

Abstract
An increased ratio of the glutamatergic drive to the overall glutamatergic/GABAergic drive characterizes the chronic stage of temporal lobe epilepsy (TLE), but it is unclear whether this modification is present during the latent period that often precedes the epileptic stage. Using the pilocarpine model of TLE in rats, we report that this ratio is decreased in hippocampal CA1 pyramidal cells during the early phase of the latent period (3–5 days post pilocarpine). It is, however, increased during the late phase of the latent period (7–10 days post pilocarpine), via cell domain-dependent alterations in synaptic current properties, concomitant with the occurrence of interictal-like activity in vivo. During the late latent period, the glutamatergic drive was increased in somata via an enhancement in EPSC decay time constant and in dendrites via an increase in EPSC frequency and amplitude. The GABAergic drive remained unchanged in the soma but was decreased in dendrites, since the drop off in IPSC frequency was more marked than the increase in IPSC kinetics. Theoretical considerations suggest that these modifications are sufficient to produce interictal-like activity. In epileptic animals, the ratio of the glutamatergic drive to the overall synaptic drive was not further modified, despite additional changes in synaptic current frequency and kinetics. These results show that the global changes to more glutamatergic and less GABAergic activities in the CA1 region precede the chronic stage of epilepsy, possibly facilitating the occurrence and/or the propagation of interictal activity.
Temporal lobe epilepsy (TLE) is the most common form of partial epilepsy in adults (Engel, 1996). Mesial TLE, a specific TLE syndrome associated with hippocampal sclerosis, often involves an initial brain insult (head trauma, meningitis, febrile seizures, etc.) followed by a latent (seizure-free) period that can last several years before the occurrence of spontaneous recurrent seizures during the chronic phase (Engel, 1996; Herman, 2002). The process that leads to epilepsy (i.e. epileptogenesis) involves structural and functional modifications within neuronal networks, which are immediate or delayed with respect to the initial insult and which ultimately lead to the first spontaneous seizure (Herman, 2002). Since retrospective studies clearly establish a causal relationship between various brain insults and the occurrence of epilepsy, the development of preventative drug treatments necessitates a better understanding of when and where critical plastic modifications occur in neuronal networks during the latent period (Herman, 2002). Such an issue can only be addressed experimentally at the cellular level in animal models of TLE.
Two morphological alterations in the hippocampus tipify the chronic stage in human TLE and animal models: the sprouting of excitatory axons and the loss of specific populations of GABAergic interneurons. The sprouting is clearly associated with an increased excitatory drive in principal cells and interneurons in a region-independent fashion (e.g. Bausch, 2005). In contrast, the functional consequences of the loss of GABAergic interneurons appear to be not only region dependent but also cell domain specific (Cossart et al. 2005). Thus, the GABAergic drive is decreased in CA1 pyramidal cell dendrites, whilst it is increased in pyramidal cell somata as a result of the hyperactivity of the surviving perisomatic interneurons (Cossart et al. 2001).
These observations are consistent with the hypothesis that imbalance between the glutamatergic and GABAergic drives characterizes epileptic networks and that such imbalance may be causally related to seizure genesis and/or propagation (Bernard, 2005). This view has recently been challenged, since some of these alterations are already present during the latent period. In the pilocarpine experimental model, the loss of GABAergic interneurons is an early outcome of the initial insult (Obenaus et al. 1993; Dinocourt et al. 2003; Kobayashi & Buckmaster, 2003), and the GABAergic drive is already decreased in dentate granule cells during the latent period (Kobayashi & Buckmaster, 2003). Whether interneuron loss in the CA1 region (Dinocourt et al. 2003) also leads to a reduced GABAergic drive in pyramidal cells in a cell domain-dependent manner (Cossart et al. 2001) is not known. During the latent period, an increase in network excitability in both hippocampal regions has also been linked to an impaired inhibition (Mangan & Bertram, 1998; Doherty & Dingledine, 2001; Dinocourt et al. 2003; Kobayashi & Buckmaster, 2003; Cobos et al. 2005). In contrast, an increased excitatory drive appears to be specific to the chronic stage, since the sprouting of excitatory axons has not been found during the latent period (Smith & Dudek, 2001). The global picture that emerges from these studies is that imbalance between the glutamatergic and GABAergic drives should already be present during the latent period. This scheme remains to be tested by measurement of both synaptic drives in individual cells, since homeostatic mechanisms could compensate a loss of inhibition by a loss of excitation (Liu, 2004).
Here, we have measured the glutamatergic and GABAergic synaptic drives received by pyramidal cells of the CA1 region of the hippocampus during the latent period and the chronic phase in pilocarpine-treated rats, a relevant model of TLE. We asked the following questions. Do the cell domain-dependent changes in GABAergic drive precede or follow the onset of spontaneous seizures? How does the glutamatergic drive evolve at the same time? Is there a modification of the respective GABAergic and glutamatergic contributions to the overall synaptic drive received by pyramidal cell somata and dendrites? What could be the functional outcome of such alterations?
Methods
Animal treatment
All experiments were carried out according to methods approved by INSERM. Pilocarpine hydrochloride (340 mg kg−1) was injected intraperitoneally (i.p.) into adult male Wistar rats (180 g) 30 min after the i.p. administration of N-methyl scopolamine (1 mg kg−1). Diazepam (8 mg kg−1, i.p.) was administered to stop status epilepticus (SE) after 3 h. Animals were then observed periodically in the vivarium for the occurrence of spontaneous behavioural seizures. Animals were housed in individual cages; water and food were available ad libitum. Twenty-eight matched sham-treated rats received saline injection. Fifty-six and 42 animals were used for the latent and chronic periods, respectively. Drugs were obtained from Sigma.
In vivo depth EEG recordings
Six adult rats were anaesthetized with a mixture of ketamine (1 mg kg−1) and xylazine (0.5 mg kg−1). One bipolar steel hippocampal electrode (3.5 mm posterior, 2.5 mm lateral and 3.7 mm deep with respect to bregma) and four monopolar stainless-steel cortical electrodes (screwed on the skull, above the right and left frontal and occipital cortex) were stereotaxically implanted. After 1 week recovery, animals were continuously recorded with a video-EEG system (Deltamed, Paris, France) 3 days before SE (control period) and over 1 month after SE. Animals were housed in individual cages; water and food were available ad libitum. At the end of the recording time, animals were anaesthetized as above and intracardiacally perfused with 4% paraformaldehyde, and the position of the hippocampal electrode was checked histologically. Its position within the CA1 region of the hippocampus was confirmed.
EEG analysis
Electroencephalogram was analysed post hoc. For each rat, it was divided into 86 400 contiguous epochs, each of 30 s, for a total duration of 24 h before (control period), during the latent period, and up to 10 days after the first spontaneous seizure (chronic period). Within each epoch, sharp waves were visually detected. They were considered as epileptic sharp waves if their amplitudes were more than twice the amplitude of the biggest sharp waves recorded during the control period. Epochs containing movement artefacts (confirmed using the video that was recorded concomitantly with the EEG) were removed. Epileptic seizures were characterized by an initial increase in the sharp wave frequency, followed by a rapid low-voltage activity that evolved into sustained high-frequency oscillations and high-voltage polyspikes. The first spontaneous seizure involved the limbic structures first, since motor behaviour was either absent or occurred more than 5 s after the beginning of the electrographic seizure (not shown). Data are presented as means ± s.e.m and analysed with Student's paired t test.
In vitro electrophysiology
Hippocampal slices (380 μm thick) were prepared according to local INSERM guidelines from sham, latent and chronic animals, which were anaesthetized by i.p. injection of chloral hydrate (800 mg kg−1). Animals were perfused intracardially with cold artificial cerebrospinal fluid (ACSF) in which NaCl was substituted with an equimolar concentration of choline. Animals were then killed by decerebration, and slices were cut in modified ACSF. Slices were then transferred to a holding chamber at room temperature in normal ACSF. Artificial CSF contained (mm): NaCl, 126; KCl, 3.5; CaCl2, 2; MgCl2, 1.3; NaH2PO4, 1.2; NaHCO3, 26; and d-glucose, 10; and was continuously aerated with 95% O2 and 5% CO2 (pH 7.3). Neurons were visualized by infrared video microscopy using an upright Leica DM LFS microscope equipped with a ×40 objective (Leica, Bensheim, Germany) or a Zeiss FSII microscope equipped with a ×60 objective (Zeiss, Jena Germany). Patch pipettes were pulled from borosilicate glass tubing (2.0 mm outer diameter, 0.5 mm wall thickness) and filled with internal solutions containing (mm): Cs-gluconate, 135; MgCl2, 10; CaCl2, 0.1; EGTA, 1; Na2- adenosine triphosphate, 2; Hepes, 10; and 0.5% biocytin, pH 7.25. For whole-cell somatic and dendritic recordings, the pipette resistance was 3–8 and 6–12 MΩ, respectively. Uncompensated series resistances were 6–30 (soma) and 15–35 MΩ (dendrites). Series resistances and membrane properties were not different between sham, latent and chronic animals (not shown), as previously reported (Bernard et al. 2004). Access resistance and holding current were continuously monitored for stability; a 20% variation led to a rejection of the experiment. The recording temperature was 32–34°C. Signals were fed to an EPC9 (HEKA, Heidelberg, Germany) or Multiclamp 700A (Axon Instruments), digitized (10 kHz) with a Labmaster interface card to a personal computer and analysed with MiniAnalysis 5.1 program (Synaptosoft, Decatur, GA, USA). Spontaneous GABAA receptor-mediated currents (IPSCs) were measured at the reversal potential for glutamatergic events (+10 mV). Bicuculline, a GABAA receptor antagonist, was applied at the end of the experiments to verify that the currents were indeed GABAergic, as described (Cossart et al. 2001). Spontaneous glutamatergic currents (EPSCs) were measured at the reversal potential for GABAA receptor-mediated events (−60 mV) and were blocked by 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and d-aminophosphonovalerate (d-APV). Single events recorded (∼200 events per cell during a continuous recording session) were fully characterized: rise times (10–90%), amplitudes and decay time constants were calculated using MiniAnalysis 5.1. More than 95% of the synaptic events were fitted with a single exponential decay. In some instances, the frequency of sIPSCs was high enough for individual synaptic events to overlap most of the time. The analysis was performed on events that could be clearly isolated, which involved longer duration recordings.
