

Abstract
The range of actions of the second messenger Ca2+ is a key determinant of neuronal excitability and plasticity. For dendritic spines, there is on-going debate regarding how diffusional efflux of Ca2+ affects spine signalling. However, the consequences of spino-dendritic coupling for dendritic Ca2+ homeostasis and downstream signalling cascades have not been explored to date. We addressed this question by four-dimensional computer simulations, which were based on Ca2+-imaging data from mice that either express or lack distinct endogenous Ca2+-binding proteins. Our simulations revealed that single active spines do not affect dendritic Ca2+ signalling. Neighbouring, coactive spines, however, induce sizeable increases in dendritic [Ca2+]i when they process slow synaptic Ca2+ signals, such as those implicated in the induction of long-term plasticity. This spino-dendritic coupling is mediated by buffered diffusion, specifically by diffusing calbindin-bound Ca2+. This represents a central mechanism for activating calmodulin in dendritic shafts and therefore a novel form of signal integration in spiny dendrites.
Neuronal information processing, associative learning in particular, requires spatial summation of synaptic inputs in the postsynaptic cell (Augustine et al. 2003). Classically, spatial summation is viewed as the integration of electrical signals and a subsequent generation of action potentials (Kandel & Siegelbaum, 2000). Spatial summation of second messengers, such as Ca2+, originating from neighbouring spines and flooding the dendritic shaft, seems to be an unlikely mechanism of neuronal integration because spine necks are assumed to represent substantial diffusion barriers for messenger molecules (Gamble & Koch, 1987; Zador et al. 1990; Müller & Connor, 1991; Svoboda et al. 1996).
While spill-over of spinous Ca2+ into the dendritic shaft has been observed by several groups (Majewska et al. 2000; Holthoff et al. 2002; Schmidt et al. 2003b), these experimental findings were significantly influenced by the buffering action of the Ca2+ indicator dye (Sabatini et al. 2002; Schmidt et al. 2003b). Recently, however, Noguchi et al. (2005) analysed spinous Ca2+ dynamics under conditions that perturbed endogenous Ca2+ handling minimally. They found that, indeed, most spines allow a sizeable Ca2+ efflux, which is tightly regulated by the geometry of the spine neck.
Here we studied the impact that specific endogenous Ca2+-binding proteins (CaBPs) have on Ca2+ diffusion across the spine neck. We also explored the implications of spino-dendritic coupling for neuronal signal integration. Our analysis was based on kinetic computer simulations that were adjusted to fit high-resolution Ca2+ imaging data from wild-type mice and mutant mice that lack parvalbumin (PV) and/or calbindin D28k (CB; Schmidt et al. 2003b). Fluorescence recovery after photobleaching (FRAP) complemented the analysis by providing the diffusional mobility of the Ca2+ sensor calmodulin (CaM). We found that slow synaptic Ca2+ signals are associated with a substantial efflux of Ca2+ from the spine into the dendrite that relies on mobile CaBPs. This diffusional coupling is capable of driving a spatial summation process in which coincident activity of neighbouring spines is integrated in the dendrite. Spatial summation by mobile CaBPs appears to be a prime mechanism for activating calmodulin in dendritic shafts.
Methods
Fluorescence recordings
As previously described (Schmidt et al. 2003b), Ca2+ imaging experiments were performed in acute cerebellar slices from wild-type, PV null-mutant (Schwaller et al. 1999) and CB/PV null-mutant mice (Vecellio et al. 2000; decapitated under isoflurane (Baxter) anaesthesia). Purkinje neurons (PNs) were equilibrated with the dye-containing pipette solution (200 or 50 μm Oregon-Green BAPTA-1; OGB) in the whole-cell patch-clamp configuration. Experiments were performed 60–120 min after the whole-cell configuration was established, a time period in which the dendritic dye concentration reaches 70–80% of that in the pipette solution (Eilers et al. 1995a; Müller et al. 2005). Fluorescence signals were recorded with a confocal laser-scanning microscope (Fluoview-300, Olympus) equipped with a 60×/0.9 NA water immersion objective. For FRAP recordings (Schmidt et al. 2003a, 2005), OGB was replaced by 100 μm Alexa 488-labelled CaM and the confocal scanner was converted to a two-photon system by adapting a mode-locked Ti–sapphire laser (Tsunami; Spectra Physics, Darmstadt, Germany set to 765 nm) and a Pockels cell (Conoptics, Danbury, CT, USA).
Data analysis
Fluorescence data were analysed with custom-written routines in Igor Pro 5.0 (Wavemetrics, Lake Oswego, OR, USA). Calcium ion imaging data were expressed as normalised relative fluorescence increase (ΔF/F0) and converted to free intracellular Ca2+ concentrations ([Ca2+]i) based on a cuvette calibration (Schmidt et al. 2003b). Median decays were computed as previously described (Schmidt et al. 2003b). Data for FRAP were analysed as described by Schmidt et al. (2005). In brief, the FRAP time course was converted to the mean apparent diffusion coefficient, D, according to the equation:
| (1) |
Where 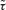 denotes the time median constant of FRAP, r the radius, l the length of the spine neck, and V the volume of the spine head (for values see Supplementary Table 1). The immobilized fraction of CaM was estimated from the offset after the bleach pulse (see Fig. 4 A and Schmidt et al. 2005).
denotes the time median constant of FRAP, r the radius, l the length of the spine neck, and V the volume of the spine head (for values see Supplementary Table 1). The immobilized fraction of CaM was estimated from the offset after the bleach pulse (see Fig. 4 A and Schmidt et al. 2005).
