

Abstract
Epidemiological studies reveal increased incidence of lung infection when air pollution particle levels are increased. We postulate that one risk factor for bacterial pneumonia, prior viral infection, can prime the lung for greater deleterious effects of particles via the γ-interferon (IFN-γ) characteristic of successful host anti-viral responses. To test this postulate, we developed a mouse model in which mice were treated with γ-interferon aerosol, followed by exposure to concentrated ambient particles (CAPs) collected from urban air. The mice were then infected with Streptococcus pneumoniae and the effect of these treatments on the lung innate immune response was evaluated. The combination of IFN-γ priming and CAPs exposure enhanced lung inflammation, manifest as increased polymorphonuclear granulocyte (PMN) recruitment to the lung, and elevated expression of pro-inflammatory cytokine mRNAs. Combined priming and CAPs exposure resulted in impaired pulmonary bacterial clearance, as well as increased oxidant production and diminished bacterial uptake by alveolar macrophages (AMs) and PMNs. The data suggest that priming and CAPs exposure lead to an inflamed alveolar milieu where oxidant stress causes loss of antibacterial functions in AMs and recruited PMNs. The model reported here will allow further analysis of priming and CAPs exposure on lung sensitivity to infection.
Keywords: γ-interferon priming, air pollution particles, Streptococcus pneumoniae, AMs, PMNs
INTRODUCTION
Epidemiological studies have shown a strong correlation between elevated concentrations of air pollution particles and increased mortality and morbidity (Dockery et al., 1993). One specific harmful effect of increased levels of particles in the air is increased hospitalization for pneumonia, both in children and in elderly populations (Schwartz, 1994b; Schwartz, 1994a; Lin et al., 1999; Wong et al., 1999; Farhat et al., 2005). The basis for particle-mediated increased susceptibility to infection is unknown, but the epidemiology of pneumonia offers some clues since both pediatric and elderly populations are affected. These groups both experience one of the well recognized risk factors for pneumonia antecedent viral respiratory tract infection (Warr and Jakab, 1983; Cate, 1998). This suggests that particles have especially deleterious effects in lungs primed by inflammatory mediators, e.g., the γ-interferon characteristic of anti-viral responses (Mo et al., 1995; van Schaik et al., 2000).
Animal models support the concept that air particles increase susceptibility to lung infection. Hatch et al. found that particle exposure increased susceptibility to bacterial pneumonia in mice (Hatch et al., 1985). Similarly, diesel exhaust particles (DEP) can impair the pulmonary clearance of Listeria monocytogenes in rats (Yang et al., 2001), and increase the severity of influenza virus infection in mice (Hahon et al., 1985). However, the effect of air particles on the most common form of community-acquired pneumonia, pneumococcal pneumonia, has not been studied, nor has the possible effect of ‘priming’ been investigated.
We postulate that lung priming (e.g., recent viral infection) followed by particle exposure induces an exacerbated inflammatory response, causing oxidant-mediated damage to both alveolar macrophages (AMs) and neutrophils (polymorphonuclear granulocytes: PMNs), and resulting in impaired bacterial phagocytosis and killing. To test this hypothesis, we developed a mouse model in which the animals were treated with IFN-γ aerosol, followed by exposure to concentrated ambient particles (CAPs) collected from the urban air of Boston, MA. The mice were then infected with S. pneumoniae and the effect of these treatments on the lung immune response was evaluated. We show that the combination of IFN-γ priming and CAPs exposure enhances lung inflammation, causes oxidative damage in the lung, and results in a loss of antibacterial functions by AMs and PMNs.
METHODS
Animals and animal exposures
8 to 10 week-old male BALB/c mice (Jackson Laboratory; Bar Harbor, ME) were exposed to phosphate buffered saline (PBS) or interferon-gamma (IFN-γ, 20,000 U/ml in PBS) aerosol for 15 minutes in individual compartments of a mouse “pie” chamber (Braintree Scientific, Braintree, MA). Aerosols were generated using a Pari IS2 nebulizer (Sun Medical Supply, Kansas City, KS) connected to an air compressor (PulmoAID; DeVilbiss, Somerset, PA). Particle exposures were performed 3h later by intranasal instillation after light anesthesia with halothane. A total volume of 50 μl PBS was introduced in both nostrils, with or without 50 μg of titanium dioxide (TiO2) or concentrated ambient particles (CAPs) produced using the Harvard Ambient Particle Concentrator (Sioutas et al., 1995). All animal experimentation was conducted under protocols approved by Harvard University’s animal care and use committee and conformed to National Institutes of Health standards as defined by the U.S. Department of Agriculture and Animal Welfare.
Chemicals and reagents
All reagents were from Sigma (St. Louis, MO) except where indicated. CAPs of respiratory size (<2.5 μm) were collected from ambient Boston air onto Teflon filters (47 mm diameter, 2 μm pore size; Gelman Sciences, Ann Arbor, MI) by the Harvard concentrator (Sioutas et al., 1995). The CAPs used in this study were a mixture of samples collected on individual days from all four seasons during 1997-1998, and prepared and stored as previously described (Imrich et al., 1999; Imrich et al., 2000).
Bronchoalveolar lavage (BAL)
Mice were euthanized with an overdose of sodium pentobarbital (Veterinary Laboratories, Lenexa, KS). The trachea was cannulated and BAL was performed: 1 ml of sterile PBS was instilled into the lung and harvested gently five times, and lavage fluid was collected after each instillation. These samples were kept on ice during lavage and then the lavage fluid was centrifuged at 1200 rpm (300 × g) for 10 min. The cell pellet was resuspended in 0.5 ml buffered salt solution (BSS+), and total cell counts and viability were determined using a hemocytometer and trypan blue dye exclusion.
Polymorphonuclear granulocyte (PMN) enumeration
Differential cell counts were determined by centrifuging 50,000 - 100,000 cells/lavage onto cytocentrifuge slides at 800 rpm for 5 min (Cytospin 2; Shandon, Pittsburgh, PA). Slides were fixed in methanol and stained with Diff-Quik (VWR, Boston, MA), a modified Wright-Giemsa stain. 200 cells were counted for each sample by microscopy. PMN number was calculated by multiplying the percentage of neutrophils in the BAL by the total cell yield.