Data treatment
The average synaptic drive was measured as the charge transfer (CT) from baseline using 10 × 1 s periods randomly taken from the recordings of spontaneous postsynaptic currents (10 epochs each for glutamatergic and GABAergic activity in each cell). The baseline was determined by epochs without synaptic activity, which were always present even when the rate of synaptic currents was high (e.g. Fig. 2C). The ratio between the glutamatergic drive and the overall synaptic drive (GABAergic + glutamatergic) in a given cell was defined as the ratio [EPSC CT]/([EPSC CT] + [IPSC CT]). Data are presented as means ± s.e.m. and analysed with Student's unpaired t test. Probability distribution functions were compared with Kolmogorov–Smirnov (KS) test.
Figure 2. Gradual increase of the glutamatergic drive and increase of the GABAergic drive in CA1 pyramidal cell somata during epileptogenesis.

A, photomicrograph of a biocytin-filled pyramidal cell during the late latent period. The arrow indicates the recording site. Scale bar represents 5 μm. B, traces at the top are recordings of spontaneous EPSCs in somata in sham, late latent (cell shown in A) and epileptic animals. Note the transient decrease in EPSC frequency during the early latent period and the increase in frequency during the chronic period, as shown in the histogram on the bottom right (*P < 0.002, Student's unpaired t test). The graphs are cumulative probability plots of amplitude, 10–90% rise time, decay time constant and charge transfer of EPSCs in all recorded cells. Although the EPSC charge transfer is decreased during the early latent period owing to a decrease in amplitude, it is increased during the late latent period as a result of the increase in rise time and decay time constant. The charge transfer is further increased during the chronic period owing to an increase in amplitude and rise time. Normalized average EPSCs are displayed in the inset. C, traces at the top are recordings of spontaneous IPSCs in somata in sham, late latent (cell shown in A) and epileptic animals. Note the transient decrease in frequency during the early and late latent periods as shown in the histogram on the bottom right (*P < 0.05, Student's unpaired t test). The graphs are cumulative probability plots of amplitude, 10–90% rise time, decay time constant and charge transfer of IPSCs in all recorded cells. There was no modification in the distribution in amplitudes, whereas rise times and decay time constants, hence IPSC charge transfers, were increased during the early and late latent periods. Decay time constants and IPSC charge transfers reversed to near control levels during the chronic phase. Normalized average IPSCs are displayed in the inset.
Morphology
Slices were processed for the detection of biocytin-filled neurons. They were fixed overnight at 4°C in a solution containing 4% paraformaldehyde in 0.12m phosphate buffer (PB, pH 7.4). After fixation, slices were rinsed in PB, cryoprotected in sucrose and quickly frozen on dry ice. To neutralize endogenous peroxidase, slices were pretreated for 30 min in 1% H2O2. After several rinses in saline potassium phosphate buffer (0.01 m KPBS, pH 7.4), slices were incubated for 24 h at room temperature, in 1:100 avidin–biotin peroxidase complex (Vector Laboratories, Inc., Burlingame, CA, USA) diluted in KPBS containing 0.3% Triton X-100. After 30 min rinses in KPBS, slices were processed with 0.04% 3,3′- diaminobenzidine-HCl (DAB; Sigma, St Louis, MO, USA) and 0.006% H2O2 diluted in KPBS. Based on their characteristic morphological features, neurons were morphologically identified post hoc as CA1 pyramidal cells. All dendritic recordings were performed farther than 300 μm from the soma (range, 300–450 μm). The recording sites in the dendrites appeared as holes (Fig. 3A) or dents, and were measured post hoc at a distance of 320 ± 28 (sham, n = 8), 342 ± 47 (latent, n = 6) and 335 ± 72 μm (chronic, n = 8) from the soma, i.e. close to the stratum radiatum–lacunosum moleculare border. CA1 pyramidal cells did not display axonal sprouting during the latent period (not shown), as reported in the kainic acid model (Smith & Dudek, 2001). Sprouting was present in epileptic animals, as reported previously (Esclapez et al. 1999; Smith & Dudek, 2001).
Figure 3. Persistent increase of the excitatory drive during the late latent period and gradual erosion of the GABAergic drive in CA1 pyramidal cell dendrites.

A, photomicrograph of a biocytin-filled pyramidal cell during the late latent period. The recording site (hole) is shown in the inset. Abbreviations: O, stratum oriens; P, stratum pyramidale; R, stratum radiatum; LM, stratum lacunosum moleculare; and M, molecular layer of the dentate gyrus. Scale bars represent 50 μm in main panel and 5 μm in the inset. B, traces are recordings of spontaneous EPSCs in dendrites in sham, late latent (cell shown in A) and epileptic animals. Note the increase in frequency during the late latent period, an increase that persists during the chronic period, as shown in the histogram on the right (*P < 0.002, **P < 0.01 versus sham). The graphs are cumulative probability plots of amplitude, 10–90% rise time, decay time constant and charge transfer of EPSCs in all recorded cells. Note the permanent increase in EPSC charge transfer during the late latent period as a result of the increase in amplitude, whilst rise times and decay time constants remain unaltered. Normalized average EPSCs are displayed in the inset. C, the traces at the top are recordings of spontaneous IPSCs in dendrites in sham, late latent (cell shown in A) and epileptic animals. Note the permanent decrease in frequency during the late latent period, as shown in the histogram on the right (***P < 0.05 versus sham, Student's unpaired t test). The graphs are cumulative probability plots of amplitude, 10–90% rise time, decay time constant and charge transfer of IPSCs in all recorded cells. There was no modification in the distribution of amplitudes and rise times, whereas decay time constants, hence IPSC charge transfers, were transiently increased during the late latent period. Normalized average IPSCs are displayed in the inset.
Computational modelling of field potential activity recorded in vivo during control, latent and chronic periods
Sufficient conditions to reproduce signals recorded during control, latent and chronic periods were studied in a neuronal population model described in detail previously (Wendling et al. 2002). Modelling at the neuronal population level was chosen for two reasons. First, this macroscopic level allows for direct comparison of model output (average postsynaptic activity generated by the subset of pyramidal cells) with field activity recorded in vivo and arising from large assemblies of cells. Second, this level of modelling is deep enough to provide insights into the relationship between excitation/inhibition-related model parameters and extracellular field activity recorded in vivo during control, latent and chronic periods.
The model design is based on experimental data describing the neuronal organization and connectivity of the CA1 region. It includes recurrent excitatory connections from pyramidal cells to pyramidal cells (Thomson & Radpour, 1991; Whittington et al. 1997). Pyramidal cells receive two types of GABA receptor-mediated currents: slow dendritic and faster perisomatic inhibitory postsynaptic currents (IPSCs). As previously proposed (Miles et al. 1996; White et al. 2000; Banks & Pearce, 2000; Banks et al. 2002), two separate classes of interneurons (possibly basket cells and dendrite-projecting interneurons, respectively, called, for simplicity, GABAA,fast interneurons and GABAA,slow interneurons) give rise to these two types of IPSCs. Both classes of interneurons interact, in that GABAA,slow cells inhibit not only pyramidal cells but also GABAA,fast interneurons (Banks et al. 2000).
The model was designed to represent this functional organization of interacting subsets of principal cells and interneurons. It consists in three subsets of neurons, namely the main cells (i.e. pyramidal cells), the slow dendritic-projecting inhibitory interneurons (GABAA,slow receptors) and the fast somatic-projecting inhibitory interneurons (GABAA,fast receptors). Interneurons receive an excitatory input (AMPA and NMDA receptor mediated) from pyramidal cells. The influence from neighbouring or more distant populations is represented by an excitatory input n(t) (modelled by a positive mean Gaussian white noise) that globally describes the average density of afferent action potentials.