Figure 4. Activation of dendritic calmodulin (CaM) by buffered Ca2+ diffusion.

A, spiny dendrite (upper panel) loaded with dye-labelled CaM. The red cross-hair marks the spine on which the FRAP recording shown in the lower panel was performed. The red line in the lower panel represents an exponential fit to the normalized fluorescence (F/F0) recovery curve. The time constant of recovery (τ) is related to the apparent diffusion coefficient of CaM, and the offset reflects immobilization of CaM in the spine. B, simulated increase in Ca2+-bound CaM (activated CaM, CaM*) relative to baseline activation (Δ[CaM*]/[CaM*]0, upper panel) and total CaM activation (i.e. integrated time evolution of CaM activation, lower panel) during fast and slow synaptic Ca2+ signals occurring simultaneously in neighbouring spines (compare Figs 2 and 3D). The dotted and continuous green lines overlap in the lower panel. Note that the total CaM activation in the inactive dendrite during slow synaptic Ca2+ signals is much larger than the CaM activation in the active spine during fast signalling. C, spatially resolved simulation as in Fig. 3D but for CaM activation resulting from concurrent slow synaptic Ca2+ signals in neighbouring spines. The spine signal is saturated on the chosen scale. Note that dendritic CaM* increases by more than 60% compared with the resting level. D, total dendritic CaM activation with CB and PV being either mobile (blue, as in lower panel of B) or immobilized (red). Note that buffered Ca2+ diffusion is required for dendritic CaM activation.
Calmodulin expression and labelling
The mouse CaM coding sequence was amplified by PCR and ligated into the pGEX-2TK vector (Amersham) using EcoR1 and BamH1 restriction sites in the sense and antisense primers. After expression in the E.coli host BL21 (DE3), the protein was purified using glutathione sepharose and the GST (glutathione S-transferase) fusion domain cleaved off with thrombin (Amersham). Fluorescence labelling was performed with Alexa-488 TFP (2,3,5,6-tetrafluorophenyl) ester (Invitrogene, Carlsbad, CA, USA). The labelling product was purified by HPLC.
Kinetic, two-compartment model
Ca2+ dynamics were simulated by ordinary differential equations in Mathematica 5.0 (Wolfram Research, Champaign, IL, USA) according to previously published formalisms (Markram et al. 1998; Schmidt et al. 2003b) and parameters (Schmidt et al. 2003a,b; 2005). For CaM, which binds four calcium ions, only the kinetics of the first, rate-limiting step (Linse et al. 1991; Holmes, 2000; Sabatini et al. 2002; Faas et al. 2004) were considered. The spine and the parent dendrite were modelled as two well-mixed compartments optionally coupled by diffusion across the spine neck. The spine geometry and diameter of the adjacent dendrite were set to the corresponding mean values of spiny dendrites of PNs (Harris & Stevens, 1988). In order to simulate the impact of Ca2+ signals from neighbouring coactive spines, the length of the dendritic compartment was set to 0.3 μm according to the following line of argument. Assuming homogeneous Ca2+ signals in the coactive spines, the volume-averaged Ca2+ signals in the adjacent dendritic compartments will also be identical and, thus, diffusional net fluxes between the spino-dendritic units do not take place. The spine density of distal dendrites (3.4 μm−1; Vecellio et al. 2000) then determines that the length of the dendritic compartment proprietary to individual spines is 0.3 μm. The amplitude and time course of metabotropic glutamate receptor (mGluR)-mediated Ca2+ signals were adjusted according to published data (Takechi et al. 1998; Barski et al. 2003). For fitting the model to experimental data, the concentrations of CB and PV were corrected for the wash-out that occurs during patch-clamp experiments (Schmidt et al. 2003b). The only free variables in the model were the amplitude of the Ca2+ influx and the maximum pump velocity. In simulations that reflect the situation in unperturbed cells, OGB and the wash-out correction for CB and PV were omitted. A detailed description of the model and full listing of all parameters are given in the Supplementary Methods and Supplementary Table 1.
Kinetic, spatially resolved models
For analysing spatial aspects of spino-dendritic Ca2+ signalling, the above formalism was extended to partial differential equations with the Cartesian co-ordinates as additional independent variables. Two models with one spine each were constructed. One model reflected simultaneous activity in neighbouring spines, in which the dendritic length was set, as in the two-compartment model, to 0.3 μm (Figs 2C, 3D and 4C). While the above-mentioned assumption of identical Ca2+ signals in neighbouring dendritic disks will only apply to the spatially resolved model if spines are located on identical radial positions, interdisk variations in [Ca2]i owing to radially staggered spines can be expected to be minimal. These variations were ignored in order to minimize the computational load of the simulations, which otherwise would have to incorporate dozens of spines and a long dendritic branch. The second model reflected activity in a single spine. Here, inactive spines were omitted for simplicity and the length of the dendritic segment was set to 10 μm (Fig. 3C). The simulations were carried out in the ‘Chemical Engineering Module’ of the finite element modelling environment FEMLAB 3.1 (Comsol, Stockholm, Sweden) using the following reaction diffusion equation:
| (2) |
Figure 2. Spino-dendritic coupling in the absence of indicator dye.

