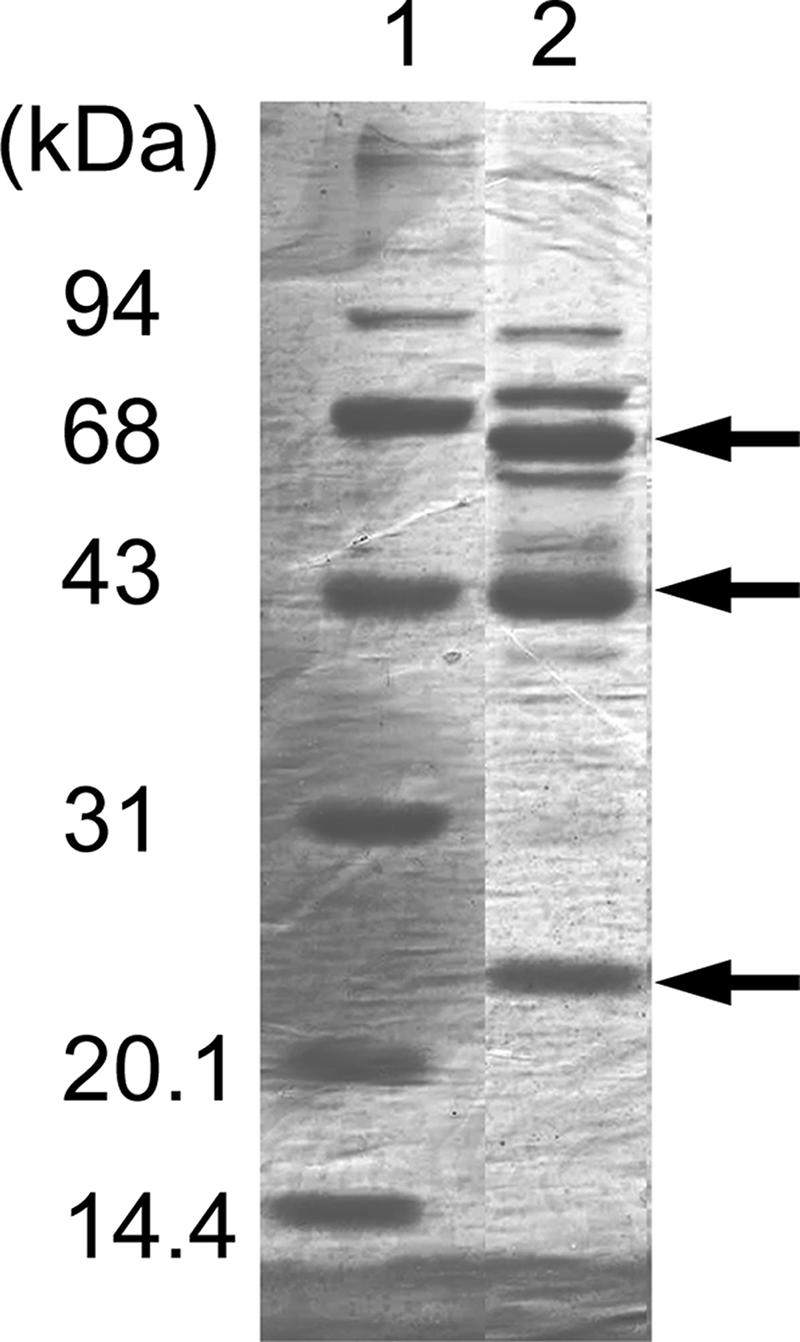
SDS-PAGE analysis of purified FADH-GADH from “G. dioxyacetonicus” IFO 3271. Ten micrograms of purified FAD-SLDH was applied, and the gel was stained with Coomassie brilliant blue R-250. The arrows indicate the large, the cytochrome c, and the small subunits of FAD-GADH, from top to bottom.
Sequence analysis of cloned gene fragments.
In total, a 9,696-bp nucleotide sequence was obtained, and nine open reading frames (ORFs) were predicted (Table 3), three of which corresponded to the structural genes of FAD-GADH, named gndS, gndL, and gndC. Recently, the genome sequence of G. oxydans 621H was reported (15), and in the sequence, GOX1232 to GOX1230 showed high levels of identity to GndS, GndL, and GndC: 39, 60, and 45%, respectively. GndS, GndL, and GndC showed 33, 61, and 45% identity to the small, large, and cytochrome subunits, respectively, of GADH from E. cypripedii (25).
