

Abstract
We have recently shown that the IK1 and maxi-K channels in parotid salivary gland acinar cells are encoded by the KCa3.1 and KCa1.1 genes, respectively, and in vivo stimulated parotid secretion is severely reduced in double-null mice. The current study tested whether submandibular acinar cell function also relies on these channels. We found that the K+ currents in submandibular acinar cells have the biophysical and pharmacological footprints of IK1 and maxi-K channels and their molecular identities were confirmed by the loss of these currents in KCa3.1- and KCa1.1-null mice. Unexpectedly, the pilocarpine-stimulated in vivo fluid secretion from submandibular glands was essentially normal in double-null mice. This result and the possibility of side-effects of pilocarpine on the nervous system, led us to develop an ex vivo fluid secretion assay. Fluid secretion from the ex vivo assay was substantially (about 75%) reduced in animals with both K+ channel genes ablated – strongly suggesting systemic complications with the in vivo assay. Additional experiments focusing on the membrane potential in isolated submandibular acinar cells revealed mechanistic details underlying fluid secretion in K+ channel-deficient mice. The membrane potential of submandibular acinar cells from wild-type mice remained strongly hyperpolarized (−55 ± 2 mV) relative to the Cl− equilibrium potential (−24 mV) during muscarinic stimulation. Similar hyperpolarizations were observed in KCa3.1- and KCa1.1-null mice (−51 ± 3 and −48 ± 3 mV, respectively), consistent with the normal fluid secretion produced ex vivo. In contrast, acinar cells from double KCa3.1/KCa1.1-null mice were only slightly hyperpolarized (−35 ± 2 mV) also consistent with the ex vivo (but not in vivo) results. Finally, we found that the modest hyperpolarization of cells from the double-null mice was maintained by the electrogenic Na+,K+-ATPase.
Mammals express three major paired salivary glands, the parotid, submandibular and sublingual. The morphology, histology and saliva composition are unique for each gland type (Young & Van Lennep, 1977). These differences are most evident in the secretory endpieces, also known as acini, the cells which secrete the water and most of the protein found in saliva. In rodents, the parotid acinar cells are serous, while submandibular acinar cells are seromucous. In contrast, the secretory endpieces of sublingual glands are mixed in nature, containing both mucous and serous demilune cells. The composition of the saliva collected from individual glands varies according to the acinar cell type. Parotid glands produce a watery fluid and sublingual glands secrete a thick, ropey saliva due to the exocytosis of heavily glycosylated mucins, whereas submandibular saliva has an intermediate consistency.
The mechanism of fluid secretion is generally studied at two levels: (i) the cellular level gives detailed information under well controlled conditions but is an over-simplified model and (ii) the in vivo level provides access to whole organ function, but the interpretation of results is complicated by systemic effects. In the latter case, saliva secretion is often induced by cholinergic agonists such as pilocarpine. It is well established that pilocarpine can stimulate saliva secretion through glandular muscarinic receptors. However, systemic administration of a cholinergic agonist also activates receptors in the brain and peripheral nervous system, stimulating saliva secretion through efferent innervation of the glands (Renzi et al. 1993; Cecanho et al. 1999; Renzi et al. 2002; Takakura et al. 2003). These systemic effects can be circumvented by studying surgically isolated salivary glands. Such ex vivo assays give control over the composition of vascular perfusate including the agonist concentration. Ex vivo, perfused submandibular glands from the rabbit and the rat are widely used model to study the mechanism of salivary secretion (e.g. Lau et al. 1990; Ishikawa et al. 1994), but this ex vivo model has not been previously applied to mice. Here we implement and directly compare the in vivo and ex vivo models in mice to confirm the details of the fluid secretion mechanism predicted at the single cell level.
The currently accepted model for Cl−-dependent fluid secretion by acinar cells (reviewed by Cook et al. 1994; Melvin et al. 2005) postulates a key role for Ca2+-activated K+ channels for maintaining the membrane potential negative to the Nernst potential for Cl−. Muscarinic stimulation initiates fluid secretion by activation of apical Ca2+-dependent Cl− channels, which mediate Cl− efflux into the acinar lumen. The associated charge movement produces a lumen-negative transepithelial potential that drives paracellular Na+ transport through tight junctions. The resulting luminal accumulation of NaCl generates an osmotic gradient necessary for transepithelial movement of water to produce a plasma-like primary secretion. Sustained secretion requires that the intracellular [Cl−] remains above its electrochemical equilibrium. Cytosolic Cl− is accumulated predominantly through a basolateral electroneutral Na+/K+/2Cl− cotransport mechanism, which is dependent on the inwardly directed Na+ gradient created by the Na+,K+-ATPase. Efflux of Cl− via apical channels depolarizes the cell membrane. Thus, it has been proposed that coordinated opening of basolateral Ca2+-activated K+ channels mediates outward K+ current to maintain the electrical driving force necessary for sustained Cl− secretion.
Previous studies provided molecular and electrophysiological evidence for two types of Ca2+-activated K+ channels in parotid salivary gland acinar cells, the intermediate conductance (IK1) and large conductance (maxi-K) channels (Maruyama et al. 1983; Wegman et al. 1992; Park et al. 2001; Nehrke et al. 2003; Takahata et al. 2003; Begenisich et al. 2004; Romanenko et al. 2006). The properties of IK1 channels are: (i) time- and voltage-independent currents, (ii) activation by submicromolar intracellular Ca2+ and by dcEBIO, (iii) inhibition by clotrimazole and TRAM-34, and (iv) 20–40 pS unitary conductance (Ishii et al. 1997; Logsdon et al. 1997; Jorgensen et al. 1999; Begenisich et al. 2004). The properties of maxi-K channels are also characteristically unique: (i) strong time dependence of the currents, (ii) strong outward rectification regulated by intracellular Ca2+, (iii) inhibition by paxilline, and (iv) large single-channel conductance of 100–300 pS (Latorre et al. 1989; Pallanck & Ganetzky, 1994; Nehrke et al. 2003; Salkoff et al. 2006) (150–200 pS in mouse parotid acinar cells; Nehrke et al. 2003; Thompson & Begenisich, 2006). Another distinctive feature of the maxi-K channel is its inhibition by IK1 current activation, which was demonstrated in parotid acinar cells as well as in a heterologous expression system (Romanenko et al. 2006; Thompson & Begenisich, 2006). The molecular identities of IK1 and maxi-K channels in parotid acinar cells were confirmed in mice in which the KCa3.1 and KCa1.1 genes, respectively, were disrupted (Begenisich et al. 2004; Romanenko et al. 2006). Surprisingly, loss of either IK1 or maxi-K channel expression did not impair parotid gland fluid secretion. However, disruptions of both the KCa3.1 and KCa1.1 genes produced a substantial (> 70%) reduction in secretion by mouse parotid glands (Romanenko et al. 2006).
Two Ca2+-activated K+ currents with properties similar to maxi-K channels are observed in mouse submandibular acinar cells (e.g. Maruyama et al. 1983). IK1-like currents have also been characterized in rat submandibular acinar cells (Ishikawa et al. 1994; Ishikawa & Murakami, 1995; Hayashi et al. 2004), and maxi-K currents were found in human submandibular acinar cells (Morris et al. 1987). Here we have determined the physiological and pharmacological properties of the Ca2+-activated IK1 and maxi-K channels in mouse submandibular acinar cells and verified their molecular identities using KCa3.1-, KCa1.1- and double-null mice. We found that the membrane potential (Vm) of submandibular cells from KCa3.1-null, KCa1.1-null, and wild-type mice remained strongly hyperpolarized during stimulation. In contrast, the Vm in the double KCa3.1/KCa1.1-null animals was only slightly more negative than the Cl− equilibrium potential. The ex vivo fluid secreted by perfused submandibular glands, but not in vivo secretion, correlated with the electrophysiological measurements in the acinar cells from the K+ channel-deficient mice, i.e. secretion was significantly impaired only in double-null mice. Finally, in the absence of both K+ channels, the electrogenic Na+,K+-ATPase generated a limited hyperpolarization sufficient to drive minimal fluid secretion.