In each subset, a linear transfer function is used to transform the average presynaptic density of afferent action potentials (the input) into an average postsynaptic membrane potential (the output). This transfer function models synaptic transmission in a simplified way but still takes into account both the amplitude and kinetics of synaptic responses. Its impulse response
and
determines the excitatory (E), dendritic inhibitory (DI) and somatic inhibitory (SI) average postsynaptic membrane potential, respectively. Lumped parameters EPSP, IPSPD and IPSPS define the amplitude of the average postsynaptic membrane potential. Although the exact relationship is not yet established, it can reasonably be assumed that these parameters aggregate both the rate and amplitude of spontaneous postsynaptic potentials (sPSP). Lumped parameters τE, τD and τS are time constant parameters that account for both the average decay time of sPSP and average distributed delays in the dendritic tree. In each subset, in turn, a static non-linear function (asymmetric sigmoid curve 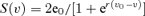 , where 2e0 is the maximum firing rate, ν0 is the post-synaptic potential corresponding to a firing rate of e0, and r is the steepness of the sigmoid) is used to model threshold and saturation effects in the relationship between the average postsynaptic potential of a given subset and the average pulse density of potentials fired by the neurons. Interactions between main cells and local neurons are summarized in the model by connectivity constants which account for the average number of synaptic contacts.
, where 2e0 is the maximum firing rate, ν0 is the post-synaptic potential corresponding to a firing rate of e0, and r is the steepness of the sigmoid) is used to model threshold and saturation effects in the relationship between the average postsynaptic potential of a given subset and the average pulse density of potentials fired by the neurons. Interactions between main cells and local neurons are summarized in the model by connectivity constants which account for the average number of synaptic contacts.
Finally, in order to model the electric extracellular field potential, we used the classical approximation based on the equivalent current dipole theory. As described by Lopes da Silva (2002), we assumed that the neuronal events that cause the generation of electric fields in the neuronal mass consist of ionic currents, the origin of which is mainly postsynaptic. Indeed, at the level of the single cell, synaptic activation of a neuron causes changes in membrane conductance that lead to the generation of primary currents through the membrane and cause a variation of the postsynaptic membrane potential. In both excitatory and inhibitory postsynaptic cases, extracellular currents (volume currents flowing in the surrounding medium) orientated in the same direction are generated. At the level of neuronal assemblies, these extracellular currents can only be measured by macroelectrodes at a distance from the sources if they sum together. This is typically the case for pyramidal cells, which are organized both in space and in time (parallel orientation and quasi-synchronous activation). These considerations allow us to approximate the electrical contribution of the whole neuronal population by an equivalent current dipole whose time-varying moment depends on the time variations of the average postsynaptic potential at the subset of pyramidal cells. Consequently, the temporal dynamics of the extracellular field potential activity seen by the electrode are represented, in the model output, by the summation of average excitatory and inhibitory postsynaptic potentials given, respectively, by the functions hE, hDI and hSI.
A parameter sensitivity study was conducted using an exhaustive procedure aimed at quantifying the frequency of interictal-like spikes detected in simulated signals as a function of the aforementioned model parameters. The amplitudes of glutamatergic average postsynaptic potentials (parameter EPSP) and GABAergic average postsynaptic potentials (parameters IPSPD and IPSPS) were varied step by step. At each step, a 15 s duration signal was simulated, in which the frequency of epileptic spikes was determined using an automatic detection method. The procedure was reiterated for increased average IPSP time constants (parameters τD and τS). All other parameters were kept constant. Finally, results were represented as colour-coded ‘activity maps’ to obtain a complete view of the model behaviour with respect to parameter variations. These maps were used to determine necessary conditions to reproduce, in the model, signals observed in control (normal background activity), latent and chronic periods (background activity mixed with interictal-like spikes). In addition, this procedure was carried out in a blind manner, i.e. without knowledge of the evolution of excitation and inhibition determined experimentally.
Results
Pathological activity during the latent period in vivo
Hippocampal EEG displayed continuous electrical activity during the control period, i.e. 3 days before pilocarpine injection, mostly within the theta band during awake states and rapid eye movemtent (REM) sleep, as previously reported (Leung et al. 1982; Buzsaki, 2002). Two to three days after status epilepticus (SE), the electrical activity was discontinuous, with no recognizable background activity (not shown). Three to five days after SE, awake states, REM sleep and slow-wave sleep (SWS) were again detectable, and background activity returned to normal visually. The first spontaneous seizure occurred 14.5 ± 1.2 days after SE (range, 12–18 days; n = 6) and always occurred first in the hippocampus versus the cortex.
Between day 3 and the first spontaneous seizure (latent period), EEG abnormalities, which were not present before SE, occurred in the form of epileptiform patterns, characterized by large-amplitude spike and sharp waves (Fig. 1A). Interictal-like activity was very infrequent between 3 and 5 days post SE (27 ± 3 events h−1), increased to 42 ± 5 events h−1 between 5 and 7 days and to 164 ± 29 events h−1 between 7 and 10 days post SE (n = 6, Fig. 1). A similar pattern of activity was also observed during the chronic phase, but at a higher frequency (Fig. 1B, +60% versus 7–10 days post SE, n = 6). This network activity was called interictal activity because it occurred between seizures. As in human TLE, these epileptiform patterns were observed during SWS and wakefulness, but not during REM sleep. There is thus an abrupt build up of interictal-like activity between 7 and 10 days post SE.
Figure 1. Pathological activity recorded in vivo in the CA1 region during epileptogenesis.

A, examples of depth EEG recordings performed in the same freely moving animal during wakefulness in the control (3 days before SE), early latent (3 days post SE), late latent (10 days post SE) and chronic periods (1 month post SE) displayed on two different time scales. Note the presence of frequent large-amplitude spikes during the late latent period. A similar pattern is recorded during the chronic phase between seizures. B, histogram of the frequency of large-amplitude spikes during epileptogenesis (n = 6 animals). Note the large increase in frequency during the late latent and chronic periods compared with the early latent period (3–5 and 5–7 days post SE). *P < 0.01, **P < 0.001, Student's paired t test.
Based on these in vivo recordings, we defined two phases of the latent period: the early latent period when interictal-like activity starts to appear (3–5 days after SE) and the late latent period when interictal-like activity is firmly established (7–10 days after SE). Animals were not assessed before day 3 because of the disorganization of the EEG. Glutamatergic and GABAergic synaptic activities were then recorded in vitro in pyramidal cells in sham, early latent, late latent and epileptic animals, and the corresponding charge transfers associated with these activities were determined.
Time-dependent modifications of the glutamatergic drive during epileptogenesis in CA1 pyramidal cell somata
The frequency of EPSCs was decreased during the early latent period (−55% versus sham; Fig. 2 and Table 1). This decrease was transient, since it returned to control values during the late latent period (+26% versus sham, statistically non-significant), whilst EPSC frequency was significantly increased (+134% versus sham) during the chronic phase (Fig. 2 and Table 1). The glutamatergic activity received by pyramidal cells depends not only upon the frequency of synaptic currents but also upon their individual properties, i.e. their kinetics and amplitude. The analysis of individual EPSCs revealed a 170% increase in rise time and a 90% increase in decay time constant during the early latent period versus sham, whilst their average amplitude was significantly decreased by 12% (Fig. 2 and Table 1). These modifications resulted in a decrease of the individual charge transfer of EPSCs (−25% versus sham; Fig. 2 and Table 1) During the late latent period, rise times and decay time constants remained increased (70% versus sham), whilst the amplitudes returned to sham values. Rise time and amplitudes were increased during the chronic phase versus the late latent period and sham (Fig. 2 and Table 1). As a result, there was a gradual increase of the individual charge transfer during the late latent period (+40% versus sham) and the chronic phase (+60% versus late latent period; Fig. 2 and Table 1). These findings show that EPSC frequencies, amplitudes, rise times, decay time constants and charge transfers are modified at different rates during epileptogenesis.
Table 1.
Properties of spontaneous EPSCs received by pyramidal cells
| Frequency (Hz) | Amplitude (pA) | Rise time (ms) | Decay time (ms) | Area (pC ms) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Soma | ||||||||||
| Sham | 3.8 ± 0.8 | (13) | 23.3 ± 0.5 | (13) | 0.80 ± 0.03 | (13) | 4.61 ± 0.02 | (13) | 322 ± 13 | (13) |
| Early latent (3–5 days) | 1.7 ± 0.5* | (12) | 20.7 ± 0.5* | (12) | 2.15 ± 0.07* | (12) | 8.9 ± 0.3* | (12) | 236 ± 16* | (12) |
| Late latent (7–10 days) | 4.8 ± 1.8† | (12) | 23.7 ± 0.5† | (12) | 1.38 ± 0.04*† | (12) | 7.87 ± 0.03* | (12) | 452 ± 28*† | (12) |
| Chronic | 8.9 ± 1.2*† | (9) | 54.8 ± 5.4*† | (9) | 1.61 ± 0.06*† | (9) | 8.49 ± 0.3* | (9) | 736 ± 33*† | (9) |
| Dendrites | ||||||||||
| Sham | 0.5 ± 0.2 | (8) | 22.9 ± 0.9 | (8) | 1.89 ± 0.08 | (8) | 7.39 ± 0.4 | (8) | 220 ± 17 | (8) |
| Latent | 3.7 ± 1.0* | (6) | 27.7 ± 1.0* | (6) | 1.90 ± 0.08 | (6) | 7.54 ± 0.5 | (6) | 268 ± 21 | (6) |
| Chronic | 1.8 ± 0.6* | (8) | 28.6 ± 0.6* | (8) | 1.88 ± 0.06 | (8) | 7.55 ± 0.4 | (8) | 315 ± 11 | (8) |
Values represent means ±s.e.m.