TABLE 3.
gndSLC and neighbors obtained from “G. dioxyacetonicus” in this study
| Gene | Size of gene product (amino acids) | Function | Locationa |
|---|---|---|---|
| ORF-1 | 372 | Putative regulatory protein, LacI family | C |
| ORF-2 | 391 | Hypothetical protein | C |
| gnlA | 342 | Glucono-δ-lactonase | P |
| ORF-3 | 218 | Hypothetical, Asp-Glu-hydantoin racemase | P |
| gndS | 242 | Small subunit of FAD-GADH | P |
| gndL | 593 | Catalytic large subunit of FAD-GADH | P |
| gndC | 441 | Cytochrome c subunit of FAD-GADH | P |
| ORF-4 | 256 | Hypothetical protein | C |
| ORF-5 | (201)b | Putative regulatory protein, LysR type | C |
C, cytoplasm: P, periplasm.
Incomplete ORF.
GndS was shown to contain a twin-arginine-translocation (tat)-dependent signal sequence (3). The amino acid sequence determined from the 23-kDa subunit of the purified enzyme was identical to the sequence from Ala43 to Glu58. Several genes obtained from the genome sequence database showed significant identity to GndS: for example, 44% identity to PA2264 from P. aeruginosa PAO1 (GenBank accession number AAG05652). These genes are composed of clusters of genes similar to gndL and gndC, indicating that they also encode FAD-GADH, in accordance with the existence of FAD-GADH in Pseudomonas species (11).
The sequence from Val2 to Val19 of GndL was identical to the N-terminal amino acid sequence determined from the 65-kDa subunit of the purified enzyme. The N-terminal region up to Gly328 contained the conserved domain sequence found in the so-called flavoprotein glucose-methanol-choline (GMC)-oxidoreductase family.
The sequence from Ala30 to Lys57 of GndC was identical to the amino acid sequence determined from the 44-kDa subunit of the purified enzyme, except at Cys47 and Cys50. Three heme c-binding motifs (CXXCH) were found. GndC showed identity to the cytochrome c subunits of several other membrane-bound dehydrogenases, for example, 46% identity to alcohol dehydrogenase (GenBank accession number CAA70689) and 30% identity to aldehyde dehydrogenase (CAA69955) of Gluconacetobacter europaeus. These cytochrome c subunits are supposed to be an anchor to the cytoplasmic membrane, although there is no hydrophobic membrane-spanning region, and to be the site of ubiquinone reduction (13).
Effects of gene disruption on growth and enzyme activities.
Three mutants of “G. dioxyacetonicus” IFO 3271 were prepared by insertion of antibiotic resistance genes, gndL::Km, sldA::Km, and gndL::Tc sldA::Km, which were defective in FAD-GADH, PQQ-GLDH, and both enzymes, respectively. Gene disruption in each strain was confirmed by PCR (Fig. 3). Growth properties of the three mutants on G-GA medium were not significantly different from those of the wild-type strain (data not shown).
FIG. 3.

PCR products to confirm gene disruption. PCR was performed with sets of primers, namely, GADH1-4 and GADH1-5 (A and C) and SldA-1 and SldA-2 (B), and genomic DNA from the wild-type (lanes 1) and mutant (lanes 2) strains. PCR products were analyzed by agarose gel electrophoresis. (A) gndL::Km strain; (B) sldA::Km strain; (C) gndL::Tc sldA::Km strain. Lanes M, λ DNA/StyI marker DNA.
FAD-GADH and PQQ-GLDH activities in the membrane fraction were measured. FAD-GADH has strict specificity (21); however, PQQ-GLDH has relatively broad specificity and reacts with glycerol, d-arabitol, and d-sorbitol better than with GA (10); thus, activity with GA is expected to be the sum of activities of PQQ-GLDH and FAD-GADH. In the gndL::Km strain, activity with GA decreased, while activities with the other three substrates were similar to those of the wild-type strain (Table 4). Indistinguishable results were obtained from the gndL::Tc strain (data not shown). The sldA::Km strain did not decrease activity with GA and lost activity with the other three substrates completely, according to the loss of PQQ-GLDH. Unexpectedly, activity with GA was still detected in the membrane fraction of the gndL::Tc sldA::Km strain and was about 80% of that of the wild-type strain (Table 4), although the activities of PQQ-GLDH with three substrates were abolished.