Methods
Mouse strains
The KCa1.1-null mice (Meredith et al. 2004) were a generous gift from R. W. Aldrich. The KCa3.1-null and the double-null, KCa1.1/KCa1.1-null mice generated by us have been previously described (Begenisich et al. 2004; Romanenko et al. 2006). Results obtained with these various K+-channel deficient mice were compared with those found in their age- (2–6 months old) and sex-matched wild-type littermates. Mice were rendered unconscious either by exposure to CO2 or by intraperitoneal injection of chloral hydrate (400 mg (kg body weight)−1). Unconscious mice were then killed by exsanguination prior to removal of the submandibular glands or after in vivo saliva collection. All experimental procedures were approved by the Animal Resources Committee of the University of Rochester. All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise stated.
In vivo stimulated fluid secretion
Mice were anaesthetized by intraperitoneal injection of chloral hydrate prior to saliva collection. Secretion was stimulated by intraperitoneal injection of the cholinergic receptor agonist pilocarpine-HCl (10 mg kg−1). Gland-specific saliva was collected by isolating the ducts from the submandibular glands and inserting their distal ends into individual calibrated glass capillary tubes (Sigma-Aldrich) as described by Evans et al. (2000). Body temperature was maintained at 37°C using a regulated blanket (Harvard Apparatus, Holliston, MA, USA) and the trachea was incised to insure a patent airway. The progression of saliva within the capillary tube was recorded at 5 min intervals. At the end of the saliva collection period, the glands were dissected, blotted dry and weighed. Secreted saliva was expressed as microlitres of saliva per 100 mg gland weight. For each individual mouse the saliva secreted from left and right submandibular glands was averaged.
Ex vivo perfused submandibular glands
The mouse submandibular gland was perfused as previously described for rat and rabbit submandibular glands (Lau et al. 1990; Murakami et al. 1990; Ishikawa et al. 1994). Briefly, mice were anaesthetized with chloral hydrate (400 mg kg−1i.p.) and all branches of the common carotid artery were ligated with the exception of the submandibular artery. Blood vessels supplying the main duct of the submandibular gland were also ligated to avoid contamination of the collected saliva by the perfusate. The gland was transferred to a perfusion chamber where the common carotid was cannulated (Microfil; 31 G WPI) and perfused with a solution containing (mm): 4.3 KCl, 125 NaCl, 25 NaHCO3, 5 glucose, 10 Hepes, 1 CaCl2 and 1 MgCl2. The solution was gassed with 95% O2–5% CO2 for at least 30 min and then titrated to pH 7.4 with NaOH just prior to perfusion. The final solution was perfused at a flow rate of ∼0.8 ml min−1 using a peristaltic pump (Ismatec IPC). Measurements were done at room temperature (20–22°C). Saliva was collected from the perfused gland by inserting the main duct into a calibrated glass capillary tube (Sigma-Aldrich). Salivation was stimulated by perfusion with the cholinergic receptor agonist carbachol (0.5 μm CCh). The progression of saliva within the capillary tube was recorded every minute. At the end of saliva collection, the glands were blotted dry and weighed. Flow rate was expressed as microlitres of saliva per 100 mg gland weight per minute. For each individual mouse the saliva flow rate from left and right submandibular glands was averaged.
Dispersed salivary cells
Single salivary gland acinar cells were prepared essentially as previously reported (Nehrke et al. 2003). Briefly, mice were killed by exsanguination following exposure to CO2. Glands were finely minced and digested for 5 min in Earle's minimal essential medium (EMEM, Biofluid) containing 0.02% trypsin and 0.17 mg ml−1 Liberase RI Enzyme (Roche Applied Science, Indianapolis, IN, USA), then centrifuged and the cell pellet rinsed in medium containing trypsin inhibitor, followed by an additional 20 min of digestion in Liberase RI. Acinar cells were attached to poly l-lysine-coated glass coverslips.
Electrophysiological recording
Electrophysiological data were acquired using Axopatch 200B amplifier and Digidata 1320A digitizer (Axon Instruments, Union City, CA, USA). Pipettes from Corning 8161 patch glass (Warner Instruments, Hamden, CT, USA) were pulled to give a resistance of 2–3 MΩ in the solutions described below. Macroscopic K+ currents were measured in whole-cell and perforated-patch experiments. In both types of experiments the external solution contained (mm): 150 Na-glutamate, 5 K-glutamate, 2 CaCl2, 2 MgCl2, 10 Hepes, pH 7.2. For whole-cell experiments the pipette solution contained (mm): 135 K-glutamate, 5 EGTA, 3 CaCl2, 10 Hepes, pH 7.2. The level of free Ca2+ was 250 nm as calculated by WEBMAXC (http://www.stanford.edu/~cpatton/webmaxcS.htm). For perforated-patch experiments the pipette solution contained (mm): 135 K-glutamate, 2 CaCl2, 2 MgCl2, 10 Hepes, pH 7.2. Liquid junction potential for such buffers was small (< 4 mV; Romanenko et al. 2006) and no corrections were made. For perforated-patch experiments K+ currents were measured in cells at rest or during stimulation with 0.3 μm CCh. Single channel experiments were done in the inside-out excised patch configuration. The bath solution consisted of (mm) 135 K-glutamate, 5 EGTA, 3 CaCl2, 10 Hepes, pH 7.2 and the pipette solution contained (mm): 135 K-glutamate, 2 CaCl2, 2 MgCl2, 10 Hepes, pH 7.2.
The membrane potential of the acinar cells was recorded using the perforated-patch in current-clamp mode. The tip of the pipette was front-filled with (mm): 95 K-methanesulphonate, 45 KCl, 15 NaCl, 1 MgCl2, 5 BAPTA, 10 Hepes (pH 7.2) and then back-filled with the same solution supplemented with 250 μg ml−1 nystatin and 2 mm Lucifer Yellow. Initially, the bath was filled with the pipette solution, and with the pipette in this solution, the voltage was zeroed. Immediately after obtaining the gigaseal the cells were equilibrated in (mm): 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 10 Hepes, pH 7.2. The access resistance declined to 5–15 MΩ in less than 10 min. With this method, there will be an insignificant uncorrected junction potential. Exclusion of Lucifer Yellow fluorescence from the patched cells confirmed that the perforated patch remained intact throughout the experiment. In some cells, total membrane conductance was determined by application of short current pulses (10 or 20 pA, 10 ms long, 2.5 Hz) and measuring the Vm deflections. The membrane potential and membrane conductance were measured in cells at rest or during stimulation with 0.3 μm CCh.
The mean cell capacitances of cells from single- and double-null mice were not significantly different from the capacitance of the wild-type cells: 19 ± 1 pF (n = 10), 17 ± 3 pF (n = 10), 20 ± 2 pF (n = 10) and 19 ± 1 pF (n = 10) for cells from wild-type, KCa3.1-, KCa1.1- and double-null mice, respectively.