Streptococcus pneumoniae infection
An encapsulated virulent strain of S. pneumoniae Serotype 3 (ATCC 6303, American Type Culture Collection, Manassas, VA) was used in this study. Bacteria were grown at 37°C on blood agar plates overnight, collected in sterile saline solution, and their concentration evaluated by spectrophotometry (OD600). A more precise CFU enumeration was conducted a posteriori by plating serial dilutions of these solutions on blood agar and incubating the plates for 24h at 37°C. Mice were infected with 105 CFU diluted in 25 μl saline solution by intranasal instillation after light anesthesia with halothane.
Bacterial load quantification after IFN-γ-priming and particle exposure
In experiments in which mice were primed with IFN-γ and subsequently exposed to particles and S. pneumoniae, bacterial load in the lungs was quantified twenty four hours after infection. After euthanasia, the lungs were removed, homogenized in sterile water, and serial dilutions were plated on blood agar. CFU were enumerated after overnight incubation at 37°C.
Histopathology after IFN-γ-priming and particle exposure
In experiments in which mice were primed with IFN-γ and subsequently exposed to particles and S. pneumoniae, histopathology was performed on a subset of the animals (4 per group) twenty-four hours after infection. After euthanasia, mouse lungs removed and fixed in formalin for histopathological assessment after preparation of hematoxylin- and eosin-stained slides.
In vivo bacterial uptake
S. pneumoniae was stained with SYTO 9 according to the manufacturer’s protocol (Molecular Probes Inc., Eugene, OR). 105 CFU of stained S. pneumoniae were diluted in 25 μl saline solution and instilled intranasally into each mouse after light anesthesia with halothane. 3h later, BAL was performed as described above. After centrifugation of the collected lavage, cells were resuspended in BSS+ and incubated for 30 minutes on ice with a 1:100 dilution of anti-Gr-1 PE-conjugated antibody (Pharmingen, San Diego, CA), which binds to PMNs but not to macrophages (Fleming et al., 1993; Conlan and North, 1994). After incubation, the cells were washed twice, resuspended in 2% formaldehyde and fixed overnight at 4°C. Green (SYTO 9-labeled bacteria) and red (anti-Gr-1 antibody) fluorescence of the cells was analyzed on a flow cytometer. The bacterial content (green fluorescence) of AMs and PMNs was differentiated by gating red- negative (AMs) and positive (PMNs) cells.
In vitro bacterial uptake
S. pneumoniae was heat-inactivated and stained with fluorescein isothiocyanate (FITC) according to the manufacturer’s protocol (Molecular Probes Inc., Eugene, OR). Cells collected by BAL were incubated with FITC-labeled bacteria at a bacteria:cell ratio of 50:1 in BSS+ at 37°C for 90 minutes and with constant rotation. Subsequently, the cells were washed twice with BSS+ and then incubated for 30 minutes on ice with a 1:100 dilution of anti-Gr-1 PE-conjugated antibody (Pharmingen, San Diego, CA), which binds to PMNs but not to alveolar macrophages (AMs), as described above. After this incubation, the cells were washed twice with BSS+ and the green (FITC-labeled bacteria) and red (anti-Gr-1 antibody) fluorescence of the cells was analyzed on a flow cytometer. The bacterial content (green fluorescence) of AMs and PMNs was differentiated by gating red- negative (AMs) and positive (PMNs) cells.
Measurement of intracellular reactive oxygen species
2.5 × 105 cells from BALs were incubated with a 40 μM solution of dichlorofluorescein diacetate (DCFH-DA) in BSS+ at 37°C for 20 minutes. DCFH-DA diffuses into cells where it is hydrolyzed by esterases to dichlorofluorescein (DCFH), which is trapped within the cell. This nonfluorescent molecule is then oxidized to fluorescent dichlorofluorescein (DCF) in the presence of intracellular oxidants. After incubation with DCFH-DA, the cells were transferred to ice and incubated for 10 minutes with a 1:100 dilution of anti-Gr-1 PE-conjugated antibody (Pharmingen, San Diego, CA) before analysis on a flow cytometer. Intracellular reactive oxygen species content (green fluorescence) of AMs and PMNs was differentiated by gating red- negative (AMs) and positive (PMNs) cells.
RNA analysis
Total RNA was isolated from mouse lungs using the Qiagen RNeasy Mini kit according to the manufacturer’s protocol (Qiagen, Valencia, CA). The Mouse Inflammatory Cytokines & Receptors Gene Array (GEArray Q Series, SuperArray, Frederick, MD), a nylon membrane-based cDNA array, was used to compare the relative expression profile of genes of interest. The array contained 96 cytokine and receptor genes associated with the inflammatory response, and 4 housekeeping genes and negative controls. The array was used according to the manufacturer’s protocol. Briefly, cDNA was synthesized by reverse transcription of 2 μg of the RNA sample using random primers. The cDNA was amplified by PCR with gene-specific primers and labeled with [α-32P]-dCTP during amplification. The probes produced were hybridized overnight with the array. Radioactivity on the array membrane was detected and quantified with a phosphoimager (Molecular Devices, Sunnyvale, CA). The results were analyzed with the software provided by SuperArray, normalizing each membrane to the background noise and the level of expression of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Each experiment was repeated 3 times and the expression levels averaged.
Flow cytometry
Flow cytometry was performed using a Coulter ELITE flow cytometer (Coulter Coporation, Miami, FL) equipped with an air-cooled argon laser (Cyonics/Uniphase) (488 nm excitation line, 15 mW output) and a dedicated data acquisition and analysis system (Coulter, Hialeah, FL), with analysis of > 5,000 cells per sample.
Statistical analysis
The experiments were performed with at least 3 mice for each treatment group, and repeated at least 3 times (n ≥ 9). Data are reported as mean ± the standard error of the mean (SEM). Differences among groups were evaluated using factorial Analysis of Variance (ANOVA) with adjustment for multiple groups (Fisher’s PLSD) using and Statview 4.5 software (Abacus Concepts, Berkeley, CA). A value of p < 0.05 was considered to be significant.
RESULTS
Combined IFN-γ priming and CAPs exposure generates inflammation
We first investigated the effect in vivo of priming by aerosol exposures to IFN-γ, followed by CAPs exposure. Titanium dioxide (TiO2), which is considered an inert particle in the lung (Driscoll et al., 1990), was used as a control to determine if any effects of CAPs were specific.