A, Ca2+ transient in a spine (black) and the adjacent dendritic compartment (grey; enlarged in the inset) simulated in the two-compartment model in the absence of indicator dye and with the Ca2+ influx being restricted to spines. B, diffusional flux of buffer-bound and free Ca2+ across the spine neck for the simulation in A. The inset shows the temporal integral of the flux. C, spatially resolved simulation of the situation in A, generated by a finite element approach. Note that the spine Ca2+ signal is saturated on the chosen colour scale and that even at the exit of the spine neck there is no significant elevation in dendritic [Ca2+]i.
Figure 3. Spatial integration of slow synaptic Ca2+ signals in dendrites.

A–C, simulated mGluR-mediated Ca2+ release in a single spine with the resulting spinous Ca2+ transient (A), the diffusional flux and its integral between the activated spine and the dendrite (B) and, in C, the spatially resolved Ca2+ changes in the neighbouring dendrite (length 10 μm; inactive spines were not considered, see icon in C). Note that dendritic [Ca2+]i is almost unaffected, despite the pronounced efflux from the spine. D, [Ca2+]i changes during concurrent mGluR-mediated Ca2+ release in neighbouring spines. The situation was simulated with a single active spine located on a proprietary dendritic segment of 0.3 μm length as in Figs 1 and 2 (see the dashed lines in the icon and text for detail). Note the significant increase in dendritic [Ca2+]i in this condition.
Where Ri is the reaction rate (–kon× educts +koff× products) for the ith reactant with concentration ci and diffusion coefficient Di. Using a conventional desktop computer (Pentium 4; CPU, 2.8 GHz), the computing times were ∼72 and ∼3 h for the two types of spatially resolved models, respectively.
Results
Kinetic model of spino-dendritic Ca2+ signalling based on Ca2+-imaging data
Ca2+ transients, elicited by stimulating the suprathreshold climbing fibre (CF) input, were recorded in the linescan mode in spiny dendrites of cerebellar PNs (Fig. 1A and B). As reported previously (Schmidt et al. 2003b), both the dendritic and the spinous Ca2+ transients relied on voltage-operated Ca2+ channels (VOCCs) and showed complex decay kinetics (Fig. 1C). From a total of 252 spine and 131 dendritic recordings, median spinous and dendritic decays were calculated. These averaged transients were fitted with a two-compartment model (representing the spine and the adjacent dendritic shaft) that optionally included diffusional coupling across the spine neck. During fitting, only the Ca2+ influx and the maximum pump velocity were variables in the model; the remaining parameters (up to 43, see Supplementary Methods) were taken from the literature.
Figure 1. Spino-dendritic coupling under experimental conditions.

A, Purkinje neuron filled with the Ca2+ indicator Oregon Green BAPTA-1 (OGB; ∼160 μm) via a somatic patch pipette. The box indicates the region in which climbing fibre (CF)-evoked Ca2+ transients were recorded. The inset shows the corresponding complex spike (scale bar represents 20 mV, 20 ms). B, higher magnification of the spiny branchlets marked in A. The grey line and arrowheads indicate the location for a linescan recording. C, CF-evoked spinous and dendritic Ca2+ transients (left and right, respectively, black traces). The grey lines represent double-exponential fits to the decays of the transients. D, median decays (black lines) in spines and dendrites (large and small transient, respectively, n = 252 and 131). The red and green lines represent decays simulated with a kinetic, two-compartment model that excluded (left) or included (right) diffusional coupling between spine and dendrite via the spine neck. The size of the dendritic compartment was calculated according to the spine density of distal dendrites. The insets show the temporal development of the integrated deviation of the spinous simulation from the measured data. E, corresponding diffusional flux (left) of free and buffer-bound Ca2+ across the spine neck. The temporal integration is shown on the right. Abbreviations: CB, calbindin D28k; and PV, parvalbumin.
The geometrical parameters of the model reflected the experimental situation in two crucial aspects. First, the length of the dendritic segment was set according to the spine density of distal dendrites such that each spine was coupled to its proprietary dendritic volume. Thus, assuming homogeneous Ca2+ transients during CF activation (Schmidt et al. 2003b), diffusion along the dendrite could be neglected. Second, the two compartments represent the resolvable structures from which volume-averaged fluorescence signals where obtained by confocal live cell imaging.
In total, ∼4700 and ∼35 000 Ca2+ ions entered the spine and the dendrite, respectively, during the CF-evoked Ca2+ transient; ∼95% of these ions were buffered by calbindin (CB), parvalbumin (PV), or the Ca2+ indicator dye (Oregon-Green BAPTA-1, OGB, ∼160 μm). As expected (Sabatini et al. 2001; Schmidt et al. 2003b; Noguchi et al. 2005), incorporation of diffusion improved the overlap between the spinous data and the simulation substantially, especially during the initial 200 ms of the decay, but had little effect on the accuracy of the dendritic simulation (Fig. 1D). Extracting the fluxes across the spine neck from the simulation (Fig. 1E) revealed that diffusional exchange between the spine and the dendritic shaft was pronounced during this time window. Approximately 20% of the Ca2+ ions that entered the spine diffused into the dendritic compartment. This spino-dendritic Ca2+ flux was dominated by buffered diffusion of Ca2+, mainly bound to the indicator dye, while the contribution of free Ca2+ ions was negligible (compare Sabatini et al. 2002). Despite its lower diffusional mobility (Schmidt et al. 2003a, 2005), CB transported about three times more Ca2+ than PV across the neck. This behaviour is explained by the Mg2+ saturation of PV under physiological conditions, which substantially limits the Ca2+ buffering capacity of PV (Lee et al. 2000; Schmidt et al. 2003b).