Most of the cells stimulated with CCh responded with oscillating membrane potentials (see text). Membrane potential data were recorded at 2.5 kHz and the slowly oscillating signals (near 0.3 Hz) were resampled at 40 ms intervals. The oscillating membrane voltage was averaged (omitting the initial ‘spike’– see text and Fig. 5) over the period of the CCh application (20–60 s). The membrane potentials of resting cells and the few cells that did not oscillate in CCh were averaged over a 20 s interval.
Figure 5. Membrane potential of submandibular acinar cells from wild-type and KCa3.1-, KCa1.1- and double-null mice.
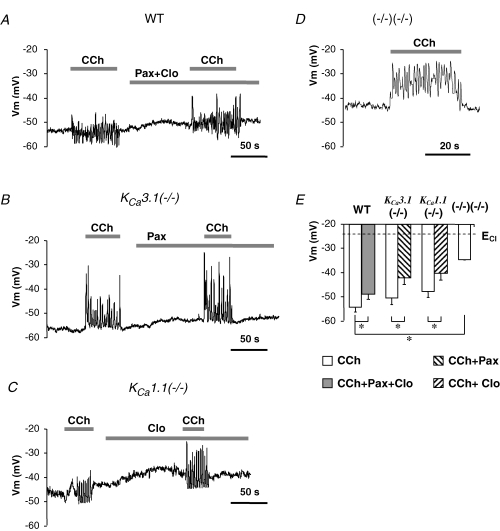
Cell were stimulated with 0.3 μm CCh with or without K+ channel blockers (with 1 μm paxillin, with 3 μm clotrimazole, or with 1 μm paxilline and 3 μm clotrimazole). A, membrane potential of a submandibular acinar cell from wild-type mice recorded in perforated-patch configuration. B–D, membrane potentials recorded in cells from KCa3.1-, KCa1.1- and double-null mice. E, time-averaged mean membrane potentials in cells from wild-type or mutant mice as indicated. *P < 0.05.
In some experiments, the K+ channel inhibitors paxilline (1 μm), TRAM-34 (1 μm) and clotrimazole (3 μm), or the Na+,K+-ATPase inhibitor ouabain (1 mm) were added to the external solutions. All data are presented as the mean ±s.e.m. Differences between means were determined by Student's paired t test with P < 0.05 considered significant. Electrophysiological experiments were done at room temperature. The osmolarity of solutions was measured using a Wescor 5500 vapour pressure osmometer (Wescor, Logan, UT, USA) and adjusted to isotonic with sucrose.
RNA isolation and RT-PCR analysis
Submandibular glands were dissected from wild-type and knockout mice and immediately frozen in liquid N2. Total RNA was extracted from about 20 mg of tissue, according to the manufacturer's protocol (RNeasy kit, Qiagen, Valencia, CA, USA). First-strand cDNA was synthesized from 1 μg of total RNA in a 20 μl reaction using the iScript™ cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). PCR was performed using primers to KCa1.1 (forward, 5-ATGCAGTTTGATGACAGCATCG-3; reverse, 5-CAGATCACCATAACAACCACCA-3) and to KCa3.1 (forward, 5-ACGTGCACAACTTCATGATGG-3; reverse, 5-TGGGTACTTAGCCTGGTTCTTCTG-3) resulting in 510 bp and 434 pb products for KCa1.1 and KCa3.1, respectively. For quantitative RT-PCR, transcripts for KCa1.1, KCa3.1 and house keeping gene L32 were quantified using gene specific primers, KCa3.1 (5′-CCCA-CTACACATATGCCCTCATC-3′, 5′-TCTCTGCAATCAG-GAGGAAACG-3′), KCa1.1 (5′-GCCCAGTTAGCCC-TGTTAGATG-3′, 5′-AGAAGATCAGGTCTGTTGGTAC-G-3′), L32 (5′-TTCATCAGGCACCAGTCAGACC-3′, 5′-ACACAAGCCATCTACTCATTTTCTTCG-3′). Quantitative RT-PCR was performed in triplicate using a 96-well iCycler IQ™ Real Time PCR Detection System (Bio-Rad) in 25 μl of SYBR Green Master Mix containing 0.5 μl of the above synthesized cDNA and 2 ng μl−1 of gene specific primers. Specificity of the amplification was determined by melting curve analysis (60°C for 10 s to 95°C in 0.5°C, 10 s increments) and by gel electrophoresis. PCR efficiency of the reaction was measured with L32 primers using serial dilution of cDNA (1, 1 : 2, 1 : 4, 1 : 8 and 1 : 16). PCR amplification curves were analysed with iCycler IQ Software version 3.1.7050. Relative quantities of each transcript were determined by comparing the cycle threshold value (Ct) of KCa1.1 and KCa3.1 in different samples after normalization to the L32 content of individual samples.
Results
Ca2+-activated K+ channels in submandibular acinar cells
Previous studies suggested that mouse submandibular acinar cells express two major K+ channels which are Ca2+ activated: the intermediate-conductance (IK1) and large-conductance (maxi-K) channels (Hayashi et al. 1996; Hayashi et al. 2004). In order to verify the molecular identities of these two channels we characterized their biophysical, pharmacological and genetic properties in native tissue. Figure 1A shows expression in the mouse submandibular gland of mRNAs from both the KCa3.1 and KCa1.1 genes, which have been proposed to encode the IK1 and maxi-K channel proteins, respectively. Additionally, using quantitative RT-PCR (data not shown) we found that the transcript levels of the KCa3.1 and KCa1.1 genes are similar in submandibular glands from all three wild-type animals used (see Methods).
Figure 1. Analysis of whole-cell and single channel K+ currents in wild-type submandibular acinar cells.
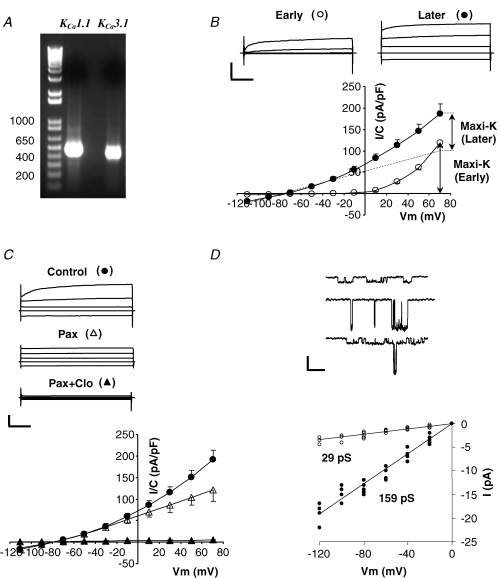
A, expression of mRNA of IK1 and maxi-K channels in submandibular glands from wild-type mice was determined using RT-PCR analysis. B, whole-cell currents recorded with 250 nm Ca2+ in the pipette. Currents were recorded immediately (10–20 s) after achieving whole-cell mode (Early) and after a steady level of IK1 current developed (Later, 5–10 min). Top panel, typical raw currents in response to 40 ms voltage pulses to −110, −30, +10 and +50 mV from −70 mV holding potential. Calibration bars are 1 nA and 10 ms. Bottom panel, mean current densities determined at the end of the 40 ms pulses to the indicated potentials at Early (^) and Later (•) times (n = 12). Dashed line represents the voltage-independent (IK1-like) component. C, sensitivity of whole-cell currents to the maxi-K channel inhibitor paxilline and the IK1 channel inhibitor clotrimazole. Raw traces and I–V plot are presented in the same format as in B; •– no inhibitors, ▵– with 1 μm paxillin, ▴– with 1 μm paxilline and 3 μm clotrimazole (n = 7). D, top panel, single channel recordings from 3 inside-out patches exposed to 250 nm Ca2+ with either IK1 (top patch) or maxi-K (middle patch), or both (bottom patch) channels present. Bottom panel, I–V plot of single channel IK1 (^) and maxi-K (•) currents at the indicated potentials (n = 7 cells).