Mice exposed to aerosolized IFN-γ (I, 20,000 U/ml) or PBS (P) were instilled intranasally 3 h later with a solution of either PBS (P), TiO2 (T, 50 μg in 50 μl PBS) or CAPs (C, 50 μg in 50 μl PBS). The 6 groups formed were designated PP, PT, PC, IP, IT, and IC, where the first letter indicates the priming agent and the second letter indicates the instilled agent. Twenty four hours later, bronchoalveolar lavage (BAL) was conducted and lavage fluids were collected and analyzed for cell content. In unprimed mice, TiO2 exposure (PT) had only a moderate effect on PMN recruitment, which was not significantly different from the untreated control (PP, Figure 1). In contrast, CAPs exposure, even in unprimed mice (PC), resulted in a significant increase in the number of PMNs in the lung -- ∼100% more than seen in unprimed mice exposed to the inert particle, TiO2.
Figure 1.
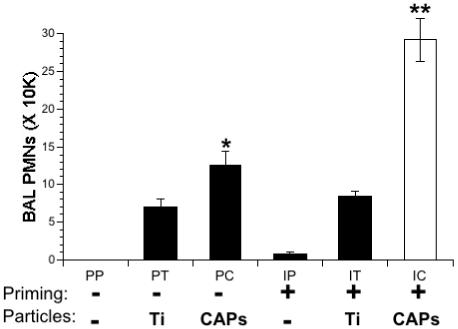
Lung inflammation in IFN-γ-primed and particle-exposed mice. Mice were primed with IFN-γ (I) or PBS (P) aerosols for 15 min. 3 hours later the same mice were exposed to intranasal solutions of PBS (P), TiO2 (T) or CAPs (C). Inflammation in the lungs was assessed by counting the number of PMNs in the BAL fluid 24 hours after the final exposure. Data represent the mean ± SEM of at least 3 independent experiments. *= p<0.05 compared to PP and PT groups. **= p<0.001 compared to all other groups.
Priming with IFN-γ produced a modest increase in the number of PMNs after both PBS and TiO2 instillations (IP and IT, respectively, Figure 1). This increase was not statistically different from the untreated controls (PP). Further, the effect of TiO2 on primed or unprimed mice was similar (IT and PT, respectively, Figure 1). In contrast, exposure of primed mice to CAPs produced a strong inflammatory response, as evidenced by the 2.5-fold increase in the number of PMNs as compared to unprimed mice (IC vs. PC, Figure 1).
To characterize the cytokines induced by the IFN-γ and CAPs treatments, we analyzed lung mRNAs for a panel of inflammatory cytokines and receptors (see Methods). The mice were divided into 4 groups receiving the following treatments: PBS or IFN-γ aerosol, followed 3 h later by intranasal PBS or CAPs. Lung RNA was analyzed at 3 time points after intranasal exposure to PBS or CAPs: 3, 6 and 24 hours. The level of expression of 96 genes relevant to inflammation was then measured.
Among the 96 genes tested, every gene that was activated by combined priming and CAPs exposure was also activated by either stimulus alone; however, several genes displayed a stronger activation when both stimuli were present as compared to either priming or CAPs alone, although this was not uniformly found. Table 1 summarizes the genes expressed in IFN-γ-primed and CAPs-treated lungs (IC) which showed an increase of at least 1.5-fold over the control group (PP) at the three time points evaluated. Since many of these highly activated genes encode chemokines that recruit PMNs or the receptors for these chemokines the data are consistent with the enhanced neutrophil influx observed in lavage samples.
Table 1.
Genes activated 3, 6 or 24 hours after treatment with PBS (P) or IFN-γ (I) aerosol for 10 min., then intranasal PBS (P) or CAPs (C) 3 hours later.
| Gene | PP* | PC | IP | IC | Fold increase (IC/PP) | Fold increase (IC/PC) | Fold increase (IC/IP) |
|---|---|---|---|---|---|---|---|
| 3 hours | |||||||
| MIP-1a | 0.047 | 0.104 | 0.091 | 0.113 | 2.38 | 1.09 | 1.24 |
| MIP-3b | 0.038 | 0.062 | 0.041 | 0.083 | 2.19 | 1.34 | 2.02 |
| MIP-1b | 0.048 | 0.083 | 0.042 | 0.101 | 2.09 | 1.22 | 2.40 |
| TECK | 0.036 | 0.053 | 0.057 | 0.066 | 1.85 | 1.25 | 1.16 |
| MDC | 0.062 | 0.083 | 0.064 | 0.103 | 1.66 | 1.24 | 1.61 |
| IL17B | 0.094 | 0.126 | 0.126 | 0.146 | 1.56 | 1.16 | 1.16 |
| 6 hours | |||||||
| TARC | 0.031 | 0.115 | 0.041 | 0.143 | 4.66 | 1.24 | 3.49 |
| MCP-5 | 0.017 | 0.063 | 0.035 | 0.073 | 4.30 | 1.16 | 2.09 |
| Lungkine | 0.474 | 0.872 | 0.743 | 1.108 | 2.34 | 1.27 | 1.49 |
| MIP-1g | 0.052 | 0.104 | 0.073 | 0.111 | 2.11 | 1.07 | 1.52 |
| IL-9 | 0.028 | 0.035 | 0.035 | 0.055 | 1.98 | 1.57 | 1.57 |
| IL-1b | 0.047 | 0.063 | 0.041 | 0.079 | 1.68 | 1.25 | 1.93 |
| 24 hours | |||||||
| IL-8Rb | 0.015 | 0.024 | 0.025 | 0.047 | 3.07 | 1.96 | 1.88 |
| MIP-1g | 0.047 | 0.084 | 0.065 | 0.124 | 2.62 | 1.48 | 1.91 |
| C-10 | 0.477 | 0.895 | 0.680 | 0.981 | 2.06 | 1.10 | 1.44 |
| IL-1b | 0.042 | 0.035 | 0.052 | 0.085 | 2.03 | 2.43 | 1.63 |
| CCR-1 | 0.020 | 0.020 | 0.022 | 0.040 | 2.03 | 2.00 | 1.82 |
| MIP-3a | 0.084 | 0.123 | 0.127 | 0.142 | 1.69 | 1.15 | 1.12 |
| CCR-2 | 0.056 | 0.045 | 0.061 | 0.084 | 1.5 | 1.87 | 1.38 |
Densitometric value Glossary: MIP1a = macrophage inflammatory protein 1 alpha; MIP3b = macrophage inflammatory protein 3 beta; MIP1b = macrophage inflammatory protein 1 beta; TECK = thymus-expressed chemokine; MDC = macrophage-derived chemokine; IL17B = interleukin 17B; TARC = thymus and activation regulated chemokine; MCP-5 = monocyte chemotactic protein 5; MIP-1g = macrophage inflammatory protein 1g; IL-9 = interleukin 9; IL-1b = interleukin 1 beta; IL-8Rb = interleukin 8 receptor b; C-10 = chemokine 10; CCR-1 = chemokine receptor 1; MIP-3a = macrophage inflammatory protein 3 alpha; CCR-2 = chemokine receptor 2.