We validated our model by performing the above analysis on mutant mice that lacked either PV or CB and PV (PV–/– and PV/CB–/–, respectively, see Supplementary Fig. 1). In the PV–/– model, all parameters were kept as in the wild-type (WT), except that PV was removed from the simulation. In the PV/CB–/– model, CB and PV were removed, and the morphology of spines was changed according to the known differences from the WT (Vecellio et al. 2000). Consistent with our previous report on dendritic Ca2+ decays (Schmidt et al. 2003b), an approximately twofold stronger clearance mechanism was required for a reliable fit of the PV/CB–/– data. In both strains, our parameter space successfully described the recorded data and underlined the importance of CB for buffered diffusion.
As a step towards the naïve, zero indicator dye situation, we analysed experiments performed with only 40 μm OGB (Supplementary Fig. 2). Without changing any parameter but the dye concentration, the simulation yielded a good overlap with the data (Supplementary Fig. 2A and B). At this dye concentration, CB dominated the Ca2+ transport across the spine neck; only one-quarter of the diffusing Ca2+ was transported by OGB (Supplementary Fig. 2C and D).
Spino-dendritic coupling during rapid spine signals
Since the model robustly reproduced the Ca2+ imaging data obtained under the different experimental conditions, we used it as a tool to analyse spino-dendritic cross-talk under unperturbed conditions, i.e. in the absence of the Ca2+ indicator dye and without the wash-out of endogenous CaBPs that occurs during prolonged whole-cell recordings (Eilers et al. 1995a; Müller et al. 2005; see Methods for details). All other parameters were kept as in the simulations of the WT data. We began by analysing rapid spine Ca2+ transients, such as those occurring during synaptic activation of VOCCs and/or ionotropic glutamate receptor channels (iGluRs; Denk et al. 1995) in neighbouring spines. Accordingly, Ca2+ influx was restricted to the spine and dendritic influx was omitted in the model. As illustrated in Fig. 2A, the spinous [Ca2+]i reached peak values of ∼2 μm before binding by CB and to a lesser extend by PV, and diffusion rapidly reduced it to ∼300 nm. The later decay phase was dominated by buffered Ca2+ diffusion and Ca2+ extrusion.
Despite the large concentration gradient between the spine and the inactive dendrite, diffusion of free Ca2+ was negligible (Fig. 2B). Only ∼2% of the Ca2+ that entered the spine (i.e. ∼100 ions) diffused unbuffered into the dendrite, a number that is in close agreement with the findings of Zador et al. (1990), as well as Sabatini et al. (2002) who studied spiny dendrites of CA1 pyramidal neurons that are endowed with a low concentration of immobile CaBPs. However, in contrast to the assumed absence of buffered transport in CA1 neurons, the mobile CaBPs of Purkinje neurons, CB and PV, shuttled ∼74% of the spinous Ca2+ load (i.e. ∼3500 of the 4700 ions) into the dendrite. This significant number did not, however, lead to a sizeable increase in the dendritic [Ca2+]i (< 10 nm, Fig. 2A inset), presumably because of the large volume difference between the spine and the dendrite and because of the effective Ca2+ extrusion mechanisms.
We wondered whether the localized ejection of Ca2+ into the dendrite may generate a Ca2+ microdomain at the exit of the spine neck. We therefore developed a spatially resolved model of the spino-dendritic Ca2+ homeostasis based on a finite element analysis. In this model, we simulated coactivity in neighbouring spines under the assumption that all spines emerge from the dendritic shaft at the same radial position. This allowed us to reduce the model to a single spine and its adjacent dendritic shaft segment of 0.3 μm length (see Methods). The corresponding four-dimensional (4-D) simulation revealed that the strong spinous Ca2+ efflux failed to build up significant Ca2+ gradients in the dendrite (Fig. 2C). Thus, in the case of rapid spinous Ca2+ transients, mobile CaBPs breach the spine limit via buffered diffusion. For free Ca2+, however, the spine represents a functional compartment that is well separated from the dendrite, even if neighbouring spines are coactive. The large dendritic shaft instead acts as a ‘clearance compartment’ for the spine.
Coupling during slow spine signals
We proceeded by simulating slower spine Ca2+ signals with the spatially resolved model, specifically those occurring during physiological synaptic activation (Chadderton et al. 2004) of mGluRs (Finch & Augustine, 1998; Takechi et al. 1998; Wang et al. 2000). Single spine mGluR-mediated responses were modelled according to published data (Takechi et al. 1998; Barski et al. 2003; see Methods). They last ∼10 times longer than VOCC/iGluR responses (Takechi et al. 1998; Barski et al. 2003; Fig. 3A) and resulted in a Ca2+ load of ∼54 000 ions for the active spine. In line with the large buffer capacity of PNs (Fierro & Llano, 1996), this strong Ca2+ influx also was largely buffered by CB and PV, leading to an ∼10 times larger diffusional efflux of buffered Ca2+ from the spine (Fig. 3B). About 75% of the spinous Ca2+ load left the spine by free and buffered diffusion.