P < 0.05, significantly different by Student's unpaired t test compared with sham.
P < 0.05, significantly different by Student's unpaired t test comparing chronic versus late latent (7–10 days) and late latent (7–10 days) versus early latent (3–5 days).
Since a change in current frequency can be compensated or amplified by alterations in current kinetics, we assessed the global charge transfer carried by EPSC activity (termed glutamatergic drive). This synaptic drive measures the current flowing through receptors during fixed time windows, thus effectively taking into account all the changes affecting frequency, amplitude and kinetics of individual EPSCs, lumping these parameters into a global parameter. Despite slower kinetics, the decrease in EPSC frequency and amplitude resulted in a large decrease of the glutamatergic drive (−70% versus sham) during the early latent period. This decrease was transient, since it switched to a large increase during the late latent period (+280% versus sham), consistent with the overall increases in amplitude, rise time and decay time constant. The glutamatergic drive was further increased during the chronic phase (+85% versus late latent; see Fig. 4 and Table 3), since both EPSC frequency and individual charge transfers were increased. We conclude that after an early decrease following SE, there is a gradual build-up of the glutamatergic drive during the course of epileptogenesis.
Figure 4. The contribution of the glutamatergic drive to the overall glutamatergic and GABAergic drives in the soma and dendrites is already increased during the late latent period and not further modified during the chronic period.

In the soma, there is a gradual increase in glutamatergic drive during the late latent and chronic periods, following an initial decrease during the early latent period. The GABAergic drive is also decreased during the early latent period, recovers to sham values during the late latent period and then increases during the chronic phase. As a result, the contribution of the glutamatergic drive is permanently increased during the late latent period, after an initial decrease during the early latent period, and not further modified during the chronic phase. In the dendrites, the glutamatergic drive is permanently increased during the late latent period and there is a gradual decrease of the GABAergic drive. As a result, the contribution of the glutamatergic drive is permanently increased during the latent period and not further modified during the chronic phase (*P < 0.05, **P < 0.05, Student's unpaired t test).
Table 3.
Permanent increase of the ratio [EPSC drive/(EPSC + IPSC) drive] during epileptogenesis
| Glutamatergic Drive (pC s−1) | GABAergic drive (pC s−1) | Ratio (%) | ||||
|---|---|---|---|---|---|---|
| Soma | ||||||
| Sham | 0.95 ± 0.22 | (13) | 13.1 ± 2.7 | (13) | 6.3 ± 1.2 | (13) |
| Early latent (3–5 days) | 0.29 ± 0.07* | (13) | 7.09 ± 1.08* | (13) | 3.8 ± (0.9)* | |
| Late latent (7–10 days) | 3.65 ± 0.66*† | (12) | 13.9 ± 1.9† | (12) | 20.9 ± 2.1*† | (12) |
| Chronic | 6.78 ± 1.44*† | (9) | 29.3 ± 5.1*† | (9) | 20.7 ± 3.8* | (9) |
| Dendrites | ||||||
| Sham | 0.56 ± 0.15 | (8) | 10 ± 3.5 | (8) | 4.9 ± 1.7 | (8) |
| Latent | 2.69 ± 0.68* | (6) | 5.3 ± 1.0* | (6) | 32.7 ± 5.1* | (6) |
| Chronic | 2.32 ± 0.57† | (8) | 3.5 ± 0.8*† | (8) | 40.9 ± 5.3* | (8) |
Values represent means ±s.e.m.
P < 0.05, significantly different by Student's unpaired t test compared with sham.
P < 0.05, significantly different by Student's unpaired t test comparing chronic versus late latent (7–10 days) and late latent (7–10 days) versus early latent (3–5 days).
Time-dependent modifications of the GABAergic drive during epileptogenesis in CA1 pyramidal cell somata
Since increased excitation can be controlled by strong operative GABAergic inhibition (Miles & Wong, 1987; Christian & Dudek, 1988), it is important to assess the state of the GABAergic drive in the soma. During both early and late latent periods, the frequency of IPSCs was similarly decreased by 40% versus sham (Fig. 2 and Table 2), in keeping with the early loss of axo-axonic cells (Dinocourt et al. 2003). The analysis of individual IPSCs during the early latent period revealed that the distribution of their amplitude was not modified, whereas both rise times and decay time constants of IPSCs were significantly increased by 25 and 45% versus sham, respectively (Fig. 2 and Table 2). Rise times and decay time constants were further increased during the late latent period (45 and 25% versus early latent period, respectively; Fig. 2 and Table 2), i.e. a 76% increase versus sham for both parameters (Fig. 2 and Table 2). The gradual increase in decay time constant and rise time resulted in a gradual increase in the individual charge transfer (Fig. 2 and Table 2).
Table 2.
Properties of spontaneous IPSCs received by pyramidal cells
| Frequency (Hz) | Amplitude (pA) | Rise time (ms) | Decay time (ms) | Area (pC ms) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Soma | ||||||||||
| Sham | 18.1 ± 2.5 | (19) | 42 ± 4 | (19) | 1.15 ± 0.04 | (19) | 8.3 ± 0.2 | (19) | 442 ± 17 | (19) |
| Early latent (3–5 days) | 10.2 ± 0.5* | (12) | 45.9 ± 2 | (12) | 1.43 ± 0.05* | (12) | 12 ± 0.3* | (12) | 579 ± 24* | (12) |
| Late latent (7–10 days) | 11.2 ± 1.2* | (15) | 55 ± 5 | (15) | 2.07 ± 0.08*† | (15) | 14.6 ± 0.4*† | (15) | 819 ± 88*† | (15) |
| Chronic | 17.4 ± 2.3† | (18) | 52 ± 4 | (18) | 1.97 ± 0.08* | (18) | 10.2 ± 0.3*† | (18) | 608 ± 42*† | (18) |
| Dendrites | ||||||||||
| Sham | 9.9 ± 1.8 | (8) | 28 ± 1.0 | (8) | 1.78 ± 0.04 | (8) | 14.2 ± 0.4 | (8) | 325 ± 17 | (8) |
| Latent | 5.0 ± 2.0* | (6) | 34 ± 1.0 | (6) | 2.36 ± 0.06* | (6) | 17.7 ± 0.5* | (6) | 542 ± 21* | (6) |
| Chronic | 5.1 ± 0.1* | (8) | 25 ± 1.0 | (8) | 1.68 ± 0.07† | (8) | 12.7 ± 0.6† | (8) | 300 ± 11† | (8) |
Values represent means ±s.e.m.
P < 0.05, significantly different by Student's unpaired t test compared with sham.
P < 0.05, significantly different by Student's unpaired t test comparing chronic versus late latent (7–10 days) and late latent (7–10 days) versus early latent (3–5 days).
As a result of these modifications, the GABAergic drive carried by spontaneous GABAergic activity was decreased by 45% during the early latent period, showing that the slowing down of kinetics of GABAergic currents did not sufficiently compensate the reduction in frequency. In contrast, the GABAergic drive returned to control values during the late latent period (see Fig. 4 and Table 3), suggesting that the increase in kinetics compensated for the drop off in frequency.
During the chronic phase, the frequency of IPSCs increased compared with both early and late latent periods, returning to control values (Fig. 2 and Table 2). Inhibitory postsynaptic current amplitudes and rise times remained unchanged (versus the late latent period), whereas IPSC decay time constants (hence individual charge transfers) decreased but remained significantly increased by 23% versus sham (Fig. 2 and Table 2). As a result of these modifications, the GABAergic drive was increased (+123% versus sham; Fig. 4 and Table 3), in keeping with a previous study (Cossart et al. 2001).
The latent period is thus characterized by a transient decrease in the GABAergic drive recorded in pyramidal cell somata during the early phase, followed by a recovery during the late phase. The recovery of the GABAergic drive is followed by its increase during the chronic phase.
Since information processing is different in dendrites and somata (Miles et al. 1996; Magee et al. 1998) and since the fate of glutamatergic and GABAergic activities is cell domain dependent (Cossart et al. 2001), we next assessed the synaptic drives in distal dendrites (>300 μm from the soma). We focused on the late latent period and chronic phase, when critical modifications seem to occur based on our somatic recordings.
Increase of the glutamatergic drive during the late latent period in pyramidal cell dendrites
During the late latent period, the frequency of EPSCs was increased (+640%; Fig. 3 and Table 1). The amplitude of individual EPSCs was increased (+21% versus sham), whilst rise times and decay time constants were not modified (Fig. 3 and Table 1). As a result, the global glutamatergic drive received by pyramidal cell dendrites was increased during the late latent period (+380% versus sham; Fig. 4 and Table 3).
During the chronic stage, EPSC frequency, amplitudes, rise times and decay time constants were not significantly modified compared to the late latent period (Fig. 3 and Table 1). As a result, the global glutamatergic drive received by pyramidal cell dendrites remained increased (+314% versus sham; Fig. 4 and Table 3).
In contrast to the soma, where a continuous build-up of the glutamatergic drive seems to occur, the increase in the dendrites is an early, persistent phenomenon.