Combined priming and CAPs exposure increases post-infection inflammation
Because priming and exposure to air pollution particles increased inflammation in the lung, we chose to test the consequence of these treatments on pneumococcal infection. As done previously, mice were exposed to PBS or IFN-γ aerosol, and, 3h later, to PBS, TiO2 or CAPs. 24h later, all the mice were infected with S. pneumoniae (B). 24h after bacterial infection, BAL was performed or lungs were harvested to assess bacterial survival.
The lungs of the mice infected by S. pneumoniae displayed acute inflammation, as shown by the presence of PMNs in the BAL of all 6 groups (Figure 2). When unprimed mice were treated with the inert particle, TiO2, prior to infection, there was no difference in the number of PMNs in this group than seen in mice infected with S. pneumoniae alone (Figure 2, PTB vs. PPB). Treatment with CAPs enhanced inflammation, causing a 2-fold increase in the number of PMNs as compared to the infected control (Figure 2, PCB vs. PPB). IFN-γ priming before infection did not affect inflammation in mice not exposed to particles, or even in mice instilled with TiO2 (Figure 2, compare unprimed vs. primed of both groups; i.e., PPB vs. IPB and PTB vs. ITB). This contrasts to what occurred when priming and CAPs treatment were combined; inflammation was exacerbated. There was a 3.5-fold increase in the number of PMNs recruited to the lung in primed and infected animals exposed to CAPs (ICB) compared to the infected control animals (PPB), and a 1.6-fold increase compared to infected animals exposed to CAPs alone (PCB).
Figure 2.
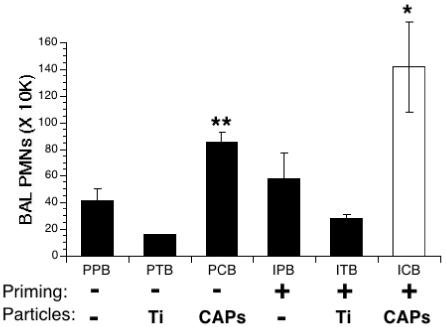
Lung inflammation in IFN-γ-primed, particle-exposed, and S. pneumoniae-infected mice. Mice were primed with IFN-γ (I) or PBS (P) aerosols for 15 min. 3 hours later the same mice were exposed to intranasal solutions of PBS (P), TiO2 (T) or CAPs (C). 24 hours later these mice were infected intranasally with S. pneumoniae (B). Inflammation in the lungs was assessed by counting the number of PMNs in the BAL fluid 24 hours after infection. Data represent the mean ± SEM of at least 3 independent experiments, * = p<0.05 compared to all other groups; **= p<0.03 compared to PPB and PTB groups.
Combined priming and CAPs exposure impairs bacterial uptake in vivo and in vitro
The bacterial load in the lungs of infected mice was assessed 24h after infection. Serial dilutions of lung homogenates were plated on blood agar plates, and the number of S. pneumoniae colonies (CFU) was determined after overnight incubation. The results are expressed as percentage of the initial inoculum (Figure 3). In the control groups, PPB, PTB, IPB and ITB, only 10 to 50 % of the bacteria were still alive in the lungs 24h after infection, showing efficient clearance of the bacteria by the lung immune system. In contrast, bacterial numbers did not decrease, but remained static over 24h, in the group treated with CAPs alone (PCB). This suggests an impairment of the bacterial clearance process in animals exposed to air pollution particles. This effect was dramatically enhanced when priming and CAPs treatments were combined: a 2.5-fold increase was observed in the bacterial population over the 24h period (Figure 3, ICB).
Figure 3.
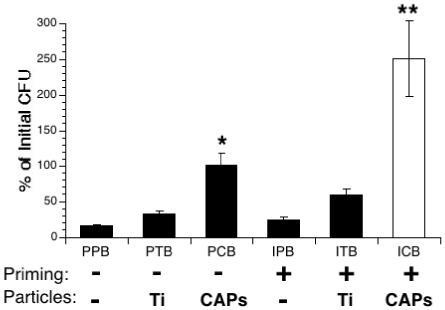
Bacterial clearance in IFN-γ-primed, particle-exposed, and S. pneumoniae-infected mice. Mice were primed with IFN-γ (I) or PBS (P) aerosols for 15 min., followed 3 hours later by intranasal exposure to PBS (P), TiO2 (T) or CAPs (C). 24 hours later mice were infected intranasally with S. pneumoniae (B). The lungs were harvested and bacterial load (CFU) was assessed 24 hours after infection, as described in Methods. Data represent the mean ± SEM of at least 3 independent experiments. * = p<0.05 compared to PPB and PTB groups. ** = p<0.001 compared to all other groups.
Histopathology of the lungs confirmed the presence of moderate pneumonia in animals infected with S. pneumoniae after CAPs exposure alone (PCB) and areas of severe pneumonia after combined priming and CAPs exposure (ICB, Figure 4, panels C and D, respectively). In contrast, little evidence of pneumonia was seen in control animals, that is, PBS only before infection (PPB) and IFN-γ only before infection (IPB, Figure 4, Panels A and B, respectively).
Figure 4.