Spatial gradients in the dendrite in response to this Ca2+ efflux from a single active spine were simulated in the 4-D model of a single spine and a 10 μm long attached dendrite from which all other, inactive spines were omitted for simplicity (Fig. 3C). Even this massive, long-lasting spino-dendritic Ca2+ transport failed to induce significant concentration gradients in the dendrite. With hindsight, this finding is explained by the fact that one-dimensional diffusion determines the supply of Ca2+ via the spine neck while more effective three-dimensional diffusion determines the drainage of Ca2+ from the exit of the neck into the dendritic compartment. Thus, owing to the large spino-dendritic volume difference, even long-lasting Ca2+ transients in individual spines must fail to induce local Ca2+ elevations in the dendrite, although they are associated with a massive Ca2+ transport along the spine neck.
Spatial integration of slow synaptic Ca2+ signals in dendrites
The powerful dilution effect of diffusion within the dendrite, however, may lose its impact, when Ca2+ from neighbouring, coactive spines enters the dendrite. In this situation, the spino-dendritic volume difference is significantly reduced because notionally each spine exhausts its Ca2+ load only into its own, small proprietary dendritic segment (see Methods). Notably, such activity patterns do play a role in certain forms of experimentally induced synaptic plasticity in PNs (Daniel et al. 1998).
We simulated this situation by modelling the average spine and its proprietary dendritic segment of 0.3 μm length (Vecellio et al. 2000), according to the spine density of distal dendrites (see Methods). While this approach ignores the morphological variability of spines (Harris & Stevens, 1988) and the corresponding impact on diffusional coupling (Noguchi et al. 2005), the exact spine position on the dendritic circumference and the corresponding spatial variations in dendritic Ca2+ gradients, as well as anomalous diffusion along the dendrite (Santamaria et al. 2006), it drastically simplified the simulation and kept computation times within a reasonable frame. Furthermore, given the high spine density of PNs, the impact of spine variability can be expected to average out in a given spiny branchlet.
As illustrated in Fig. 3D, concurrent mGluR-mediated responses in neighbouring spines indeed caused a significant build-up of Ca2+ in the dendrite. The dendritic Ca2+ reached a maximum amplitude of ∼80 nm, i.e. about one-quarter of the peak amplitude in the spine (Fig. 3A). This dendritic Ca2+ signal relied on the massive and long-lasting efflux of Ca2+ from the spine (Fig. 3B). Approximately 74% of the spinous Ca2+ load was ejected into the dendrite, with the majority of Ca2+ ions being ferried by CB, while diffusion of free Ca2+ ions was again negligible.
Activation of dendritic calmodulin
In order to reveal functional consequences of the spino-dendritic cross-talk during concurrent mGluR responses, we studied the resulting activation of the Ca2+-dependent regulatory protein calmodulin (CaM; Fig. 4; Xia & Storm, 2005). Since the diffusional mobility of CaM was unknown, we quantified this parameter by two-photon fluorescence recovery after photobleaching (FRAP; Fig. 4A). According to our previously described formalism (Schmidt et al. 2003a), the average diffusion coefficient of CaM was determined to be 21 ± 4 μm2 s–1 (median ±s.e. of medians; n = 65, 6 cells), a value similar to that reported in HEK 293 cells (Kim et al. 2004). Twenty-three per cent (± 4%, median ±s.e.m.) of CaM was immobile, indicating binding to intracellular targets (Schmidt et al. 2005).
Inclusion of CaM into our kinetic simulation revealed that rapid, VOCC/iGluR-mediated spinous Ca2+ signals were not associated with a significant increase in the total amount of activated CaM (CaM*), not even in active spines (Fig. 4B, lower panel), a finding that is consistent with the simulations of Sabatini et al. (2002). In contrast, mGluR-mediated Ca2+ signals substantially increased the total concentration of CaM* in the spine but not in the dendrite if only a single spine was active (data not shown). Only mGluR-mediated activity in neighbouring, coactive spines led to a significant increase of CaM* in both the activated spines and the adjacent dendritic compartments (Fig. 4B and C). Remarkably, the total CaM* in the dendrite reached about one-fifth of the spinous value.
Finally, we analysed the origin of dendritic calmodulin activation. Both diffusion of CaM* from the spine into the dendrite and diffusion of Ca2+ with subsequent activation of dendritic CaM may be involved. We distinguished between these two possibilities by immobilizing CB and PV in our simulation, thereby minimizing Ca2+ transport across the spine neck (compare Fig. 3B). In this condition, activation of dendritic CaM was negligible (Fig. 4D). Thus, in dendrites, CaM activation is a local event, requiring summation of slow synaptic Ca2+ signals from neighbouring spines.
Discussion
We established a kinetic, spatially resolved simulation of Ca2+ and calmodulin signalling in spiny dendrites of cerebellar PNs that allowed us to study the functional role of mobile CaBPs in synaptic integration. The simulations were based on high-resolution Ca2+-imaging data and on a first-time determination of the diffusional mobility of CaM in spiny dendrites. We found that CB and, to a lesser extent, PV transport a major fraction of postsynaptic Ca2+ loads from the spine into the dendrite. This spino-dendritic coupling has the potential to spatially summate slow synaptic Ca2+ signals at the level of dendritic branchlets. The resulting activation of dendritic CaM reaches about 20% of the corresponding spinous signal, by far surpassing spinous CaM activation during fast synaptic Ca2+ signals. These findings suggest that spino-dendritic coupling by CaBPs may serve as a potent form of spatial signal integration in spiny dendrites.