Gradual decrease of the GABAergic drive in pyramidal cell dendrites
In keeping with the early loss of O-LM interneurons that project to the distal dendrites of pyramidal cells (Dinocourt et al. 2003), the frequency of IPSCs was decreased (−49% versus sham; Fig. 3 and Table 2) during the late latent period. The parameters characterizing individual IPSCs recorded in the dendrites followed the same pattern as those recorded in somata. The distribution of IPSC amplitudes was not significantly modified, but rise times (+33%) and decay time constants (+25%) were significantly increased (Fig. 3 and Table 2). The GABAergic drive assessed by the global charge transfer was decreased (−47% versus sham) during the late latent period (Fig. 4 and Table 3). This suggests that the increase of the rise times and decay time constants did not compensate enough for the large decrease in IPSC frequency, in contrast to what happened in somata.
During the chronic phase, there was no further modification in IPSC frequency, which remained decreased versus sham (−51%). However, both rise times and decay time constants returned to control values. Since the only modification found during the chronic period compared with the late latent period was faster kinetics, the GABAergic drive was further decreased in the dendrites (−34% versus latent and −47% versus sham; Fig. 4 and Table 3).
In contrast to the soma, where the GABAergic drive is not modified during the late latent period and increased during the chronic phase, there is a gradual erosion of the GABAergic drive recorded in pyramidal cell dendrites during the course of epileptogenesis.
Persistent increased contribution of the glutamatergic drive to the overall synaptic drive
The previous observations show that the modifications affecting the GABAergic and glutamatergic drives are dependent on time and cell compartment. To obtain a better assessment of the overall impact of these alterations, we computed the ratio between the glutamatergic drive and the summed glutamatergic and GABAergic drives in each recorded cell. This ratio represents the contribution of the glutamatergic drive to the overall synaptic drive received by a given cell. In pyramidal cell somata, the ratio was transiently decreased (−40% versus sham) during the early latent period. In contrast, during the late latent period, the ratio was increased by 231% versus sham) and was not further modified in epileptic animals (Fig. 4 and Table 3). The glutamatergic drive, which represents 6% of the synaptic drive measured in the soma of sham animals, amounts to 20% in both late latent and epileptic animals (Fig. 4 and Table 3). The same pattern was found in pyramidal cell dendrites. The ratio was increased during the late latent period (+567% versus sham) and was not significantly further modified during the chronic stage (Fig. 4 and Table 3). The contribution of the glutamatergic drive measured in distal apical dendrites is thus considerably reinforced during both late latent and epileptic periods, from 5 to more than 33%. Like the situation in somata, this modification occurred during the late latent period and persisted during the chronic phase without further modification.
Increased contribution of the glutamatergic drive may be sufficient to induce interictal-like activity
Pharmacological and theoretical studies suggest that increasing excitation and/or decreasing inhibition are sufficient to drive a neuronal network into a hyperexcitable state (Traub & Miles, 1991; Whittington et al. 1995). Although the CA1 region and the dentate gyrus display signs of hyperexcitability (bursting activity and loss of paired pulse inhibition) during the latent period (Mangan & Bertram, 1998; Doherty & Dingledine, 2001; Kobayashi & Buckmaster, 2003), including the interictal-like activity reported here, it is difficult to establish a causal relationship between these macroscopic observations and the more microscopic modifications we and others report (i.e. changes in frequency, amplitude and kinetics of glutamatergic and GABAergic currents).
To obtain some insight into the possible functional consequences of these alterations, we took advantage of a computer model that has been developed to mimic the hippocampal architecture to generate EEG activity (Wendling et al. 2002). Modelling was performed blind regarding the experimental results. The only experimental data provided (see Methods) was the depth EEG recordings performed before and after SE (interictal-like activities during latent and chronic periods).
Model parameters (related to EPSC/IPSC frequency/amplitude and decay time constants) were comprehensively varied in somatic/dendritic compartments (parameter sensitivity study). Necessary conditions to produce normal background and interictal-like activities were then determined from colour-coded activity maps representing the frequency of interictal-like events detected in the model ouput. For standard parameter values (including those corresponding to experimental ones, as assessed after running the simulations), the model produced an EEG background activity similar to that observed in animals before SE (Fig. 5A). Realistic interictal-like activity (Fig. 5A and B) was obtained when the glutamatergic drive (represented in the model by the average EPSP, which lumps together EPSP frequency and kinetics) was increased by 100% and the GABAergic drive (represented in the model by the average IPSP, which lumps together IPSP frequency and kinetics) was decreased by 54% in the dendrites and by 25% in the soma (Fig. 5C, top panels). The frequency of interictal-like events remained, however, low (1–3 sporadic events for the 15 s simulated activity).
Figure 5. Increasing the glutamatergic drive and decreasing the GABAergic drive may constitute sufficient conditions for the genesis of interictal-like activity.

A, examples of depth EEGs generated by the model in control, latent and chronic periods. Note the similarity in EEG patterns with the experimental results (cf. Fig. 1). B, frequency content of interictal-like events. Note the similarity between the recorded and simulated signals. C, the frequency of interictal-like events is presented as a colour map (dark blue, 0 Hz; red, 1.66 Hz). Average dendritic and somatic GABAergic drives are presented in each colour map on the abscissa and ordinate, respectively. The excitatory drive is increased from left to right and the decay time constant of IPSPs is increased from top to bottom. The numbers in the colour maps correspond to the activity presented in A, as follows: (1) background activity; (2) interictal-like activity during the latent period; and (3) interictal activity during the chronic period. Although a very large increase in the excitatory drive is sufficient by itself to produce interictal-like activity (top right map), the latter is readily induced when the GABAergic drive is decreased. Note that the occurrence of interictal-like activity is more sensitive to a decreased GABAergic drive in the dendrites versus the soma. An increase in the decay time constant of IPSPs acts as a limiting factor, decreasing the frequency of interictal-like events.
Interestingly, the network was more sensitive to a decrease of the dendritic GABAergic drive than to a decrease of the somatic GABAergic drive, as previously reported (Wendling et al. 2002). When the glutamatergic drive was increased, interictal-like events became more frequent for a given value of the GABAergic somatic and dendritic drives. The sensitivity to a decrease in dendritic GABAergic drive was more apparent for the greater values of the glutamatergic drive (as demonstrated by maps reading from left to right in Fig. 5C). These simulations suggest that the occurrence of interictal-like events is particularly sensitive to an increase in the excitatory drive and a parallel decrease in the dendritic, and to a lesser extent, somatic GABAergic drives.
We then explored the consequences of increasing the decay time constant of IPSPs (Fig. 5C, middle and bottom panels). The frequency of interictal activity decreased for increased IPSP decay time constants, confirming that this phenomenon may act as a compensatory mechanism to increase the GABAergic drive, hence dampening the hyperexcitability.
The same parametric study was used to explore the chronic phase, which is characterized by an increase in the frequency of interictal events (Fig. 1). In the model, this phenomenon could be reproduced for parameter settings compatible with experimental observations: (i) further increase of the glutamatergic drive with respect to the latent period; (ii) decay time constants returning back to control values (‘pro-epileptic’ effect); and (iii) an increased somatic GABAergic drive with respect to the latent period (‘antiepileptic’ effect).
The model predicts that there exist a large number of solutions (no unique set of parameters) to mimic the activity recorded in vivo, including a set of parameters that takes experimental values. Nevertheless, the global analysis of activity maps reveals the main relationships between model parameters and model output in terms of tendencies; thus, increased glutamatergic drive and decreased GABAergic drive (with differential roles at the soma and dendrites) lead to a higher frequency of interictal-like events, while increased IPSC decay time constants tend to reduce this frequency.
Discussion
The purpose of this study was to investigate whether the alterations of the glutamatergic and GABAergic drives found during the chronic phase in CA1 pyramidal cell dendrites and somata were already present during the latent period. We found that the glutamatergic and GABAergic inputs are modified in a time- and cell domain-dependent manner during epileptogenesis. There are four principal findings. First, there is a transient decrease of the contribution of the excitatory drive to the overall synaptic drive during the early latent period, 3–5 days following the initial status epilepticus. Second, the contribution of the excitatory drive is increased during the late latent period (7–10 days) and is not further modified during the chronic phase. Third, EEG is already abnormal during the late latent period, displaying interictal-like activity. Fourth, computer simulations suggest that the imbalance between the glutamatergic and GABAergic drives constitutes a sufficient condition for the occurrence of interictal-like activity.