Histopathology after S. pneumoniae infection with and without antecedent priming and particle exposure. Panels A and B: Little or no acute inflammation (PMN influx) was seen 24 hours after S. pneumoniae infection in control animals (PBS only before infection, PPB, Panel A, and IFN-γ only before infection, IPB, Panel B). Panels C and D: Moderate to severe inflammation (PMN influx) was seen 24 hours after S. pneumoniae infection in particle treated animals (no priming and CAPs exposure before infection, PCB, Panel C, and IFN-γ-priming and CAPs-exposure before infection, ICB, Panel D).
It is noteworthy that despite increased recruitment of PMNs in response to combined IFN-γ and CAPs treatments (Figure 2), the lungs were unable to clear the S. pneumoniae infection (Figures 3 and 4). This suggests that the phagocytic cells, the PMNs, and perhaps the AMs, had lost their ability to efficiently phagocytize and kill the bacteria. To test that hypothesis, we evaluated the bacterial uptake of AMs and PMNs, both in vivo and in vitro.
For the in vivo experiments, S. pneumoniae were labeled prior to infection with SYTO 9, a green fluorescent nucleic acid stain that can be used to label live bacteria. Mice were infected with SYTO 9-labeled bacteria, and BAL was performed 3h after infection. The collected cells were then stained with a red fluorescent anti-Gr-1 antibody specific for PMNs. The bacterial content of AMs and PMNs was determined by flow cytometry analysis.
A decrease in green fluorescence, and therefore in bacterial content, was observed in AMs in all the treated groups, with a more marked decrease in the IFN-γ-primed and CAPS exposed (IC) group (Figure 5, top panel). However, the decreases observed did not reach statistical significance. A similar decrease in bacterial content was observed for PMNs in all treatment groups (Figure 5, bottom panel). The decreased bacterial content was statistically significant in both groups exposed to CAPs (PC and IC), indicating that bacterial uptake by PMNs is impaired in the presence of air pollution particles,
Figure 5.

In vivo bacterial uptake of phagocytes recovered from IFN-γ-primed and particle-exposed mice. Mice were exposed to IFN-γ (I) or PBS (P) aerosol for 15 min., then to intranasal PBS (P) or CAPs (C) 3 hours later. 24 hours later mice were infected intranasally with S. pneumoniae labeled with SYTO 9 (a green fluorophore). 3 hours after infection, BAL was performed and cells analyzed by flow cytometry.. Top panel: AMs. Bottom panel: PMNs. Data represent the mean ± SEM of at least 2 independent experiments. * = p<0.05 compared to PP group.
To confirm this result, in vitro experiments were performed using another staining method. The surface of heat-inactivated S. pneumoniae was labeled with the fluorophore FITC. Mice were treated as in the previous experiment, and BAL was performed 24h after PBS or CAPs instillation. The cells collected from lavage were incubated in vitro with the FITC-labeled bacteria for 90 minutes. The cells were then stained with the red fluorescent anti-Gr-1 antibody, specific to PMNs, and cell fluorescence analyzed on a flow cytometer.
AMs lavaged from mice treated with CAPs showed a trend towards reduced bacterial uptake (PC, Figure 6, top panel) as compared to the control group, PP, or to the IP group. Similar to the in vivo results, the decrease was not statistically significant. In contrast, the fluorescence of PMNs was dramatically reduced in the presence of CAPs, both in the unprimed (PC) and the primed (IC) groups (Figure 6, bottom panel).
Figure 6.

Ex vivo bacterial uptake in IFN-γ-primed and particle-exposed mice. Mice were exposed to IFN-γ (I) or PBS (P) aerosol for 15 min., then to intranasal PBS (P) or CAPs (C) 3 hours later. BAL was performed 24 hours later and the collected cells were cultured in the presence of heat-inactivated, FITC-labeled S. pneumoniae for 90 minutes at 37°C. After incubation the cells were transferred to ice and labeled with Gr-1 antibody, which binds specifically to PMNs. Flow cytometry followed, measuring green fluorescence (FITC-labeled S. pneumoniae) and gating the cells by their red fluorescence (Gr-1 antibody). Top panel: AMs. Bottom panel: PMNs. Data represent the mean ± SEM of at least 3 independent experiments; * = p<0.05 compared to PP and IP group.
Combined priming and CAPs exposure increases oxidative stress in AMs and PMNs
The previous experiment showed that PMNs, and possibly AMs, were impaired in their capacity to phagocytize bacteria. As part of the inflammation caused by IFN-γ-priming and CAPs exposure, high levels of potentially deleterious reactive oxygen species (ROS) are likely produced in the alveolar milieu by the activated AMs and recruited PMNs. To determine if this was occurring in the environment created by priming and particle exposure, mice were treated as in the previous experiment, and BAL was performed 24 h after the PBS or CAPs instillation. Lavaged cells were incubated for 30 minutes with anti-Gr-1 antibody and DCFH-DA, a non-fluorescent compound that diffuses into cells and is metabolized into a fluorescent compound, DCF, in the presence of intracellular ROS. After this incubation, the cells were analyzed on a flow cytometer.
The level of basal oxidant production (as measured by generation of fluorescent reporter DCF) within AMs was significantly increased when priming and CAPs treatment were combined, approximately 50% greater than controls (IC vs. PP, Figure 7, top panel). A greater increase in intracellular ROS in response to combined priming and CAPs was also observed in PMNs, fully 100% greater than controls (IC vs. PP, Figure 7, bottom panel).
Figure 7.

Intracellular oxidative stress in cells collected via BAL from IFN-γ-primed and particle-exposed mice. Mice were exposed to IFN-γ (I) or PBS (P) aerosol for 15 min., then to intranasal PBS (P) or CAPs (C) 3 hours later. BAL was performed 24 hours later. The collected cells were incubated for 30 min. at 37°C with DCFH and anti-Gr-1 antibody, which binds specifically to PMNs. Intracellular oxidative stress was assessed via flow cytometry, with green fluorescence indicative of intracellular oxidation of DCFH. The cells were gated by their red fluorescence (anti-Gr-1 antibody). Top panel: AMs. Bottom panel: PMNs. Data represent the mean ± SEM of at least 3 independent experiments. * = p<0.05 compared to PP group.