TABLE 4.
GADH activity in membrane fractions of mutant strains
| Membrane | Ferricyanide reductase activity (U/mg of protein)a
|
|||
|---|---|---|---|---|
| GA | Glycerol | d-Arabitol | d-Sorbitol | |
| Wild type | 0.71 ± 0.13 | 0.55 ± 0.05 | 0.37 ± 0.08 | 0.41 ± 0.10 |
| gndL::Km mutant | 0.44 ± 0.18 | 0.51 ± 0.10 | 0.34 ± 0.09 | 0.38 ± 0.09 |
| sldA::Km mutant | 0.77 ± 0.10 | ND | ND | ND |
| gndL::Tc sldA::Km mutant | 0.56 ± 0.12 | ND | ND | ND |
ND, not detected. Data were obtained from three independent experiments.
2KGA and 5KGA that accumulated in the culture medium were also measured (Fig. 4). Under the growth conditions used, the wild-type strain produced both 2KGA and 5KGA. The sldA::Km and gndL::Tc sldA::Km strains completely lost 5KGA production according to the absence of PQQ-GLDH, as reported previously (10); however, the gndL::Km strain still produced both 2KGA and 5KGA, although 5KGA production increased about 1.5-fold. Moreover, the sldA::Km and gndL::Tc sldA::Km strains produced a larger amount of 2KGA than the other strains, since the competing 5KGA production was abolished. These results clearly indicate that there is another enzyme reacting with GA and yielding 2KGA.
FIG. 4.

2KGA and 5KGA production in culture medium by mutant strains. (A) Culture supernatant (4 μl) after cells were grown on G-GA medium for 30 h was analyzed by thin-layer chromatography as described in Materials and Methods Lanes: 1 to 4, 4 μl of a 100 mM standard solution (d-glucose [lane 1], NaGA [lane 2], 2KGA [lane 3], or 5KGA [lane 4]); 5 to 9, culture medium (before inoculation [lane 5] or with the wild type [lane 6], gndL::Km strain [lane 7], sldA::Km strain [lane 8], or gndL::Tc sldA::Km strain [lane 9]). (B) The culture supernatant was also quantified using 2KGR and 5KGR as described in Materials and Methods. Data were obtained from three independent experiments. ND, not detected.
Reaction product of FAD-GADH.
To determine the product of FAD-GADH purified in this study, GA was oxidized with the purified enzyme by coupling it with the purified ubiquinol oxidase of Acetobacter (9) in the presence of quinol (Q2H2). Analysis by thin-layer chromatography showed the same color and Rf value (0.24) as those of authentic 2KGA (data not shown). Moreover, by using 2KGA reductase, 2KGA was stoichiometrically detected in the initial amount of the substrate (data not shown). Thus, it was clearly shown that FAD-GADH purified in this study produces 2KGA from GA.
DISCUSSION
In this study, the gene for FAD-GADH of “G. dioxyacetonicus” IFO 3271 was cloned by PCR and the colony hybridization of gene libraries. The genes gndSLC showed identity to those from E. cypripedii (25), and corresponding genes were found in genome sequences of several bacteria, including G. oxydans 621H. Surprisingly, disruption of gndL did not result in the termination of 2KGA production, which is inconsistent with G. oxydans 621H (5). This result indicates that there is another 2KGA-producing, membrane-bound GADH in “G. dioxyacetonicus” IFO 3271. At this moment, it is not clear that FAD-GADH purified in this study is identical to the enzyme previously purified by Shinagawa et al. (21), although they showed almost identical molecular properties, including covalent attachment of FAD on the large subunit. During the purification of FAD-GADH, no active fractions clearly separated from the enzyme were observed (data not shown).
FAD-GADH is a periplasmic, membrane-bound protein; therefore, each subunit is required to be translocated to the periplasm. GndC has a sec signal sequence required for translocation to the periplasm (7). GndS has a tat signal sequence, and GndL has no signal sequence; therefore, the large subunit (GndL) seems to be translocated together with the small subunit (GndS) by the “hitchhiker” mechanism after the prosthetic group, FAD, is incorporated in the cytoplasm (3). The mechanism of covalent attachment of FAD is not certain; however, it is suggested that this occurs spontaneously after incorporation into apoprotein (20). A gene with similarity to glucono-δ-lactonase was found near gndSLC, and it also has a signal sequence; thus, this gene might work for 2KGA production.