Kinetics of synaptic events in somata and dendrites
Synaptic currents were recorded in somata and dendrites because the geometry of the cell and the recording conditions make it impossible for a recording electrode, whatever its location along the somato-dendritic tree, to record all the synaptic inputs a cell is receiving at a given time. For example, the ability of a somatic recording electrode to detect dendritically generated events varies as a function of the distance between the activated synapse and the soma, its position in the dendritic tree (main apical branch, oblique dendrites, etc.), the series resistance, etc. (Cossart et al. 2000). It is therefore important to assess what is recorded by our somatic and dendritic recording electrodes. Being separated by more than 300 μm, there should be little cross-talk between soma and distal dendrite recording electrodes (Soltesz et al. 1995; Cossart et al. 2000), although large-amplitude, distally generated events may be picked up by the recording electrode (Magee & Cook, 2000; Cossart et al. 2000). This issue was directly assessed with simultaneous recordings of the soma and the apical dendrite of the same pyramidal cell (n = 3). Although large-amplitude events or events generated midway were detected by somatic and dendritic recording electrodes, most of the synaptic events recorded by one electrode were not detected by the other (supplemental Fig. 1). Furthermore, if there is a high level of cross-talk, increased activity at the distal site would be reflected at the recording site, with slower and smaller events (e.g. supplemental Fig. 1). However, despite the fact that EPSC and IPSC frequencies measured in the soma were increased during the chronic phase versus the latent period, EPSC and IPSC frequencies and kinetics measured in dendrites were neither increased nor slower. Together, these results suggest that we sampled synaptic events generated mostly in the vicinity of the recording electrode. For example, spontaneous EPSCs recorded in the soma are most likely to reflect EPSCs generated in the proximal apical and basal strata radiatum and oriens dendrites, since the soma is devoid of symmetrical synapses (Megias et al. 2001). Conversely, EPSCs recorded in the dendrites are most likely to reflect the activation of synapses located in the distal part of the dendritic tree, in particular in the stratum lacunosum moleculare. Dendritic and somatic recordings thus allow different synaptic pathways to be sampled.
The kinetics of synaptic events is different from its actual value at the postsynaptic site because it is altered by two factors. The first factor is space clamp problems, which may arise from the quality of the recording. However, series resistance and membrane properties were not different between sham, latent and epileptic animals. The second factor is dendritic filtering, which could be specifically altered during epileptogenesis. However, we could not identify major morphological alterations in the dendritic tree between sham, latent and epileptic animals. Therefore, kinetic modifications are most likely to reflect postsynaptic modifications. However, we cannot rule out a spatial redistribution of active synapses (farther from the recording site), which would result in slower kinetics (e.g. supplemental Fig. 1).
Aetiology of glutamatergic neurotransmission alterations
The glutamatergic drive was considerably decreased during the early latent period, probably a direct consequence of the initial insult (the pilocarpine-induced status epilepticus). The frequency of EPSCs recorded in dendrites was significantly increased during the late latent period, most likely reflecting enhanced activity within the glutamatergic pathways that target the distal dendrites, e.g. proximal CA3 pyramidal cells (Ishizuka et al. 1990; Bernard & Wheal, 1994) and the perforant path (Witter et al. 2000; Cossart et al. 2001; Wozny et al. 2005). The increased cell excitability in the entorhinal cortex is consistent with this scheme (Shah et al. 2004; Wozny et al. 2005).
The frequency of EPSCs recorded in somata was not modified during the late latent period, suggesting that a lack of modification of the presynaptic glutamatergic pathways projecting in the perisomatic region, in particular mid/distal CA3 pyramidal cells and the CA1 associational pathway (Ishizuka et al. 1990; Bernard & Wheal, 1994). However, during the chronic phase, the EPSC frequency measured in the soma was increased, most likely reflecting the sprouting of the CA1 associational pathway is stratum oriens, and to a lesser extent in stratum radiatum (Esclapez et al. 1999; Perez et al. 1996), and perhaps CA3 axonal sprouting (Siddiqui & Joseph, 2005). Increased activity within the presynaptic glutamatergic pathways may also be a consequence of a decrease of the inhibitory tone received by presynaptic cells (as reported here) and/or an increase of their excitability (Sanabria et al. 2001; Bernard et al. 2004). Alternatively, a greater susceptibility to slicing-induced irritation during epileptogenesis may explain the increase in EPSC frequency. The fact that miniature activity is decreased in epileptic animals (Esclapez et al. 1999; Hirsch et al. 1999; Cossart et al. 2001), argues against such a scheme. Finally, a change in the tonic control of transmitter release by presynaptic receptors (mGlu, GABAB, cannabinoid, etc.) cannot be ruled out (Yang et al. 2006), an issue that remains to be investigated. Postsynaptic alterations also occurred during epileptogenesis, with slower EPSC kinetics, a property only found for somatic recordings, suggesting that these modifications are pathway specific. The mechanisms underlying kinetics and amplitude modifications (spatial distribution of synapses, subunit composition, receptor number/distribution and phosphorylation states) was beyond the scope of this study.
An intriguing aspect of our results is the time-, pathway- and parameter-dependent modifications of the properties of presynaptic glutamatergic networks and postsynaptic receptors. Following a postinsult depression, although these alterations take different forms in somata and dendrites, they all concur to increase the glutamatergic drive. We conclude that the glutamatergic drive is already increased in both cell compartments during the seizure-free late latent period, but, whereas it is not subsequently modified in dendrites, it is further increased in somata in epileptic animals.
Aetiology of GABAergic neurotransmission alterations
During the early latent period, the GABAergic drive was decreased, like the glutamatergic drive, further supporting a post-insult modification (Goodkin et al. 2005). During the late latent period, IPSCs were less frequent in somata and dendrites than in sham animals, probably reflecting the loss of axo-axonic and O-LM interneurons, respectively (Dinocourt et al. 2003). Decreased interneuron excitability (Kobayashi et al. 2003) and decreased miniature GABAergic activity (Hirsch et al. 1999) could also contribute to the reduction in IPSC frequency. This decrease was transient in the soma, since IPSC frequency returned to control values during the chronic phase, reflecting the increased excitability of perisomatic interneurons (Esclapez et al. 1997; Cossart et al. 2001). Axonal sprouting of surviving interneurons cannot be ruled out, but the issue remains controversial (Bausch, 2005). The fact that IPSC frequency measured in the dendrites remained decreased during the chronic phase suggests a lack of compensatory mechanisms in dendrite projecting interneurons. Postsynaptic modifications also occurred with the increase in IPSC kinetics, which could be explained in part by a change in subunit composition, as in the dentate gyrus (Brooks-Kayal et al. 1998; Shao & Dudek, 2005), or by receptor internalization (Goodkin et al. 2005). In contrast to EPSCs, kinetics alterations affecting IPSCs occurred in both cell compartments and were transient, since they returned to control values during the chronic period. These results suggest that complex intracellular mechanisms differentially alter GABAergic and glutamatergic receptors, according to their location along the somato-dendritic axis and the time window during epileptogenesis.
We conclude that the GABAergic drive in the dendrites is already decreased during the late latent period (the increase in kinetics does not sufficiently compensate the decrease in frequency) and further decreased during the chronic stage owing to a return to control kinetics. In contrast, the somatic GABAergic drive remained unaltered during the late latent period after a post-insult transient depression, since the increase in IPSC kinetics compensated for the decrease in IPSC frequency. The GABAergic drive was, however, increased during the chronic period, via an activity-dependent process, as reported previously (Cossart et al. 2001). These results further confirm the dichotomy between the two cell compartments (Cossart et al. 2001), with a gradual erosion of the GABAergic drive in dendrites and a gradual increase in somata.
Although a gradual build-up of the excitatory drive during the course of epileptogenesis may constitute a general rule of reorganization (i.e. independent of model and region), the fate of the GABAergic drive appears to be model specific. During the latent period, IPSCs were less frequent in dentate gyrus granule cells in the pilocarpine model (Kobayashi & Buckmaster, 2003), although they were not modified in the kainic acid model (Shao & Dudek, 2005). Interestingly, IPSC decay time constants were also increased in dentate gyrus granule cells during the latent period in the kainic acid model, but this increase persisted during the chronic phase (Shao & Dudek, 2005), whilst it was transient in the CA1 region (present data). These model- and region-dependent modifications may have different consequences in terms of network behaviour.
Functional implications of altered glutamatergic and GABAergic drives
One can only speculate about the functional outcome of the multiple time-dependent and domain-dependent synaptic alterations found within the glutamatergic and GABAergic pathways. The decreased GABAergic drive found during the late latent period may explain why the CA1 region is already hyperexcitable during the latent period (Mangan & Bertram, 1998; Gorter et al. 2002), a property also found in the dentate gyrus (Doherty & Dingledine, 2001; Gorter et al. 2002). It is important to note that we have analysed only one aspect of synaptic physiology, namely the focusing of the synaptic drives. Other mechanisms, including changes in cell excitability, GABA action (depolarizing versus hyperpolarizing) and glial function, also contribute to hyperexcitability (Cohen et al. 2002; Su et al. 2002; Bernard et al. 2004; Tian et al. 2005). In acute in vitro models of epilepsy, hippocampal networks can support a large loss of GABAergic activity before generating epileptiform discharges (Whittington et al. 1995). In this context, it is not surprising to find that some erosion of inhibition during the latent period is not associated with seizures. The same conclusion applies to the increased excitatory drive found during the late latent period, which is not associated with seizures.