A direct observation of buffered diffusion of Ca2+ via fluorescence imaging is, at present, not possible. Available Ca2+ indicator dyes, such as OGB, inevitably distort Ca2+ signals by reducing their peak amplitudes, prolonging their decays, affecting Ca2+ diffusion and by competing with endogenous CaBPs (Neher, 1999; Sabatini et al. 2002). Distortion-free measurements would require either dyes that sense Ca2+ not by binding but by collision quenching (as in Cl–-sensitive dyes; Lakowicz, 1999) or genetically encoded, CB/PV-based reporter proteins (see, for example, troponin C-based Ca2+ sensors; Heim & Griesbeck, 2004). As long as such sensors remain unavailable, studies on buffered diffusion must rely on indirect methods, i.e. on simulations that reflect the kinetics and the diffusional mobility of all Ca2+-binding molecules.
It has been argued that Monte-Carlo simulations are more appropriate for simulations in spines than deterministic/finite element methods (Franks & Sejnowski, 2002; Bhalla, 2004b). We felt, however, that a simulation of the movement of tens of thousands of particles (Ca2+, PV, CB and CaM) would overstrain current stochastic simulation environments, although there exist algorithms that accelerate Monte-Carlo simulations (Gillespie, 2001; Puchalka & Kierzek, 2004; Santamaria et al. 2006). Furthermore, it has been demonstrated that in the limit of large molecule numbers the solutions of deterministic and stochastic models are asymptotically equivalent (Bhalla, 2004a; Blackwell, 2006). In compartments approximately 10 times larger than spines, even 100 ions are sufficient to give a good overlap between stochastic and deterministic solutions to the diffusion equation in most cases; with 1000 ions, even the fluctuations around the deterministic value become negligible (Blackwell, 2006). We therefore relied on the finite element approach, which yielded acceptable computation times even for Ca2+ signals lasting several seconds, a prerequisite for fitting models to experimental data. The latter was achieved with a model of two well-mixed compartments, reflecting the experimental situation of line-scan recordings. The two-compartment and the finite element model yielded almost identical results for all situations analysed.
Our findings challenge the common notion that the spine represents a well-separated biochemical compartment (Gamble & Koch, 1987; Zador et al. 1990; Müller & Connor, 1991; Denk et al. 1995; Svoboda et al. 1996; Sabatini et al. 2002; Augustine et al. 2003). For neurons that express significant amounts of mobile CaBPs, such as cerebellar PNs, the spine neck does not impose a functional diffusion barrier. We show that about ∼75% of the Ca2+ load of active spines is discharged into the dendritic shaft. The apparent compartmentalization in spines, as measured with Ca2+ indicator dyes, could be attributed to rapid diffusional redistribution and dilution of free and buffer-bound Ca2+ within the large dendritic compartment. Thus, it is the large volume difference between spines and dendrites, not limited diffusion across the spine neck, that leads to an apparent restriction of synaptic Ca2+ signals to spines.
In agreement with a powerful spino-dendritic cross-talk, we find that spatial summation of slow Ca2+ signals from neighbouring, coactive spines results in sizeable Ca2+ increases in the dendritic shaft and a concomitant on-site activation of CaM. Indeed, we may even have underestimated the activation of CaM, since its Ca2+ affinity is increased substantially in the presence of certain CaM target proteins (Xia & Storm, 2005).
Besides mobile Ca2+-binding proteins, the morphology of individual spines can be expected to influence spino-dendritic coupling (Noguchi et al. 2005). In particular, spines with short and wide necks will facilitate dendritic CaM activation and allow this type of spatial signal integration to be triggered by a smaller number of coactive spines. Future work, based on high-resolution spine reconstructions (Vecellio et al. 2000), may shed light on the functional role of specific spine morphologies for signal integration in cerebellar PNs.
Activated CaM may regulate, among other targets (Xia & Storm, 2005), the voltage dependence of dendritic A-type (Varga et al. 2004) as well as SK-type potassium channels (Bond et al. 2005). Given the fact that parallel fibres fire in high-frequency bursts during physiological activation (Chadderton et al. 2004) and that such bursts reliably induce mGluR-mediated spinous Ca2+ transients (Takechi et al. 1998), dendritic CaM-dependent targets may be triggered whenever a beam of parallel fibres becomes active (Eccles, 1967). This situation is comparable to the requirements for subthreshold spatial integration of electrical signals in spiny dendrites (Eilers et al. 1995b). As with this latter, electrical form of integration (Eilers et al. 1997), spino-dendritic cross-talk driven by mobile CaBPs may serve as a powerful coincidence detector in the induction of synaptic plasticity (Xia & Storm, 2005). Since it potentially also includes nearby inactive spines, it may be specifically involved in certain forms of heterosynaptic plasticity (Hartell, 2002; Xia & Storm, 2005). Taken together, the presence or absence of mobile CaBPs is a critical determinant of spine compartmentalization and dendritic integration.
Acknowledgments
We thank B. Schwaller for providing null-mutant mice and G. Bethge for technical assistance. This work was supported by grants from the Human Frontier Science Program and the Bundesministerium für Bildung und Forschung to J.E.