Using the whole-cell and single-channel techniques, we examined whether two Ca2+-activated K+ channels are functionally active in mouse submandibular acinar cells. Figure 1B shows currents recorded with the pipette solution buffered to 250 nm of free Ca2+, a concentration within the physiological range of the free Ca2+ levels observed in acinar cells during stimulation (Foskett et al. 1989; Foskett & Melvin, 1989; Tojyo et al. 1998). The currents recorded immediately after the whole-cell configuration was established (‘Early’) had strong time and voltage dependence (open circles), characteristic for maxi-K channels. At potentials more negative than −30 mV the strongly rectifying maxi-K-like current was not active. The mean value of the maxi-K-like current immediately after breakthrough was 119 ± 8 pA pF−1 at +70 mV (n = 12). Within 2–5 min (‘Later’) voltage- and time-independent IK1-like currents developed (filled circles). Previously we have demonstrated a unique interaction of IK1 and maxi-K channels in mouse parotid acinar cells and in a heterologous expression system: activation of IK1 channels inhibits maxi-K activity (Thompson & Begenisich, 2006). Accordingly, examination of the voltage-dependent component of the currents before and after development of IK1-like current (see the current–voltage (I–V) plot) demonstrated the expected current interaction in the acinar cells from mouse submandibular glands. The mean value of the maximally developed IK1-like current determined 8 ± 2 min after break-in was −14 ± 4 pA pF−1 at −110 mV (n = 12). Because IK1 has time-independent current, the maxi-K-like current was estimated as the time-dependent component over a 40 ms voltage pulse to +70 mV. After stable development of IK1-like current, the maxi-K-like current was significantly down-regulated by 27 ± 9% to 87 ± 15 pA pF−1 (n = 12, P < 0.05).
As illustrated in Fig. 1C, the time- and voltage-dependent current component was highly sensitive to the specific maxi-K channel inhibitor paxilline (open triangles), whereas the remaining time- and voltage-independent current was effectively inhibited by the specific IK1 inhibitors clotrimazole (filled triangles) and TRAM-34 (not shown). The time- and voltage-independent current component could also be activated by an IK1-specific activator dcEBIO when intracellular free Ca2+ was kept low (80 nm; not shown).
We determined single channel conductances of these K+ currents in the excised inside-out patch configuration. Two conductances in the expected range for IK1 (29 pS) and maxi-K (159 pS) were observed (Fig. 1D). Altogether, our results are consistent with the presence of two Ca2+-activated K+ currents in mouse submandibular acinar cells with the characteristic pharmacological and electrophysiological properties of IK1 and maxi-K channels.
To verify the molecular identities of the Ca2+-activated K+ currents in submandibular acinar cells we analysed the currents obtained from KCa3.1-, KCa1.1- and double-null mice. Disruption of these genes has been previously verified in these mice (Begenisich et al. 2004; Romanenko et al. 2006). Figure 2 shows representative K+ current traces and I–V plots in submandibular acinar cells from the three strains of mice. Figure 2A illustrates K+ currents in cells from KCa3.1-null mice with time- and voltage-dependent maxi-K-like properties that were apparent immediately after breakthrough (open circles). No IK1-like current was present in the KCa3.1-null mice even up to 22 min after establishing the whole-cell configuration (filled circles): the observed current of −2.1 ± 0.6 pA pF−1 at −110 mV (n = 7) is close to the minimal background current expected in patch clamp experiments with an average cell capacitance of 17 pF. The mean values for maxi-K-like currents were 231 ± 12 pA pF−1 and 268 ± 6 pA pF−1 at +70 mV (n = 7) in the beginning and at the end of the experiment, respectively, showing no decrease with time (i.e. inhibition due to IK1 activation; compare to Fig. 1B). Paxilline inhibited the current by more that 90% (filled triangles; n = 7). A comparison of maxi-K-like currents in cells from wild-type mice (Fig. 1B‘Early’) to the currents in cells from KCa3.1-null animals shows a 94% increase (119 ± 8 pA pF−1versus 231 ± 12 pA pF−1, respectively), possibly indicating compensation for the loss of IK1 expression.
Figure 2. Whole-cell submandibular acinar K currents in KCa3.1- and KCa1.1- and double-null mice.
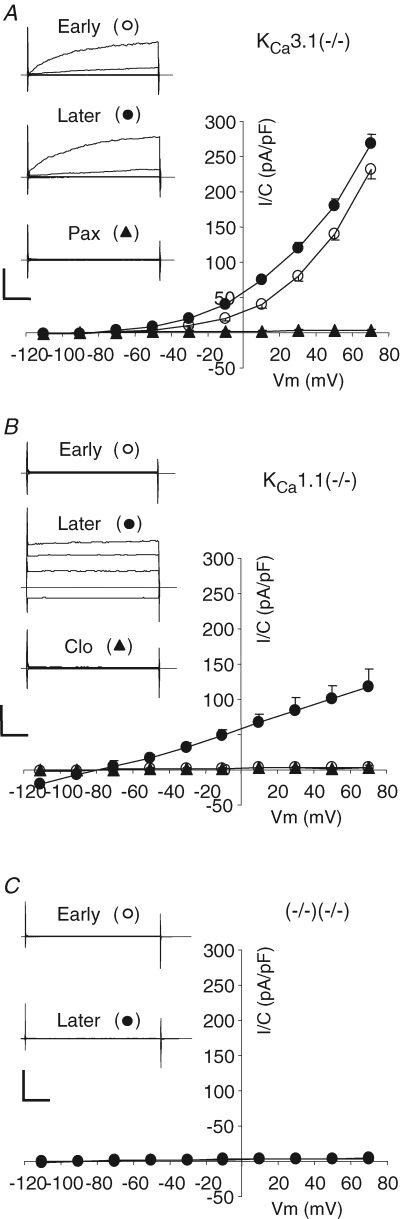
A, whole-cell currents in a cell from KCa3.1-null mouse recorded with 250 nm Ca2+ in the pipette. Currents were recorded immediately after achieving whole-cell mode (Early and ^) and after a steady level of IK1 current developed (Later and •) and after addition of 1 μm paxillin, a maxi-K channel inhibitor (▴). Typical raw traces and average I–V plot are presented in the same format as in Fig. 1B (n = 7). B and C, same as described in A but currents recorded in cells from KCa1.1-null mice (n = 5) and from double-null mice (n = 7). B, the identity of the developed current was tested by addition of 3 μm clotrimazole, the specific inhibitor of IK1 channel (▴).
The maxi-K-like current was absent in cells from KCa1.1-null mice; instead, only time- and voltage-independent and clotrimazole-sensitive K+ current was observed (Fig. 2B). Essentially no K+ current was apparent immediately after breakthrough (open circles) or in the presence of clotrimazole (filled triangles), and the mean value for fully developed IK1-like current was −19 ± 2 pA pF−1 at −110 mV (filled circles; n = 5), which was not significantly different from the current in the wild-type mice (−14 ± 4 pA pF−1; P = 0.17). Finally, Ca2+-activated K+ currents were essentially absent in the acinar cells from double-null mice (Fig. 2C). The absence of K+ currents was observed both immediately after establishing whole-cell configuration and more that 15 min afterwards. The mean values of currents measured at −110 and +70 mV were −1.9 ± 0.4 pA pF−1 and 2.9 ± 0.7 pA pF−1 (n = 7), respectively. Taken together, the results shown in Fig. 2 confirm the molecular identities of the Ca2+-activated IK1- and maxi-K-like currents in submandibular acinar cells as KCa3.1 and KCa1.1, respectively.