DISCUSSION
We sought to test the interaction of priming and air pollution particles on lung sensitivity to pneumococcal infection in a mouse model. The results show that IFN-γ-priming enhances the inflammatory effect of CAPs, and that this leads to impaired lung defense (reduced phagocytosis and clearance) against S. pneumoniae. The data also suggest that excessive ROS production and overexpression of chemokines and their receptors play important roles in the impaired lung defense. These findings provide a possible mechanism to explain the association between episodes of increased particle pollution and increased hospital admissions for pneumonia.
The choice of IFN-γ aerosols to prime the lungs of mice in our experiments merits discussion. Although particle exposures increase lung infection rates, only a subpopulation develops pneumonia, suggesting effects on susceptible subpopulations. We reasoned that one possible common cause of increased susceptibility to pneumonia in both pediatric and elderly populations is antecedent viral infection. We chose to model the altered post-inflammatory milieu of such lungs using IFN-γ aerosols since this is a major cytokine generated in viral infections. Also, this approach provides more uniform and less complex priming than viral infection per se, allowing simpler analysis of the effect of particles. CAPs exposure of mice with a different type of priming, allergic inflammation due to ovalbumin sensitization and aerosol challenge, has also proven useful for analysis of particle effects asthma (Goldsmith and Kobzik, 1999; Goldsmith et al., 2002; Gavett et al., 2003). While epidemiological data indicate that pneumococcal pneumonia risk is augmented after viral infection, the mechanisms for this increased risk are not entirely clear. Animal models used to elucidate these mechanisms have provided contradictory results. For example, mice infected by viruses, such as influenza or murine cytomegalovirus, show an increased sensitivity to either bacteria or bacterial products (Jones et al., 1983; Leung and Hashimoto, 1986). These results were supported by Nguyen et al, who reported increased mortality in mice infected by the lymphocytic choriomeningitis virus (LCMV) prior to lipopolysaccharide (LPS) injection (Nguyen and Biron, 1999). Furthermore, these authors showed that γ-interferon (IFN-γ) is an essential mediator of the effects of the antecedent viral infection: both anti-IFN-γ antibody-treated-mice and IFN-γ gene knockout mice were protected from the effect of prior LCMV infection (Nguyen and Biron, 1999). On the other hand, IFN-γ gene knockout mice are more sensitive than wild type mice to Klebsiella pneumoniae and pneumococcal lung infection (Rubins and Pomeroy, 1997; Moore et al., 2002). An interpretation of these apparently conflicting results is that IFN-γ can induce either an enhanced or a dysregulated immune response, depending on the immunological context,
In our experiments, the adverse effects were specific to exposure to air pollution particles and were not seen with exposure to an inert particle, TiO2. For example, mice exposed to CAPs, but not TiO2, had a large influx of PMNs into the alveoli (Figure 1), a well known effect of CAPs-induced acute lung inflammation that occurs in both animal models and in humans (Costa and Dreher, 1997; Clarke et al., 1999; Ghio and Devlin, 2001). When CAPs exposure was preceeded by IFN-γ-priming, a marked enhancement in inflammatory cell recruitment was seen (2.5-fold increase, Figure 1). This increase is especially remarkable, as priming alone created virtually no inflammation; the effects of priming and air pollution particle exposure were synergistic, not additive. Comparison of gene expression levels after particles and priming identified increases in several mediators linked to acute inflammation, including leukocyte chemokines and their receptors. One limitation worth noting is our use of a relatively high dose of particles given as an intranasal bolus. While useful for proof-of-principle, additional studies using inhalation exposures and dose-response analysis would allow better extrapolation to real-world exposures.
When control mice were infected with S. pneumoniae, the expected response was observed, with recruitment of PMNs into the lungs (Figure 2). Priming alone did not significantly change this response. In contrast, CAPs exposure increased PMN recruitment, and the combination of priming and CAPs exposure further increased PMN recruitment, up to a 3-fold increase over the control. Hence, despite the presence of already large numbers of PMNs in the lungs of mice exposed to CAPs at the time of S. pneumoniae infection, further augmentation of the inflammatory response occurred. It might be expected that the presence of large numbers of PMNs in the lungs at the time of infection would increase resistance to S. pneumoniae. Indeed, priming of the immune system with endotoxin, viral infection or IFN has been reported to have a protective effect against bacterial infection in several experimental models. For example, mortality of mice infected with Streptococcus pneumoniae decreases if they are treated with IFN-γ (Weigent et al., 1986). On the other hand, IFN-γ gene knockout mice are more sensitive to pneumococcal lung infection and to Klebsiella pneumoniae infection than wild type mice (Rubins and Pomeroy, 1997; Moore et al., 2002). In general, it is thought that IFN-γ plays a protective role in host response to bacterial infection (Boehm et al., 1997).
In our experiments, treatment with IFN-γ alone produced no effect on the response to pneumococcal infection, either positive or negative. This absence of priming effect maybe due to the relatively low dose of bacteria used for our study, as compared to the higher doses used by others. Moreover, priming before infection does not always have a protective effect. For example, mice infected with influenza virus prior to pneumococcal infection led to increased mortality (McCullers and Rehg, 2002). In the present study, while priming itself had no effect, air pollution particle exposure (CAPs), alone and in concert with priming, increased sensitivity to infection. When S. pneumoniae infected mice were pre-treated with CAPs alone, bacterial clearance was impaired; if the lungs were primed with IFN-γ before CAPs exposure, the bacterial load in the lungs increased dramatically, showing a synergistic effect (Figures 3 and 4). Despite the presence of numerous AMs and PMNs in the lung at that time of infection, these cells were unable to cope with the infection and the bacteria proliferated. These findings are similar to data showing that DEP and bacterial endotoxin instilled together enhance lung injury in mice, and increase the expression of pro-inflammatory molecules (Takano et al., 2002). Taken together, our data suggest that AMs and PMNs are present in the lung at the time of infection, but as a consequence of IFN-γ and CAPs treatments, are not functioning normally.