In the genome sequence of G. oxydans 621H, two genes other than GOX1231 showed significant homology to GndL from “G. dioxyacetonicus” IFO 3271: GOX2095 and GOX1071, which have 26 and 25% identities, respectively. However, GOX1231 is the sole gene responsible for 2KGA production, which has already been confirmed by Elfari et al. (5). On the other hand, in “G. dioxyacetonicus” IFO 3271, a mutation in gndL did not result in the loss of 2KGA production; therefore, another enzyme is responsible for 2KGA production. Accumulation of 2KGA and 5KGA from GA is highly dependent on species and growth conditions (22), although all Gluconobacter species seem to produce constitutive PQQ-GLDH, responsible for 5KGA production, as discussed previously for d-sorbitol oxidation (24). With genomic DNA of “G. dioxyacetonicus” IFO 3271 as a template, PCR was performed with a set of primers designed from conserved regions of gndL, and we obtained another DNA fragment with a sequence similar to but different from that of gndL (unpublished result), indicating another FAD-GADH in this strain. Cloning and analysis of the second gene and purification of the enzyme from the double-knockout strain are now under way. Gluconobacter strains which produce 2KGA much more than 5KGA might possess duplicated genes for FAD-GADH.
Acknowledgments
This work was supported in part by a grant-in-aid for scientific research from the Ministry of Education, Science, and Culture, Japan (grant 18580078 to H.T.).
Footnotes
Published ahead of print on 24 August 2007.
REFERENCES
- 1.Ameyama, M., O. Adachi, and A. W. Willis. 1982. [33] 5-Keto-d-gluconate reductase from Gluconobacter suboxydans. Methods Enzymol. 89:198-202. [Google Scholar]
- 2.Ameyama, M., O. Adachi, and A. W. Willis. 1982. [34] 2-Keto-d-gluconate reductase from acetic acid bacteria. Methods Enzymol. 89:203-210. [DOI] [PubMed] [Google Scholar]
- 3.Berks, B. C., F. Sargent, and T. Palmer. 2000. The Tat protein export pathway. Mol. Microbiol. 35:260-274. [DOI] [PubMed] [Google Scholar]
- 4.Dulley, J. R., and P. A. Grieve. 1975. A simple technique for eliminating interference by detergents in the Lowry method of protein determination. Anal. Biochem. 64:136-141. [DOI] [PubMed] [Google Scholar]
- 5.Elfari, M., S. W. Ha, C. Bremus, M. Merfort, V. Khodaverdi, U. Herrmann, H. Sahm, and H. Gorisch. 2005. A Gluconobacter oxydans mutant converting glucose almost quantitatively to 5-keto-d-gluconic acid. Appl. Microbiol. Biotechnol. 66:668-674. [DOI] [PubMed] [Google Scholar]
- 6.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 7.Manting, E. H., and A. J. Driessen. 2000. Escherichia coli translocase: the unravelling of a molecular machine. Mol. Microbiol. 37:226-238. [DOI] [PubMed] [Google Scholar]
- 8.Marmur, J. 1961. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3:208-218. [Google Scholar]
- 9.Matsushita, K., H. Ebisuya, and O. Adachi. 1992. Homology in the structure and the prosthetic groups between two different terminal ubiquinol oxidases, cytochrome a1 and cytochrome o, of Acetobacter aceti. J. Biol. Chem. 267:24748-24753. [PubMed] [Google Scholar]
- 10.Matsushita, K., Y. Fujii, Y. Ano, H. Toyama, M. Shinjoh, N. Tomiyama, T. Miyazaki, T. Sugisawa, T. Hoshino, and O. Adachi. 2003. 5-Keto-d-gluconate production is catalyzed by a quinoprotein glycerol dehydrogenase, major polyol dehydrogenase, in Gluconobacter species. Appl. Environ. Microbiol. 69:1959-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsushita, K., E. Shinagawa, and M. Ameyama. 1982. d-Gluconate dehydrogenase from bacteria, 2-keto-d-gluconate-yielding, membrane-bound. Methods Enzymol. 89:187-193. [DOI] [PubMed] [Google Scholar]