Muscarinic stimulation of IK1 and maxi-K channels
The perforated-patch technique preserves the integrity of the cell membrane and therefore is not accompanied by washout of multivalent and macromolecular constituents of the cytosol. Importantly, this method can measure the IK1 and maxi-K currents activated by physiologically relevant cytosolic free Ca2+ by stimulation with the muscarinic agonist carbachol. First, we recorded currents in submandibular acinar cells from wild-type mice (Fig. 3A and B). The IK1 current was determined during 10 ms voltage pulses to −100 mV (Fig. 3A, upper panel) and maxi-K activity was estimated as the time-dependent current during 40 ms steps to +30 mV (Fig. 3A, lower panel). These currents were monitored every 0.4 s from a holding potential of −60 mV. The current recordings were briefly interrupted (arrows) to obtain full I–V plots when the cells were ‘at rest’ or during stimulation with 0.3 μm CCh and with or without inhibitors (paxilline and clotrimazole; Fig. 3A and B). Consistent with the whole-cell results (Fig. 2), the IK1 current in wild-type mice was absent (with only minimal background current) before cell stimulation whereas a substantial maxi-K current was observed: −0.9 ± 0.7 pA pF−1 and 45 ± 11 pA pF−1, respectively (n = 7). During stimulation, the K+ currents in 5 out of 7 cells presented an oscillatory pattern with a frequency of 0.35 ± 0.07 Hz (n = 4; one additional cell showed irregular oscillations). Oscillations of K+ currents are likely to be the result of oscillation of the cytoplasmic free Ca2+ levels, which were often observed in stimulated parotid acinar cells (Gray, 1988; Bruce et al. 2002; Harmer et al. 2005). Maxi-K current oscillations were strictly antiphase with IK1 current oscillations (see online supplemental material, Fig. S1), as previously shown for parotid acinar cells (Thompson & Begenisich, 2006). The reversal potential during stimulation was −79 ± 8 mV, close to the calculated K+ equilibrium potential (−85 mV). At the end of each experiment, the cells were superfused with paxilline and clotrimazole, which suppressed both IK1 and maxi-K currents by more than 90%.
Figure 3. K+ currents in resting and stimulated cells from wild-type mice and KCa3.1- and KCa1.1-null mice.
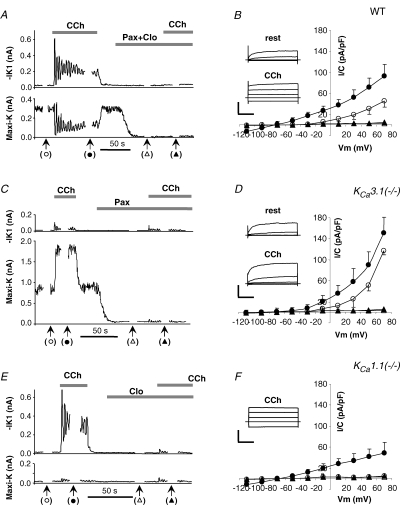
IK1 and maxi-K currents were simultaneously recorded using perforated-patch mode in cells from wild-type mice (A and B), KCa3.1-null (C and D) and KCa1.1-null (E and F) mice. For A, C and E, every 0.4 s IK1 currents (upper panels) were measured at −100 mV (10 ms voltage step) and maxi-K currents (lower panels) were determined as a time-dependent component of the current elicited by 40 ms steps to +30 mV from holding potential of −60 mV. At time points indicated by the arrows in A, C and E full I–V plots were obtained. The average I–V plots in B (n = 7), D (n = 6), and F (n = 4) show K+ currents before (^) and during stimulation with 0.3 μm CCh (•) as well as after blocking K+ channels without (▵) and with (▴) stimulation. Insets of B, D and F, raw currents in response to 40 ms voltage pulses to −110, −30, +10 and +50 mV from −70 mV holding potential. Calibration bars: 1, 2 and 0.5 nA for B, D and F, respectively, versus 10 ms.
Consistent with the above results (Fig. 2A), the IK1 current was absent in cells from KCa3.1-null mice and only maxi-K current was observed (Fig. 3C and D). The maxi-K current was 2.6 times bigger (117 ± 6 pA pF−1; n = 6) in resting KCa3.1-null cells than in wild-type cells. During stimulation with 0.3 μm CCh maxi-K current was strongly up-regulated but showed no resolvable oscillations. As in the beginning of the experiment, no IK1 current was observed when the KCa3.1-null cells were stimulated: −0.5 ± 0.8 pA pF−1 and −1.5 ± 1.1 pA pF−1, respectively (n = 6). In addition, paxilline effectively suppressed the maxi-K currents.
Next, we examined the K+ currents in the submandibular acinar cells from KCa1.1-null mice. Figure 3E and F shows a typical K+ current trace and I–V plot, respectively. Maxi-K current was absent and only IK1 current was observed. As in wild-type cells, stimulation elicited oscillations of IK1 current in KCa1.1-null cells. The time-averaged mean value of the currents was −11 ± 3 pA pF−1 (n = 4), similar to that in the wild-type cells (−14 ± 4 pA pF−1; P = 0.16) and suggesting that no compensation for loss of maxi-K occurred in KCa1.1-null mice. We completed the experiment by demonstrating strong inhibition of the IK1 currents with clotrimazole.
Finally, in the cells from the double-null mice we observed no significant K+ current both prior and during muscarinic stimulation. All currents were less than 3 pA pF−1 (n = 6, not shown).
Membrane potential and K+ and Cl− currents in submandibular acinar cells
As described in the Introduction, fluid secretion is thought to be dependent on both Ca2+-activated K+ and Cl− channels and requires that the Vm of acinar cells be negative to the Cl− equilibrium potential to provide the driving force for Cl− efflux. To understand the functional role of Ca2+-activated K+ channels in this process we used the perforated-patch technique to monitor the Vm as well as K+ and Cl− currents in the submandibular acinar cells from wild-type mice and from mice lacking IK1, maxi-K, or both channels. First, we measured the Vm of single submandibular acinar cells from wild-type mice both in the resting state and when stimulated with 0.3 μm CCh. Figure 4A and B shows representative examples of Vm traces. The mean Vm value in unstimulated cells was −55 ± 2 mV (n = 34). Stimulation with 0.3 μm CCh produced robust responses of two types: (i) 64% of the cells (22 out of 34) responded to stimulation with generally regular and sustained oscillations (Fig. 4A); (ii) the remaining cells produced a relatively flat Vm trace, with the exception of an initial depolarization spike (Fig. 4B, upper panel). The common feature of both types of responses to stimulation was an initial strong depolarization spike. After this initial depolarization, the Vm usually oscillated with an average amplitude of 8 ± 6 mV and a frequency of 0.38 ± 0.06 Hz (n = 17 cells with pronounced regular oscillations), which was similar to the K+ current oscillation frequency of 0.35 ± 0.07 Hz (see Fig. 3A). The time-averaged mean value of Vm oscillations was −55 ± 2 mV (n = 22), not significantly different from that measured prior to simulation. The Vm in non-oscillating cells, excluding the initial depolarization spike, was −58 ± 3 mV (n = 12), comparable to the mean value in oscillating cells. To insure that non-oscillating cells had activated ion channels beyond the initial depolarization response, we determined the total membrane conductance by periodic application of short current pulses and measuring the resulting Vm deflections. Figure 4B (lower panel) shows that the membrane conductance was, indeed, much higher throughout the stimulation period (210 ± 64 pS pF−1) than before and after stimulation (26 ± 3 pS pF−1) (n = 5).