Supplemental material
Online supplemental material for this paper can be accessed at:http://jp.physoc.org/cgi/content/full/jphysiol.2007.127860/DC1 and http://www.blackwell-synergy.com/doi/suppl/10.1113/jphysiol.2007.127860
References
- Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/s0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- Barski JJ, Hartmann J, Rose CR, Hoebeek F, Mörl K, Noll-Hussong M, De Zeeuw CI, Konnerth A, Meyer M. Calbindin in cerebellar Purkinje cells is a critical determinant of the precision of motor coordination. J Neurosci. 2003;23:3469–3477. doi: 10.1523/JNEUROSCI.23-08-03469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla US. Signaling in small subcellular volumes. I. Stochastic and diffusion effects on individual pathways. Biophys J. 2004a;87:733–744. doi: 10.1529/biophysj.104.040469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla US. Signaling in small subcellular volumes. II. Stochastic and diffusion effects on synaptic network properties. Biophys J. 2004b;87:745–753. doi: 10.1529/biophysj.104.040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell KT. An efficient stochastic diffusion algorithm for modeling second messengers in dendrites and spines. J Neurosci Meth. 2006;157:142–153. doi: 10.1016/j.jneumeth.2006.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond CT, Maylie J, Adelman JP. SK channels in excitability, pacemaking and synaptic integration. Curr Opin Neurobiol. 2005;15:305–311. doi: 10.1016/j.conb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Häusser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Daniel H, Levenes C, Crepél F. Cellular mechanisms of cerebellar LTD. Trends Neurosci. 1998;21:401–407. doi: 10.1016/s0166-2236(98)01304-6. [DOI] [PubMed] [Google Scholar]
- Denk W, Sugimori M, Llinás R. Two types of calcium responses limited to single spines in cerebellar Purkinje cells. Proc Natl Acad Sci U S A. 1995;92:8279–8282. doi: 10.1073/pnas.92.18.8279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC. Circuits in the cerebellar control of movement. Proc Natl Acad Sci U S A. 1967;58:336–343. doi: 10.1073/pnas.58.1.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers J, Augustine GJ, Konnerth A. Subthreshold synaptic Ca2+ signalling in fine dendrites and spines of cerebellar Purkinje neurons. Nature. 1995b;373:155–158. doi: 10.1038/373155a0. [DOI] [PubMed] [Google Scholar]
- Eilers J, Schneggenburger R, Konnerth A. Patch clamp and calcium imaging in brain slices. In: Sakmann B, Neher E, editors. Single-Channel Recording. New York: Plenum Press; 1995a. pp. 213–229. [Google Scholar]
- Eilers J, Takechi H, Finch EA, Augustine GJ, Konnerth A. Local dendritic Ca2+ signaling induces cerebellar long-term depression. Learn Mem. 1997;3:159–168. doi: 10.1101/lm.4.1.159. [DOI] [PubMed] [Google Scholar]
- Faas GC, Lee W, Vergara J, Adelman J, Mody I. Calcium binding kinetics of calmodulin. Abstract Viewer/Itinerary Planner. 2004 Program No. 165.5. Washington, DC: Society for Neuroscience. Online. [Google Scholar]
- Fierro L, Llano I. High endogenous calcium buffering in Purkinje cells from rat cerebellar slices. J Physiol. 1996;496:617–625. doi: 10.1113/jphysiol.1996.sp021713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ. Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature. 1998;396:753–756. doi: 10.1038/25541. [DOI] [PubMed] [Google Scholar]
- Franks KM, Sejnowski TJ. Complexity of calcium signaling in synaptic spines. Bioessays. 2002;24:1130–1144. doi: 10.1002/bies.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble E, Koch C. The dynamics of free calcium in dendritic spines in response to repetitive synaptic input. Science. 1987;236:1131–1135. doi: 10.1126/science.3495885. [DOI] [PubMed] [Google Scholar]
- Gillespie DT. Approximate accelerated stochastic simulation of chemically reacting systems. J Chem Physics. 2001;115:1716–1733. [Google Scholar]
- Harris KM, Stevens JK. Dendritic spines of rat cerebellar Purkinje cells: serial electron microscopy with reference to their biophysical characteristics. J Neurosci. 1988;8:4455–4469. doi: 10.1523/JNEUROSCI.08-12-04455.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartell NA. Parallel fiber plasticity. Cerebellum. 2002;1:3–18. doi: 10.1080/147342202753203041. [DOI] [PubMed] [Google Scholar]
- Heim N, Griesbeck O. Genetically encoded indicators of cellular calcium dynamics based on troponin C and green fluorescent protein. J Biol Chem. 2004;279:14280–14286. doi: 10.1074/jbc.M312751200. [DOI] [PubMed] [Google Scholar]
- Holmes WR. Models of calmodulin trapping and CaM kinase II activation in a dendritic spine. J Comput Neurosci. 2000;8:65–85. doi: 10.1023/a:1008969032563. [DOI] [PubMed] [Google Scholar]
- Holthoff K, Tsay D, Yuste R. Calcium dynamics of spines depend on their dendritic location. Neuron. 2002;33:425–437. doi: 10.1016/s0896-6273(02)00576-7. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Siegelbaum SA. Synaptic integration. In: Kandel ER, Schwartz JH, Jessell TM, editors. Principles of Neural Science. New York: Elsevier; 2000. pp. 207–229. [Google Scholar]
- Kim SA, Heinze KG, Waxham MN, Schwille P. Intracellular calmodulin availability accessed with two-photon cross-correlation. Proc Natl Acad Sci U S A. 2004;101:105–110. doi: 10.1073/pnas.2436461100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz J. Principles of Fluorescence Spectroscopy. New York: Plenum; 1999. [Google Scholar]