Indeed, we were able to show, both in vivo and ex vivo, that PMNs had lost most of their phagocytic capacities following IFN-γ and CAPs treatments (Figures 5 and 6); a similar trend was observed for AMs. One potential confounder is the possibility that flow cytometric measurements relying on fluorescent bacteria may underestimate uptake, for example, if quenching by acidification occurs. Alternate assays using stable fluorochromes or non-digestible particles may allow improved analysis. Hence, further evaluation is needed to determine whether and to what degree AMs lose phagocytic function in this model. The postulate that AM function is depressed under these conditions This is consistent with reports that AMs’ phagocytic function is markedly attenuated by particle exposure in vitro, and is further impaired by incubation with IFN-γ (Lundborg et al., 1999). We have also reported that in vitro exposure to CAPs causes diminished phagocytosis and killing of pneumococci in both unprimed and IFN-γ primed AMs (Zhou and Kobzik, 2007). These studies also showed that soluble components of CAPs, most likely soluble iron, mediate effects on AM phagocytosis. This suggests that the increased sensitivity to pneumococcal infection we observed in mice exposed to the combination of IFN-γ and CAPs is due to PMNs’, and perhaps to a lesser extent AMs’, loss of function. Whether oxidant stress mediates the additional effect of IFN-γ priming remains to be directly investigated in future experiments with antioxidants. Exposure to particles causes significant intracellular oxidative stress in AMs and PMNs (Goldsmith et al., 1998; Becker et al., 2002). Based on increased oxidant production by AMs and PMNs in the mice treated with IFN-γ and CAPs combined (Figure 7), and prior work showing oxidant-dependent impairment by CAPs of AM bacterial phagocytosis and killing, we postulate that the loss of phagocytic function observed in these experiments is caused, at least in part, by oxidative damage to the cells.
The model reported here will allow further analysis of priming and CAPs exposure on lung sensitivity to infection.
ACKNOWLEDGMENTS
Supported by NIH ES 011903 ES 00002. This supporting agency had no involvement in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
Footnotes
CONFLICT OF INTEREST STATEMENT
All authors declare they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Becker S, Soukup JM, Gallagher JE. Differential particulate air pollution induced oxidant stress in human granulocytes, monocytes and alveolar macrophages. Toxicol In Vitro. 2002;16:209–218. doi: 10.1016/s0887-2333(02)00015-2. [DOI] [PubMed] [Google Scholar]
- Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- Cate TR. Impact of influenza and other community-acquired viruses. Seminars in Respiratory Infections. 1998;13:17–23. [PubMed] [Google Scholar]
- Clarke RW, Catalano PJ, Koutrakis P, Murthy GG, Sioutas C, Paulauskis J, Coull B, Ferguson S, Godleski JJ. Urban air particulate inhalation alters pulmonary function and induces pulmonary inflammation in a rodent model of chronic bronchitis. Inhalation Toxicology. 1999;11:637–656. doi: 10.1080/089583799196781. [DOI] [PubMed] [Google Scholar]
- Conlan JW, North RJ. Neutrophils are essential for early anti-Listeria defense in the liver, but not in the spleen or peritoneal cavity, as revealed by a granulocyte-depleting monoclonal antibody. J Exp Med. 1994;179:259–268. doi: 10.1084/jem.179.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa DL, Dreher KL. Bioavailable transition metals in particulate matter mediate cardiopulmonary injury in healthy and compromised animal models. Environmental Health Perspectives. 1997;105:1053–1060. doi: 10.1289/ehp.97105s51053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockery D, Pope C, III, Xu X, Spengler J, Ware J, Fay M, Ferris B, Speizer F. An association between air pollution and mortality in six U.S. cities. N. Engl. J. Med. 1993;329:1753–1759. doi: 10.1056/NEJM199312093292401. [DOI] [PubMed] [Google Scholar]
- Driscoll KE, Lindenschmidt RC, Maurer JK, Higgins JM, Ridder G. Pulmonary response to silica or titanium dioxide: inflammatory cells, alveolar macrophage-derived cytokines, and histopathology. Am J Respir Cell Mol Biol. 1990;2:381–390. doi: 10.1165/ajrcmb/2.4.381. [DOI] [PubMed] [Google Scholar]
- Farhat SC, Paulo RL, Shimoda TM, Conceicao GM, Lin CA, Braga AL, Warth MP, Saldiva PH. Effect of air pollution on pediatric respiratory emergency room visits and hospital admissions. Braz J Med Biol Res. 2005;38:227–235. doi: 10.1590/s0100-879x2005000200011. [DOI] [PubMed] [Google Scholar]
- Fleming TJ, Fleming ML, Malek TR. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol. 1993;151:2399–2408. [PubMed] [Google Scholar]
- Gavett SH, Haykal-Coates N, Copeland LB, Heinrich J, Gilmour MI. Metal composition of ambient PM2.5 influences severity of allergic airways disease in mice. Environ Health Perspect. 2003;111:1471–1477. doi: 10.1289/ehp.6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghio AJ, Devlin RB. Inflammatory lung injury after bronchial instillation of air pollution particles. American Journal of Respiratory & Critical Care Medicine. 2001;164:704–708. doi: 10.1164/ajrccm.164.4.2011089. [DOI] [PubMed] [Google Scholar]
- Goldsmith C, Ning Y, Qin G, Lawrence J, Murthy K, Catalano P, Kobzik L. Combined air pollution particle and ozone exposure increases airway responsiveness in mice. Inhal. Toxicol. 2002;14:325–347. doi: 10.1080/08958370252870989. [DOI] [PubMed] [Google Scholar]
- Goldsmith CA, Imrich A, Danaee H, Ning YY, Kobzik L. Analysis of air pollution particulate-mediated oxidant stress in alveolar macrophages. Journal of Toxicology & Environmental Health. Part A. 1998;54:529–545. doi: 10.1080/009841098158683. [DOI] [PubMed] [Google Scholar]
- Goldsmith CA, Kobzik L. Particulate air pollution and asthma: a review of epidemiological and biological studies. Reviews on Environmental Health. 1999;14:121–134. doi: 10.1515/reveh.1999.14.3.121. [DOI] [PubMed] [Google Scholar]