Figure 4. Membrane potential, membrane conductance and K+ and Cl− currents in submandibular acinar cells from wild-type mice.
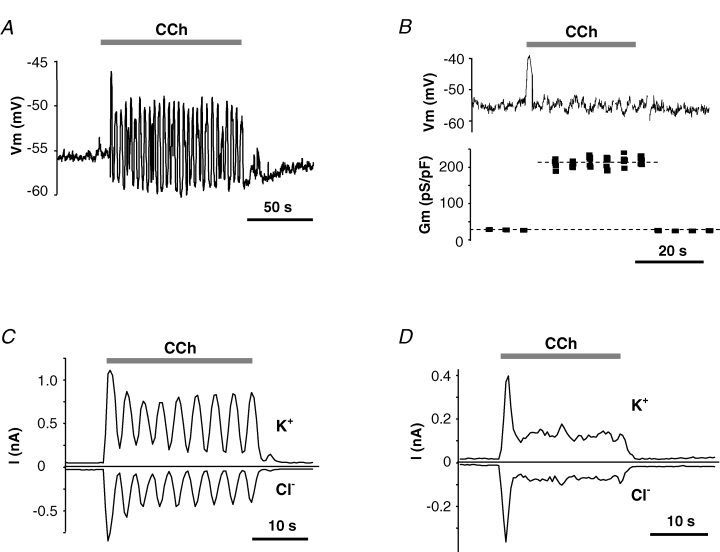
Membrane voltage and currents were recorded in perforated-patch configuration. Stimulation with 0.3 μm CCh induced either membrane oscillations (A) or non-oscillatory responses (B, upper panel) with concurrent increase in membrane conductance (B, lower panel). Typical current traces of ‘oscillatory’ (C) and ‘non-oscillatory’ (D) cells were recorded during 10 ms steps to either −25 mV (upper traces) or to −85 mV (lower traces) with 10 ms interpulse interval applied every 0.4 s from holding potential of −60 mV.
Next, we measured the currents that underlie the agonist-induced changes in Vm. K+ channels almost exclusively support cationic conductance of submandibular acinar cells as demonstrated by the near complete suppression of membrane conductance and the negative values of the reversal potentials when K+ channels were pharmacologically and/or genetically inhibited (Figs 1C, 2 and 3). Therefore, we evaluated the activity of the K+ and Cl− channels by measuring currents at the equilibrium potentials for Cl− (−24 mV) and for K+ (−85 mV), respectively. As with Vm measurements, most of the acinar cells (15 out of 24 cells) showed K+ and Cl− current oscillations (Fig. 4C and D). There was complete correlation between the presence (or absence) of Vm oscillations and K+ and Cl− current oscillations in the five cells tested (3 ‘oscillating’ and 2 ‘non-oscillating’ cells) when K+ and Cl− currents were measured immediately after Vm recording in the same cells. K+ and Cl− current oscillations were generally in phase and had a frequency of 0.31 ± 0.09 Hz (n = 5), which was similar to the frequency of Vm oscillations in these cells. The current densities oscillated around a mean value of 7.6 ± 0.7 pA pF−1 and −4.8 ± 0.4 pA pF−1 (n = 15) for K+ and Cl− currents, respectively. Figure 4D illustrates the K+ and Cl− currents in a cell with non-oscillating Vm. It shows an absence of current oscillations, indicating that synchronized cationic and anionic current oscillations did not result in the relatively flat Vm trace. The initial current spike, which probably caused the corresponding Vm spike, declined to a plateau where the average K+ and Cl− currents were 8.6 ± 2.1 pA and −5.5 ± 1.3 pA (n = 8), respectively, comparable to the time-averaged mean currents seen in oscillating cells.
The effects of pharmacological and/or genetic inhibition of IK1 and maxi-K currents on Vm are illustrated in Fig. 5. The mean values for the Vm in resting cells were −55 ± 3 mV (n = 25), −47 ± 2 mV (n = 20) and −47 ± 2 mV (n = 12) in cells from KCa3.1-, KCa1.1- and double-null mice, respectively. The Vm in KCa3.1-null and wild-type cells (−55 ± 2 mV, Fig. 4A) were the same. However, KCa1.1-null and double-null cells were slightly but significantly less hyperpolarized (P = 0.02 and P = 0.03 compared with wild-type cells). Stimulation with CCh, as in wild-type cells, elicited Vm oscillations in most cells from KCa3.1-null (86%), KCa1.1-null (86%) and double-null (91%) mice. The frequencies of oscillations were 0.32 ± 0.05 Hz (n = 11), 0.40 ± 0.10 Hz (n = 10) and 0.36 ± 0.07 Hz (n = 10) in cells from KCa3.1-, KCa1.1- and double-null mice, respectively, not significantly different from the value in wild-type animals (0.38 ± 0.06, Fig. 4A). The amplitude of oscillations increased by 55 ± 7%, 63 ± 9% and 175% in cells from KCa3.1-, KCa1.1- and double-null mice, respectively, compared to the amplitude in wild-type cells, suggesting that both K channels play a role in modulating the size of the Vm oscillation. The time-averaged mean values for the Vm in oscillating and non-oscillating cells were not statistically different and therefore were pooled. The mean Vm values during stimulation were not different amongst cells from wild-type (−54 ± 3 mV, n = 32) and single knockout mice (−51 ± 3 mV, n = 11; −48 ± 3 mV, n = 9 for KCa3.1- and KCa1.1-null cells, respectively); however, double-null cells were significantly less hyperpolarized (−39 ± 3 mV, n = 10) than cells from wild-type and single knockout mice.
At the end of each experiment, cells were superfused with the K+ channel inhibitors paxilline and/or clotrimazole. Despite strong inhibition of both IK1 and maxi-K channels (see Figs 1C, 2 and 3), the cells did not dramatically depolarize and maintained a time-average Vm significantly more negative than the equilibrium potential for Cl− in all cases (at rest and during stimulation). The mean Vm in non-stimulated cells from wild-type, KCa3.1- and KCa1.1-null mice were −50 ± 3 mV (n = 13), −49 ± 4 mV (n = 8) and −39 ± 5 mV (n = 5), respectively, and were not different from Vm values measured in the absence of the inhibitors (not shown). When treated with K+ channel inhibitors during stimulation, cells from wild-type and both single knockout mice were slightly, but significantly, less hyperpolarized compared to the cells before treatment with the inhibitors (summarized in Fig. 5E). The time-averaged mean Vm values during stimulation with K+ channels inhibited were −49 ± 3 mV (n = 9), −42 ± 3 mV (n = 5) and −40 ± 4 mV (n = 4) for wild-type, KCa3.1- and KCa1.1-null cells, respectively.
In vivo stimulated fluid secretion from submandibular glands
Taken together, the above experiments showed that simultaneous inhibition (pharmacological and/or genetic) of IK1 and maxi-K channels significantly depolarized the membrane voltage of submandibular acinar cells, although the Vm remained at least 10–15 mV more hyperpolarized than the Cl− equilibrium potential. In contrast, inhibition of only one K+ channel produced only modest Vm changes. This suggests that the driving force for Cl− efflux should be dramatically reduced in double knockouts, and thus, should appreciably decrease the amount of saliva generated by these mice. In our previous studies we demonstrated that null mutations of either the KCa3.1 or KCa1.1 gene (to disrupt expression of IK1 and maxi-K channels, respectively) did not impair saliva secretion in mouse parotid glands. However, secretion of saliva by double-null mutants, where parotid acinar cells were completely devoid of Ca2+-activated K+ currents, was severely reduced (Romanenko et al. 2006). Therefore, based on this study and the above results, we predicted that only one type of Ca2+-activated K+ channel is needed for normal fluid secretion in mouse submandibular glands. Indeed, in vivo secretion stimulated from KCa3.1-null or KCa1.1-null mice was comparable to the saliva produced by wild-type submandibular glands, as illustrated in Fig. 6A and B, respectively.