- 12.Matsushita, K., H. Toyama, and O. Adachi. 1994. Respiratory chains and bioenergetics of acetic acid bacteria. Adv. Microb. Physiol. 36:247-301. [DOI] [PubMed] [Google Scholar]
- 13.Matsushita, K., T. Yakushi, H. Toyama, O. Adachi, H. Miyoshi, E. Tagami, and K. Sakamoto. 1999. The quinohemoprotein alcohol dehydrogenase of Gluconobacter suboxydans has ubiquinol oxidation activity at a site different from the ubiquinone reduction site. Biochim. Biophys. Acta 1409:154-164. [DOI] [PubMed] [Google Scholar]
- 14.McIntire, W., T. P. Singer, M. Ameyama, O. Adachi, K. Matsushita, and E. Shinagawa. 1985. Identification of the covalently bound flavins of d-gluconate dehydrogenases from Pseudomonas aeruginosa and Pseudomonas fluorescens and of 2-keto-d-gluconate dehydrogenase from Gluconobacter melanogenus. Biochem. J. 231:651-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prust, C., M. Hoffmeister, H. Liesegang, A. Wiezer, W. F. Fricke, A. Ehrenreich, G. Gottschalk, and U. Deppenmeier. 2005. Complete genome sequence of the acetic acid bacterium Gluconobacter oxydans. Nat. Biotechnol. 23:195-200. [DOI] [PubMed] [Google Scholar]
- 16.Reece, K. S., and G. J. Phillips. 1995. New plasmids carrying antibiotic-resistance cassettes. Gene 165:141-142. [DOI] [PubMed] [Google Scholar]
- 17.Salusjarvi, T., N. Hvorslev, and A. N. Miasnikov. 2005. Characterisation of a secondary alcohol dehydrogenase from Xanthomonas campestris DSM 3586. Appl. Microbiol. Biotechnol. 66:664-667. [DOI] [PubMed] [Google Scholar]
- 18.Salusjarvi, T., M. Povelainen, N. Hvorslev, E. V. Eneyskaya, A. A. Kulminskaya, K. A. Shabalin, K. N. Neustroev, N. Kalkkinen, and A. N. Miasnikov. 2004. Cloning of a gluconate/polyol dehydrogenase gene from Gluconobacter suboxydans IFO 12528, characterisation of the enzyme and its use for the production of 5-ketogluconate in a recombinant Escherichia coli strain. Appl. Microbiol. Biotechnol. 65:306-314. [DOI] [PubMed] [Google Scholar]
- 19.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 20.Scrutton, N. S. 1999. Identification of covalent flavoproteins and analysis of the covalent link. Methods Mol. Biol. 131:181-193. [DOI] [PubMed] [Google Scholar]
- 21.Shinagawa, E., K. Matsushita, O. Adachi, and M. Ameyama. 1984. d-Gluconate dehydrogenase, 2-keto-d-gluconate yielding, from Gluconobacter dioxyacetonicus: purification and characterization. Agric. Biol. Chem. 48:1517-1522. [Google Scholar]
- 22.Shinagawa, E., K. Matsushita, O. Adachi, and M. Ameyama. 1983. Selective production of 5-keto-d-gluconate by Gluconobacter strains. J. Ferment. Technol. 61:359-363. [Google Scholar]
- 23.Stottmeister, U., A. Aurich, H. Wilde, J. Andersch, S. Schmidt, and D. Sicker. 2005. White biotechnology for green chemistry: fermentative 2-oxocarboxylic acids as novel building blocks for subsequent chemical syntheses. J. Ind. Microbiol. Biotechnol. 32:651-664. [DOI] [PubMed] [Google Scholar]
- 24.Toyama, H., W. Soemphol, D. Moonmangmee, O. Adachi, and K. Matsushita. 2005. Molecular properties of membrane-bound FAD-containing d-sorbitol dehydrogenase from thermotolerant Gluconobacter frateurii isolated from Thailand. Biosci. Biotechnol. Biochem. 69:1120-1129. [DOI] [PubMed] [Google Scholar]
- 25.Yum, D. Y., Y. P. Lee, and J. G. Pan. 1997. Cloning and expression of a gene cluster encoding three subunits of membrane-bound gluconate dehydrogenase from Erwinia cypripedii ATCC 29267 in Escherichia coli. J. Bacteriol. 179:6566-6572. [DOI] [PMC free article] [PubMed] [Google Scholar]