- Lee S-H, Schwaller B, Neher E. Kinetics of Ca2+ binding to parvalbumin in bovine chromaffin cells: implications for [Ca2+] transients of neuronal dendrites. J Physiol. 2000;525:419–432. doi: 10.1111/j.1469-7793.2000.t01-2-00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linse S, Helmersson A, Forsen S. Calcium binding to calmodulin and its globular domains. J Biol Chem. 1991;266:8050–8054. [PubMed] [Google Scholar]
- Majewska A, Brown E, Ross J, Yuste R. Mechanisms of calcium decay kinetics in hippocampal spines: role of spine calcium pumps and calcium diffusion through the spine neck in biochemical compartmentalization. J Neurosci. 2000;20:1722–1734. doi: 10.1523/JNEUROSCI.20-05-01722.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Roth A, Helmchen F. Competitive calcium binding: implications for dendritic calcium signaling. J Comput Neurosci. 1998;5:331–348. doi: 10.1023/a:1008891229546. [DOI] [PubMed] [Google Scholar]
- Müller W, Connor JA. Dendritic spines as individual neuronal compartments for synaptic Ca2+ responses. Nature. 1991;354:73–76. doi: 10.1038/354073a0. [DOI] [PubMed] [Google Scholar]
- Müller A, Kukley M, Stausberg P, Beck H, Müller W, Dietrich D. Endogenous Ca2+ buffer concentration and Ca2+ microdomains in hippocampal neurons. J Neurosci. 2005;25:558–565. doi: 10.1523/JNEUROSCI.3799-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Some quantitative aspects of calcium fluorimetry. In: Yuste R, Lenny F, Konnerth A, editors. Imaging Neurons: a Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; 1999. pp. 31.1–31.11. [Google Scholar]
- Noguchi J, Matsuzaki M, Ellis-Davies GC, Kasai H. Spine-neck geometry determines NMDA receptor-dependent Ca2+ signaling in dendrites. Neuron. 2005;46:609–622. doi: 10.1016/j.neuron.2005.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchalka J, Kierzek AM. Bridging the gap between stochastic and deterministic regimes in the kinetic simulations of the biochemical reaction networks. Biophys J. 2004;86:1357–1372. doi: 10.1016/S0006-3495(04)74207-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Maravall M, Svoboda K. Ca2+ signaling in dendritic spines. Curr Opin Neurobiol. 2001;11:349–356. doi: 10.1016/s0959-4388(00)00218-x. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca2+ ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- Santamaria F, Wils S, De Schutter E, Augustine GJ. Anomalous diffusion in Purkinje cell dendrites caused by spines. Neuron. 2006;52:635–648. doi: 10.1016/j.neuron.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Brown EB, Schwaller B, Eilers J. Diffusional mobility of parvalbumin in spiny dendrites of cerebellar Purkinje neurons quantified by fluorescence recovery after photobleaching. Biophys J. 2003a;84:2599–2608. doi: 10.1016/S0006-3495(03)75065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Schwaller B, Eilers J. Calbindin D28k targets myo-inositol monophosphatase in spines and dendrites of cerebellar Purkinje neurons. Proc Natl Acad Sci U S A. 2005;102:5850–5855. doi: 10.1073/pnas.0407855102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Stiefel K, Racay P, Schwaller B, Eilers J. Mutational analysis of dendritic Ca2+ kinetics in rodent Purkinje cells: role of parvalbumin and calbindin D28k. J Physiol. 2003b;551:13–32. doi: 10.1113/jphysiol.2002.035824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B, Dick J, Dhoot G, Carroll S, Vrbova G, Nicotera P, Pette D, Wyss A, Bluethmann H, Hunziker W, Celio MR. Prolonged contraction-relaxation cycle of fast-twitch muscles in parvalbumin knockout mice. Am J Physiol Cell Physiol. 1999;276:C395–C403. doi: 10.1152/ajpcell.1999.276.2.C395. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Tank DW, Denk W. Direct measurement of coupling between dendritic spines and shafts. Science. 1996;272:716–719. doi: 10.1126/science.272.5262.716. [DOI] [PubMed] [Google Scholar]
- Takechi H, Eilers J, Konnerth A. A new class of synaptic responses involving calcium release in dendritic spines. Nature. 1998;396:757–760. doi: 10.1038/25547. [DOI] [PubMed] [Google Scholar]
- Varga AW, Yuan L-L, Anderson AE, Schrader LA, Wu G-Y, Gatchel JR, Johnston D, Sweatt JD. Calcium-calmodulin-dependent kinase II modulates Kv4.2 channel expression and upregulates neuronal A-type potassium currents. J Neurosci. 2004;24:3643–3654. doi: 10.1523/JNEUROSCI.0154-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecellio M, Schwaller B, Meyer M, Hunziker W, Celio MR. Alterations in Purkinje cell spines of calbindin D-28 k and parvalbumin knock-out mice. Eur J Neurosci. 2000;12:945–954. doi: 10.1046/j.1460-9568.2000.00986.x. [DOI] [PubMed] [Google Scholar]
- Wang SS-H, Denk W, Häusser M. Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci. 2000;3:1266–1273. doi: 10.1038/81792. [DOI] [PubMed] [Google Scholar]
- Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci. 2005;6:267–276. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- Zador A, Koch C, Brown TH. Biophysical model of a Hebbian synapse. Proc Natl Acad Sci U S A. 1990;87:6718–6722. doi: 10.1073/pnas.87.17.6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.