Figure 6. In vivo stimulated fluid secretion from submandibular glands.
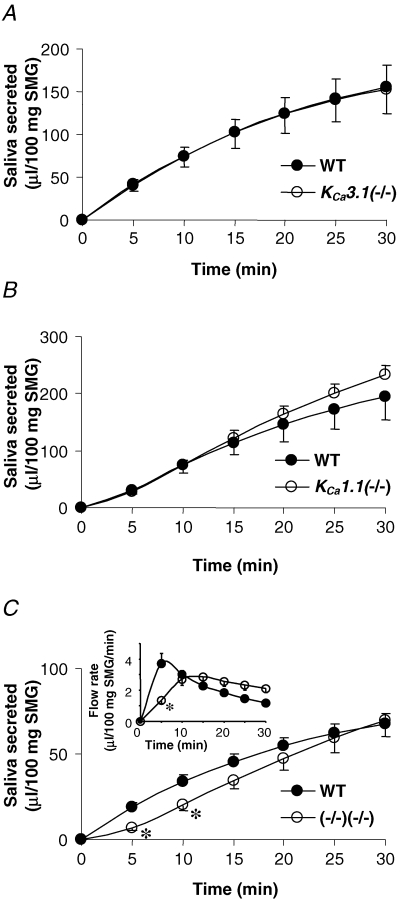
A, amount of saliva secreted over 30 min from KCa3.1-null (^) and wild-type (•) mice in response to 10 mg (kg body weight)−1 of pilocarpine (n = 11 and 10 mice, respectively). B, saliva secreted from KCa1.1-null (^) and wild-type (•) mice (n = 7 and 6 mice, respectively). C, volume of saliva secreted from KCa3.1/KCa1.1-double-null (^) and wild-type (•) mice (n = 7 and 11 mice, respectively). Inset, respective average saliva flow rates calculated for 5 min time intervals. *P < 0.05.
Surprisingly, simultaneous disruption of both the KCa3.1 and KCa1.1 genes did not result in the predicted impaired secretion by submandibular glands (Fig. 6C). This is in contrast to the > 70% inhibition of parotid gland secretion reported in KCa3.1/KCa1.1-double-null mice (Romanenko et al. 2006). While the submandibular glands in the double-null mice produced a significantly smaller quantity of saliva during the initial 10 min of stimulation, by 15 min, the saliva volume was similar to the amount in the control mice. The significant reduction of saliva volume generated during the initial 5 min of the experiment was the result of a lower flow rate in double knockout mice (Fig. 6C, inset). However, by 10 min the difference between the flow rates for wild-type and double knockout mice disappeared, and thereafter, the double knockouts secreted saliva at a slightly faster rate than the wild-type animals. Consequently, the total volume of saliva secreted by the double knockout mice over 30 min did not differ from their wild-type littermates.
It is important to note that wild-type littermates were used as the controls for the above experiments with the three mutant mice types. In the experiments with the KCa3.1/KCa1.1-double-null mice, the control was their hybrid wild-type littermates generated by crossing KCa3.1+/− and KCa1.1+/− mice. It was previously shown that the resulting hybrid wild-type mouse strain had significantly less parotid secretion than the parental wild-type stains (Romanenko et al. 2006). For submandibular glands, we also found that in vivo saliva secretion by the hybrid wild-type mouse was about half the rate of their parental wild-type stains (compare Fig. 6C with 6A and B).
Ex vivo stimulated fluid secretion from perfused submandibular glands
One interpretation of the in vivo results (Fig. 6) is that K+ channels do not play an important role in the submandibular gland secretion mechanism (in contrast to the results from our parotid gland study; Romanenko et al. 2006). Alternatively, the complex in vivo pharmacology of systemic administration of a broad-range cholinergic secretagogue (pilocarpine) may not be an appropriate method for dissection of the secretory mechanism in submandibular glands. To investigate in more detail the role of IK1 and maxi-K channels in submandibular saliva production we developed an ex vivo, perfused gland preparation and compared the fluid generated by KCa3.1-null, KCa1.1-null and double-null mice to the fluid secreted by the glands of their wild-type littermates. Vascular perfusion of the isolated submandibular permits control of the ion composition and agonist concentration, and eliminates circulating factors and neural inputs that complicated the interpretation of the in vivo experiments.
Figure 7 shows the saliva secretion rate induced by perfusion of ex vivo submandibular glands with 0.5 μm CCh. Figure 6, as well as our previous in vivo experiments (Romanenko et al. 2006), demonstrated that saliva secretion may significantly differ for different mouse strains. Therefore, each knockout strain was compared to wild-type littermates with the same genetic background. However, in contrast to our in vivo experiments, in which the wild-type mice for the double-null mice secreted about half as much saliva compared to the wild-types for KCa3.1- and KCa1.1-null animals (Fig. 6), ex vivo experiments showed that all wild-type mice, independent of the genetic background, produced saliva at a comparable rate of about 8 μl (100 mg gland weight)−1 min−1 (Fig. 7A–C).
Figure 7. Muscarinic-stimulated saliva secretion from isolated submandibular glands.
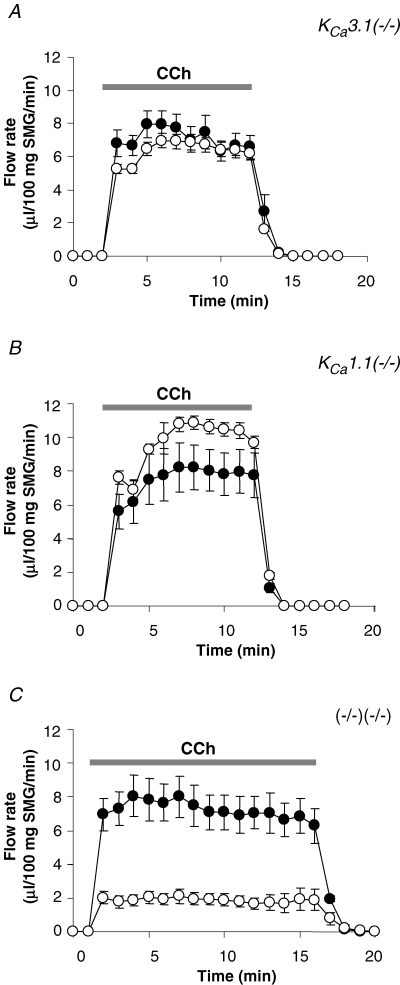
A, saliva secretion from ex vivo submandibular glands from either wild-type or KCa3.1-null mice, the artery and main excretory duct cannulated. Secretion was stimulated by vascular perfusion with 0.5 μm CCh: •– wild-type, ^–KCa3.1-null glands (n = 8 and 9 mice, respectively). B and C, same as described for A but fluid flow rates were compared in KCa1.1-null (n = 5 mice) and double-null (n = 5 mice) glands with their wild-type controls (n = 4 and n = 4 mice, respectively).