What could be the functional consequences of an increased glutamatergic drive and a decreased GABAergic drive? The model we have used to asses this issue, although not as detailed as those including networks of multicompartment neurons (Traub & Miles, 1991; Traub et al. 2005), allowed exploration of different sets of solutions to produce specific EEG patterns (Wendling et al. 2002), in particular interictal activity. In epileptic patients with TLE, interictal activity corresponds to one steady state of the system, whilst seizures are rare events, involving the recruitment of different brain regions (Bartolomei et al. 2001). The model predicts that steady-state interictal activity can result from an offset between the glutamatergic and GABAergic drives. Spontaneous synaptic activities recorded in the slice preparation also characterize the steady-state of network activity in vitro. The fact that spontaneous interictal-like activity was not measured in the slice is not surprising, since most extrinsic and intrinsic connections are severed. Such limitation is not present in the computer model, in which more complete connectivity can be achieved. Together, these arguments lead us to propose that interictal activity can result from an offset between the glutamatergic and GABAergic drives. It is interesting to note that during the early latent period, the offset is reversed compared with the late latent period (i.e. the contribution of the excitatory drive is decreased), whilst interictal-like activity is starting to appear at a very low frequency (corresponding to random transient events rather than steady-state activity, as during the late latent and chronic periods). Although one of the possible theoretical solutions to produce interictal-like activity includes the one found experimentally, other sets of parameters can produce such activity. These sets of solutions (achieving an offset between the glutamatergic and GABAergic drives) could correspond to what may occur in other limbic and non-limbic regions, giving them the possibility for generating interictal-like activity. Interestingly, EEG recordings in TLE patients show that interictal activity does not necessarily emerge from subsets of temporal structures, but can emerge in specific regions independently of the others (Bourien et al. 2005).
Although it is tempting to propose that interictal-like activity recorded in the CA1 region results from an imbalance between the glutamatergic and GABAergic drives, both observations may not be causally related. Pharmacologically induced interictal-like activity in vitro can originate from the CA3 region (Avoli et al. 2002) or the entorhinal cortex (Wozny et al. 2005), and it remains to be determined whether the interictal-like activity we record in CA1 is locally generated or propagated from other structures. Even if the imbalance between the glutamatergic and GABAergic drives in CA1 does not produce interictal-like activity, it can facilitate the spread of epileptiform activity from the entorhinal cortex to the CA1 region, as previously reported in slices from epileptic animals (Cossart et al. 2001; Wozny et al. 2005).
Which modifications are then specific to the chronic period? Among many alterations, two features can be distinguished: the increased excitatory drive associated with the formation of aberrant excitatory connections and a further erosion of the GABAergic drive in the dendrites (via a return to faster IPSC kinetics). Interestingly, the theoretical analysis suggests that these modifications are sufficient to explain the increased frequency of interictal-like events found in vivo during the chronic phase. The fact that these modifications reproduce steady-state (interictal) activity and not transient events (seizures) is consistent with our hypothesis that the offset between the glutamatergic and GABAergic drives may be causally related to the genesis of interictal-like activity. The increase of the perisomatic GABAergic drive during the chronic phase is not necessarily paradoxical if one takes into account the synchronizing action of interneurons (Freund & Buzsáki, 1996) and/or the depolarizing action of GABA in normal and epileptic tissue (Cohen et al. 2002; Szabadics et al. 2006). If the inhibitory action of GABAergic neurotransmission is solely considered (like in the model), the lengthening of the GABAergic current during the latent period acts as a protective mechanism, whilst the return to faster control level values is clearly increasing network excitability.
Conclusion
The present study provides further evidence of the considerable amount of plasticity that takes place within glutamatergic and GABAergic pathways during the latent period. These modifications are dependent on pathway, time and cell compartment, but the net effect is a larger contribution of the excitatory drive to the overall synaptic drive. Importantly, although some parameters are permanently modified as a result of the initial insult, others specifically evolve between the latent and chronic periods. As a working hypothesis, we propose that interictal-like activity is a direct early outcome of the underlying morpho-functional network modifications. The occurrence of interictal-like activity may constitute a signature of the latent period (Hellier et al. 1999; Cobos et al. 2005; Staley et al. 2005), and its functional consequences remain to be investigated. It may not be sufficient to cause seizures, as reported in humans (Cavazzuti et al. 1980), but it may constitute one key process of epileptogenesis, e.g. enabling further plastic modifications in temporal structures to produce an epileptic zone (Staley et al. 2005). It will be interesting to test whether interictal activity appears before epilepsy in patients at risk (with a previous history of brain trauma, meningitis, etc.) in prospective studies. This would open the way to preventive therapeutic treatments.
Acknowledgments
This work was supported by INSERM, Fédération pour la Recherche sur le Cerveau (FRC), Fondation pour la Recherche Médicale (FRM) and Action Concertée Incitative (ACI). M.M. was supported by an INSERM MD-PhD fellowship. We wish to express our thanks to A. Nehlig for her help with in vivo recordings and to M. Fontes for hosting us in his laboratory.
Supplementary material
The online version of this paper can be accessed at:
DOI: 10.1113/jphysiol.2006.119297
http://jp.physoc.org/cgi/content/full/jphysiol.2006.119297/DC1 and contains supplemental material consisting of a figure entitled: Simultaneous somatic and dendritic recording of a CA1 pyramidal cell during the late latent period (7 days post SE).
This material can also be found as part of the full-text HTML version available from http://www.blackwell-synergy.com
References
- Avoli M, D'Antuono M, Louvel J, Kohling R, Biagini G, Pumain R, D'Arcangelo G, Tancredi V. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog Neurobiol. 2002;68:167–207. doi: 10.1016/s0301-0082(02)00077-1. [DOI] [PubMed] [Google Scholar]
- Bartolomei F, Wendling F, Bellanger JJ, Regis J, Chauvel P. Neural networks involving the medial temporal structures in temporal lobe epilepsy. Clin Neurophysiol. 2001;112:1746–1760. doi: 10.1016/s1388-2457(01)00591-0. [DOI] [PubMed] [Google Scholar]
- Bausch SB. Axonal sprouting of GABAergic interneurons in temporal lobe epilepsy. Epilepsy Behav. 2005;7:390–400. doi: 10.1016/j.yebeh.2005.07.019. [DOI] [PubMed] [Google Scholar]
- Bernard C. Dogma and dreams: experimental lessons for epilepsy mechanism chasers. Cell Mol Life Sci. 2005;62:1177–1181. doi: 10.1007/s00018-005-5009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–535. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- Bernard C, Wheal H. A model of the connectivity patterns in the CA3 and CA1 areas of the hippocampus. Hippocampus. 1994;4:497–529. doi: 10.1002/hipo.450040502. [DOI] [PubMed] [Google Scholar]
- Bourien J, Bartolomei F, Bellanger JJ, Gavaret M, Chauvel P, Wendling F. A method to identify reproducible subsets of co-activated structures during interictal spikes. Application to intracerebral EEG in temporal lobe epilepsy. Clin Neurophysiol. 2005;116:443–455. doi: 10.1016/j.clinph.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABAA receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Cavazzuti GB, Cappella L, Nalin A. Longitudinal study of epileptiform EEG patterns in normal children. Epilepsia. 1980;21:43–55. doi: 10.1111/j.1528-1157.1980.tb04043.x. [DOI] [PubMed] [Google Scholar]
- Christian E, Dudek F. Electrophysiological evidence from glutamate microapplications for local excitatory circuits in the CA1 area of rat hippocampal slices. J Neurophysiol. 1988;59:110–123. doi: 10.1152/jn.1988.59.1.110. [DOI] [PubMed] [Google Scholar]
- Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, Rubenstein JL. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat Neurosci. 2005;8:1059–1068. doi: 10.1038/nn1499. [DOI] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Cossart R, Hirsch JC, Cannon RC, Dinoncourt C, Wheal HV, Ben-Ari Y, Esclapez M, Bernard C. Distribution of spontaneous currents along the somato-dendritic axis of rat hippocampal CA1 pyramidal neurons. Neuroscience. 2000;99:593–603. doi: 10.1016/s0306-4522(00)00231-1. [DOI] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Ben-Ari Y, Esclapez M, Bernard C. Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci. 2001;4:52–62. doi: 10.1038/82900. [DOI] [PubMed] [Google Scholar]