- Hahon N, Booth JA, Green F, Lewis TR. Influenza virus infection in mice after exposure to coal dust and diesel engine emissions. Environ Res. 1985;37:44–60. doi: 10.1016/0013-9351(85)90048-9. [DOI] [PubMed] [Google Scholar]
- Hatch GE, Boykin E, Graham JA, Lewtas J, Pott F, Loud K, Mumford JL. Inhalable particles and pulmonary host defense: in vivo and in vitro effects of ambient air and combustion particles. Environmental Research. 1985;36:67–80. doi: 10.1016/0013-9351(85)90008-8. [DOI] [PubMed] [Google Scholar]
- Imrich A, Ning Y, Kobzik L. Insoluble components of concentrated air particles mediate alveolar macrophage responses in vitro. Toxicology & Applied Pharmacology. 2000;167:140–150. doi: 10.1006/taap.2000.9002. [DOI] [PubMed] [Google Scholar]
- Imrich A, Ning YY, Koziel H, Coull B, Kobzik L. Lipopolysaccharide priming amplifies lung macrophage tumor necrosis factor production in response to air particles. Toxicology & Applied Pharmacology. 1999;159:117–124. doi: 10.1006/taap.1999.8731. [DOI] [PubMed] [Google Scholar]
- Jones WT, Menna JH, Wennerstrom DE. Lethal synergism induced in mice by influenza type A virus and type Ia group B streptococci. Infect Immun. 1983;41:618–623. doi: 10.1128/iai.41.2.618-623.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung WC, Hashimoto K. Modification of susceptibility to Klebsiella pneumoniae during murine cytomegalovirus infection. Microbiol Immunol. 1986;30:761–776. doi: 10.1111/j.1348-0421.1986.tb03003.x. [DOI] [PubMed] [Google Scholar]
- Lin CA, Martins MA, Farhat SC, Pope CA, 3rd, Conceicao GM, Anastacio VM, Hatanaka M, Andrade WC, Hamaue WR, Bohm GM, Saldiva PH. Air pollution and respiratory illness of children in Sao Paulo, Brazil. Paediatr Perinat Epidemiol. 1999;13:475–488. doi: 10.1046/j.1365-3016.1999.00210.x. [DOI] [PubMed] [Google Scholar]
- Lundborg M, Johansson A, Lastbom L, Camner P. Ingested aggregates of ultrafine carbon particles and interferon-gamma impair rat alveolar macrophage function. Environmental Research. 1999;81:309–315. doi: 10.1006/enrs.1999.3992. [DOI] [PubMed] [Google Scholar]
- McCullers JA, Rehg JE. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis. 2002;186:341–350. doi: 10.1086/341462. [DOI] [PubMed] [Google Scholar]
- Mo XY, Sarawar SR, Doherty PC. Induction of cytokines in mice with parainfluenza pneumonia. Journal of Virology. 1995;69:1288–1291. doi: 10.1128/jvi.69.2.1288-1291.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore TA, Perry ML, Getsoian AG, Newstead MW, Standiford TJ. Divergent role of gamma interferon in a murine model of pulmonary versus systemic Klebsiella pneumoniae infection. Infect Immun. 2002;70:6310–6318. doi: 10.1128/IAI.70.11.6310-6318.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KB, Biron CA. Synergism for cytokine-mediated disease during concurrent endotoxin and viral challenges: roles for NK and T cell IFN-gamma production. J Immunol. 1999;162:5238–5246. [PubMed] [Google Scholar]
- Rubins JB, Pomeroy C. Role of gamma interferon in the pathogenesis of bacteremic pneumococcal pneumonia. Infect Immun. 1997;65:2975–2977. doi: 10.1128/iai.65.7.2975-2977.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz J. Air pollution and hospital admissions for the elderly in Birmingham, Alabama. American Journal of Epidemiology. 1994a;139:589–598. doi: 10.1093/oxfordjournals.aje.a117048. [DOI] [PubMed] [Google Scholar]
- Schwartz J. Air pollution and hospital admissions for the elderly in Detroit, Michigan. American Journal of Respiratory & Critical Care Medicine. 1994b;150:648–655. doi: 10.1164/ajrccm.150.3.8087333. [DOI] [PubMed] [Google Scholar]
- Sioutas C, Koutrakis P, Burton R. A technique to expose animals to concentrated fine ambient aerosols. Environ. Health Perspect. 1995;103:172–177. doi: 10.1289/ehp.95103172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano H, Yanagisawa R, Ichinose T, Sadakane K, Yoshino S, Yoshikawa T, Morita M. Diesel exhaust particles enhance lung injury related to bacterial endotoxin through expression of proinflammatory cytokines, chemokines, and intercellular adhesion molecule-1. Am J Respir Crit Care Med. 2002;165:1329–1335. doi: 10.1164/rccm.2108122. [DOI] [PubMed] [Google Scholar]
- van Schaik SM, Obot N, Enhorning G, Hintz K, Gross K, Hancock GE, Stack AM, Welliver RC. Role of interferon gamma in the pathogenesis of primary respiratory syncytial virus infection in BALB/c mice. Journal of Medical Virology. 2000;62:257–266. doi: 10.1002/1096-9071(200010)62:2<257::aid-jmv19>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Warr GA, Jakab GJ. Pulmonary inflammatory responses during viral pneumonia and secondary bacterial infection. Inflammation. 1983;7:93–104. doi: 10.1007/BF00917815. [DOI] [PubMed] [Google Scholar]
- Weigent DA, Huff TL, Peterson JW, Stanton GJ, Baron S. Role of interferon in streptococcal infection in the mouse. Microb Pathog. 1986;1:399–407. doi: 10.1016/0882-4010(86)90071-9. [DOI] [PubMed] [Google Scholar]
- Wong TW, Lau TS, Yu TS, Neller A, Wong SL, Tam W, Pang SW. Air pollution and hospital admissions for respiratory and cardiovascular diseases in Hong Kong. Occupational & Environmental Medicine. 1999;56:679–683. doi: 10.1136/oem.56.10.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HM, Antonini JM, Barger MW, Butterworth L, Roberts BR, Ma JK, Castranova V, Ma JY. Diesel exhaust particles suppress macrophage function and slow the pulmonary clearance of Listeria monocytogenes in rats. Environmental Health Perspectives. 2001;109:515–521. doi: 10.1289/ehp.01109515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Kobzik L. Effect of Concentrated Ambient Particles on Macrophage Phagocytosis and Killing of Streptococcus pneumoniae. Am J Respir Cell Mol Biol. 2007;36:460–465. doi: 10.1165/rcmb.2006-0293OC. [DOI] [PMC free article] [PubMed] [Google Scholar]