- Cossart R, Bernard C, Ben-Ari Y. Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005;28:108–115. doi: 10.1016/j.tins.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Dinocourt C, Petanjek Z, Freund TF, Ben Ari Y, Esclapez M. Loss of interneurons innervating pyramidal cell dendrites and axon initial segments in the CA1 region of the hippocampus following pilocarpine-induced seizures. J Comp Neurol. 2003;459:407–425. doi: 10.1002/cne.10622. [DOI] [PubMed] [Google Scholar]
- Doherty J, Dingledine R. Reduced excitatory drive onto interneurons in the dentate gyrus after status epilepticus. J Neurosci. 2001;21:2048–2057. doi: 10.1523/JNEUROSCI.21-06-02048.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J., Jr Introduction to temporal lobe epilepsy. Epilepsy Res. 1996;26:141–150. doi: 10.1016/s0920-1211(96)00043-5. [DOI] [PubMed] [Google Scholar]
- Esclapez M, Hirsch JC, Ben Ari Y, Bernard C. Newly formed excitatory pathways provide a substrate for hyperexcitability in experimental temporal lobe epilepsy. J Comp Neurol. 1999;408:449–460. doi: 10.1002/(sici)1096-9861(19990614)408:4<449::aid-cne1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Esclapez M, Hirsch JC, Khazipov R, Ben-Ari Y, Bernard C. Operative GABAergic inhibition in hippocampal CA1 pyramidal neurons in experimental epilepsy. Proc Natl Acad Sci U S A. 1997;94:12151–12156. doi: 10.1073/pnas.94.22.12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Goodkin HP, Yeh JL, Kapur J. Status epilepticus increases the intracellular accumulation of GABAA receptors. J Neurosci. 2005;25:5511–5520. doi: 10.1523/JNEUROSCI.0900-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter JA, van Vliet EA, Aronica E, Lopes da Silva FH. Long-lasting increased excitability differs in dentate gyrus vs. CA1 in freely moving chronic epileptic rats after electrically induced status epilepticus. Hippocampus. 2002;12:311–324. doi: 10.1002/hipo.1100. [DOI] [PubMed] [Google Scholar]
- Hellier JL, Patrylo PR, Dou P, Nett M, Rose GM, Dudek FE. Assessment of inhibition and epileptiform activity in the septal dentate gyrus of freely behaving rats during the first week after kainate treatment. J Neurosci. 1999;19:10053–10064. doi: 10.1523/JNEUROSCI.19-22-10053.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman ST. Epilepsy after brain insult: targeting epileptogenesis. Neurology. 2002;59:S21–S26. doi: 10.1212/wnl.59.9_suppl_5.s21. [DOI] [PubMed] [Google Scholar]
- Hirsch JC, Agassandian C, Merchan-Perez A, Ben-Ari Y, DeFelipe J, Esclapez M, Bernard C. Deficit of quantal release of GABA in experimental models of temporal lobe epilepsy. Nat Neurosci. 1999;2:499–500. doi: 10.1038/9142. [DOI] [PubMed] [Google Scholar]
- Ishizuka N, Weber J, Amaral D. Organization of intrahippocampal projections originating from CA3 pyramidal cells in the rat. J Comp Neurol. 1990;295:580–623. doi: 10.1002/cne.902950407. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci. 2003;23:2440–2452. doi: 10.1523/JNEUROSCI.23-06-02440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Wen X, Buckmaster PS. Reduced inhibition and increased output of layer II neurons in the medial entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci. 2003;23:8471–8479. doi: 10.1523/JNEUROSCI.23-24-08471.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung LW, Lopes da Silva FH, Wadman WJ. Spectral characteristics of the hippocampal EEG in the freely moving rat. Electroencephalogr Clin Neurophysiol. 1982;54:203–219. doi: 10.1016/0013-4694(82)90162-6. [DOI] [PubMed] [Google Scholar]
- Liu G. Local structural balance and functional interaction of excitatory and inhibitory synapses in hippocampal dendrites. Nat Neurosci. 2004;7:373–379. doi: 10.1038/nn1206. [DOI] [PubMed] [Google Scholar]
- Lopes da Silva F. Electrical potentials. In: Ramachandran VS, editor. Encyclopedia of the Human Brain. Vol. 2. Elsevier, New York: 2002. pp. 147–167. [Google Scholar]
- Magee JC, Cook EP. Somatic EPSP amplitude is independent of synapse location in hippocampal pyramidal neurons. Nat Neurosci. 2000;3:895–903. doi: 10.1038/78800. [DOI] [PubMed] [Google Scholar]
- Magee J, Hoffman D, Colbert C, Johnston D. Electrical and calcium signaling in dendrites of hippocampal pyramidal neurons. Annu Rev Physiol. 1998;60:327–346. doi: 10.1146/annurev.physiol.60.1.327. [DOI] [PubMed] [Google Scholar]
- Mangan PS, Bertram EH., III Ontogeny of altered synaptic function in a rat model of chronic temporal lobe epilepsy. Brain Res. 1998;799:183–196. doi: 10.1016/s0006-8993(98)00411-9. [DOI] [PubMed] [Google Scholar]
- Megias M, Emri Z, Freund TF, Gulyas AI. Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience. 2001;102:527–540. doi: 10.1016/s0306-4522(00)00496-6. [DOI] [PubMed] [Google Scholar]
- Miles R, Toth K, Gulyás AI, Hajos N, Freund TF. Differences between somatic and dendritic inhibition in the hippocampus. Neuron. 1996;16:815–823. doi: 10.1016/s0896-6273(00)80101-4. [DOI] [PubMed] [Google Scholar]
- Miles R, Wong R. Inhibitory control of local excitatory circuits in the guinea-pig hippocampus. J Physiol. 1987;388:611–629. doi: 10.1113/jphysiol.1987.sp016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenaus A, Esclapez M, Houser CR. Loss of glutamate decarboxylase mRNA-conatining neurons in the rat dentate gyrus following pilocarpine-induced seizures. J Neurosci. 1993;13:4470–4485. doi: 10.1523/JNEUROSCI.13-10-04470.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Y, Morin F, Beaulieu C, Lacaille J-C. Axonal sprouting of CA1 pyramidal cells in hyperexcitable hippocampal slices of kainate-treated rats. Eur J Neurosci. 1996;8:736–748. doi: 10.1111/j.1460-9568.1996.tb01259.x. [DOI] [PubMed] [Google Scholar]
- Sanabria ER, Su H, Yaari Y. Initiation of network bursts by Ca2+-dependent intrinsic bursting in the rat pilocarpine model of temporal lobe epilepsy. J Physiol. 2001;532:205–216. doi: 10.1111/j.1469-7793.2001.0205g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao LR, Dudek FE. Changes in mIPSCs and sIPSCs after kainate treatment: evidence for loss of inhibitory input to dentate granule cells and possible compensatory responses. J Neurophysiol. 2005;94:952–960. doi: 10.1152/jn.01342.2004. [DOI] [PubMed] [Google Scholar]
- Siddiqui AH, Joseph SA. CA3 axonal sprouting in kainate-induced chronic epilepsy. Brain Res. 2005;1066:129–146. doi: 10.1016/j.brainres.2005.10.066. [DOI] [PubMed] [Google Scholar]
- Smith BN, Dudek FE. Short- and long-term changes in CA1 network excitability after kainate treatment in rats. J Neurophysiol. 2001;85:1–9. doi: 10.1152/jn.2001.85.1.1. [DOI] [PubMed] [Google Scholar]
- Soltesz I, Smetters DK, Mody I. Tonic inhibition originates from synapses close to the soma. Neuron. 1995;14:1273–1283. doi: 10.1016/0896-6273(95)90274-0. [DOI] [PubMed] [Google Scholar]
- Staley K, Hellier JL, Dudek FE. Do interictal spikes drive epileptogenesis? Neuroscientist. 2005;11:272–276. doi: 10.1177/1073858405278239. [DOI] [PubMed] [Google Scholar]
- Su H, Sochivko D, Becker A, Chen J, Jiang Y, Yaari Y, Beck H. Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus. J Neurosci. 2002;22:3645–3655. doi: 10.1523/JNEUROSCI.22-09-03645.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Molnar G, Olah S, Barzo P, Tamas G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- Thomson A, Radpour S. Excitatory connections between CA1 pyramidal cells revealed by spike triggered averaging in slices of rat hippocampus are partially NMDA receptor mediated. Eur J Neurosci. 1991;3:587–601. doi: 10.1111/j.1460-9568.1991.tb00845.x. [DOI] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Contreras D, Cunningham MO, Murray H, LeBeau FE, Roopun A, Bibbig A, Wilent WB, Higley MJ, Whittington MA. Single-column thalamocortical network model exhibiting γ oscillations, sleep spindles, and epileptogenic bursts. J Neurophysiol. 2005;93:2194–2232. doi: 10.1152/jn.00983.2004. [DOI] [PubMed] [Google Scholar]
- Traub R, Miles R. Neuronal Network of the Hippocampus. New York: Cambridge University Press; 1991. [Google Scholar]
- Wendling F, Bartolomei F, Bellanger JJ, Chauvel P. Epileptic fast activity can be explained by a model of impaired GABAergic dendritic inhibition. Eur J Neurosci. 2002;15:1499–1508. doi: 10.1046/j.1460-9568.2002.01985.x. [DOI] [PubMed] [Google Scholar]
- Whittington MA, Stanford IM, Colling SB, Jefferys JG, Traub RD. Spatiotemporal patterns of γ frequency oscillations tetanically induced in the rat hippocampal slice. J Physiol. 1997;502:591–607. doi: 10.1111/j.1469-7793.1997.591bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington MA, Traub RD, Jefferys JG. Erosion of inhibition contributes to the progression of low magnesium bursts in rat hippocampal slices. J Physiol. 1995;486:723–734. doi: 10.1113/jphysiol.1995.sp020848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP, Wouterlood FG, Naber PA, van Haeften T. Anatomical organization of the parahippocampal-hippocampal network. Ann N Y Acad Sci. 2000;911:1–24. doi: 10.1111/j.1749-6632.2000.tb06716.x. [DOI] [PubMed] [Google Scholar]
- Wozny C, Gabriel S, Jandova K, Schulze K, Heinemann U, Behr J. Entorhinal cortex entrains epileptiform activity in CA1 in pilocarpine-treated rats. Neurobiol Dis. 2005;19:451–460. doi: 10.1016/j.nbd.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Yang J, Woodhall GL, Jones RS. Tonic facilitation of glutamate release by presynaptic NR2B-containing NMDA receptors is increased in the entorhinal cortex of chronically epileptic rats. J Neurosci. 2006;26:406–410. doi: 10.1523/JNEUROSCI.4413-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.