The saliva flow rates were similar between the wild-type (filled circles) and KCa3.1-null (open circles) animals (Fig. 7A). The fluid secretion rate was somewhat faster in KCa1.1-null mice (open circles) compared to the wild-type (filled circles) animals (Fig. 7B), resulting in about a 30% increase (n = 4 wild-type mice and n = 5 KCa1.1-null mice) in the total volume of fluid secreted. However, fluid secretion was significantly decreased (∼75%) in submandibular glands from double-null mice (open circles) compared to its wild-type (filled circles) mouse strain (Fig. 7C). Therefore, ex vivo fluid secretion was impaired only in mice lacking expression of both IK1 and maxi-K channels, consistent with our conclusion in our earlier study (Romanenko et al. 2006) that only one type of Ca2+-activated K channel is needed for normal fluid secretion.
Mechanism for membrane hyperpolarization in double-null mice
As shown above, double-null mice lack both IK1 and maxi-K channels (Fig. 2C), but the ex vivo, perfused submandibular glands were still able to secrete fluid, albeit at significantly reduced flow rates (Fig. 7C). Consistent with the residual secretion detected in the double-null mice, pharmacological inhibition and/or genetic ablation of IK1 and maxi-K channels did not lead to depolarization of stimulated acinar cells to the Cl− equilibrium potential (Fig. 5E). One possible explanation for the ‘background’ hyperpolarization and the fluid secretion in the double-null glands is that an electrogenic transporter provides sufficient hyperpolarization to drive a limited amount of secretion. As would be expected for an electrogenic transporter that moves 3 Na+ out of the cell in exchange for 2 extracellular K+, it has been reported that Na+,K+-ATPase can hyperpolarize acinar cell membrane (Roberts et al. 1978; Lee et al. 2005). We tested the possibility that the Na+ pump hyperpolarizes the cell when both IK1 and maxi-K channels are blocked by measuring the membrane potential of acinar cells in the presence of ouabain, an inhibitor of Na+,K+-ATPase. Figure 8 shows that ouabain alone produced only a modest effect on stimulated wild-type cells (Fig. 8A; −48 ± 3 mV, n = 5) and cells from either KCa3.1-null (−50 ± 5 mV, n = 5) or KCa1.1-null (−45 ± 4 mV, n = 5) mice (Fig. 8C), demonstrating that K+ channels are normally responsible for setting the Vm. However, ouabain superfusion depolarized the cell membrane to near the equilibrium potential for Cl− (−24 mV) when both IK1 and maxi-K channels were simultaneously inhibited (pharmacologically or genetically): −35 ± 4 mV (n = 5), −28 ± 4 mV (n = 5), −28 ± 4 mV (n = 5) and −27 ± 3 mV (n = 5) for wild-type, KCa3.1-, and KCa1.1- and double-null cells, respectively. These results suggest that Na+,K+-ATPase has the capacity to hyperpolarize Vm in the absence of IK1 and maxi-K channels, and under these severe conditions, can support some fluid secretion by submandibular glands as demonstrated in Fig. 7C.
Figure 8. Membrane depolarization upon inhibition of Na+,K+-ATPase.
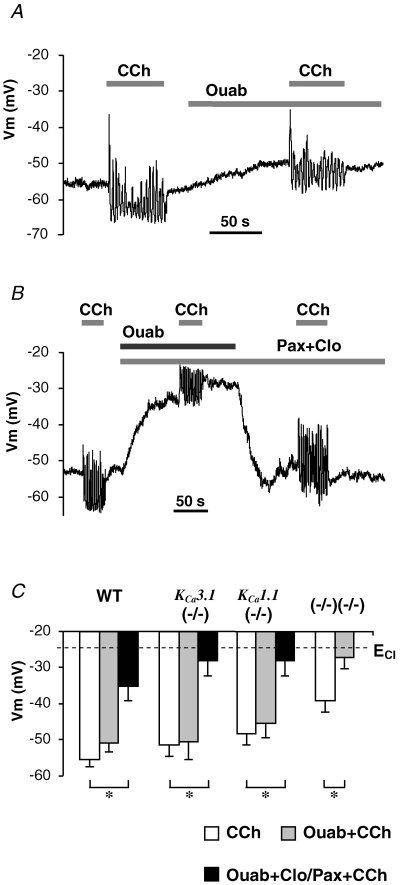
A and B, membrane potential traces recorded in wild-type cells stimulated with 0.3 μm CCh prior to or along with superfusion of ouabain alone (A) or with paxilline and clotrimazole (B). C, the mean values of Vm from cells during stimulation with no inhibitors (open bars), with K+ channel inhibitors (grey bars), or with both K+ channel inhibitors and Na+,K+-ATPase inhibitor (black bars). *P < 0.05.
Discussion
In the present study we verified the molecular identities of the two Ca2+-activated K+ currents expressed in the seromucous acinar cells of mouse submandibular glands and analysed their role in saliva secretion. We have shown that (i) transcripts were present for the KCa3.1 and KCa1.1 genes, which encode the IK1 and maxi-K channel proteins, respectively, (ii) the observed Ca2+-activated K+ currents had the biophysical and pharmacological footprints of IK1 and maxi-K channels, and (iii) these IK1- and maxi-K-like K+ currents were not present in acinar cells obtained from mice with disrupted KCa3.1 and KCa1.1 genes, respectively. Further, we determined that ablation of either the IK1 or maxi-K channel did not result in strong depolarization of the membrane potential of acinar cells and did not produce a significant effect on the volume of saliva secreted ex vivo. However, simultaneous disruption of both channels strongly depolarized the cell Vm levels and greatly impaired ex vivo saliva production. Moreover, the Na+,K+-ATPase inhibitor ouabain had only modest effects on the Vm of cells from wild-type or single knockout mice, but depolarized the double-null cells to near the Cl− equilibrium potential, suggesting that the residual saliva produced by the double knockout mice is driven by the electrogenic Na+ pump.
We also showed that muscarinic stimulation often induced synchronized oscillations of Vm, and K+ and Cl− currents in submandibular acinar cells. These oscillations are consistent with previously demonstrated oscillations of free cytosolic Ca2+ stimulation in intact salivary acinar cells (Gray, 1988, 1989; Bruce et al. 2002; Harmer et al. 2005). The Vm and current oscillations were of similar frequency in cells of all genotypes, including double knockout mice, supporting the previous proposal that cytosolic free Ca2+ regulates K+ and Cl− currents. Conversely, the stable Vm oscillations observed in cells lacking IK1 and/or maxi-K expression suggest that these channels do not play a significant part in modulating the frequency of the Ca2+ oscillations. Even though the physiological significance of these oscillations remains to be determined, we demonstrate that both oscillating and non-oscillating cells maintain a comparable average membrane hyperpolarization during stimulation, indicating tight coupling between the Ca2+-dependent K+ and Cl− currents and, thus supporting conditions favourable for secretion.
The fluid secretion mechanism appears to be qualitatively similar in mouse seromucous submandibular (present study) and serous parotid acinar cells (Nehrke et al. 2003; Begenisich et al. 2004; Melvin et al. 2005; Romanenko et al. 2006). As previously shown in the mouse parotid gland, normal fluid secretion and hyperpolarization of submandibular acinar cells requires the functional presence of only one of the two Ca2+-activated K+ channels. Therefore, the reason for the expression of two Ca2+-activated K+ channels in salivary acinar cells is unclear. It has been hypothesized that the differences in channel properties and the unique interaction between these two channels may be necessary to support efficient saliva secretion in response to various types of stimulation or to allow compensation when one channel is inactivated by (patho)physiological conditions (Melvin et al. 2005; Romanenko et al. 2006). Here we demonstrated that the properties and interaction of IK1 and maxi-K channels in mouse submandibular acinar cells are similar to those found in mouse and human parotid acinar cells (Maruyama et al. 1986; Park et al. 2001; Nehrke et al. 2003; Begenisich et al. 2004; Romanenko et al. 2006). Interestingly, we found no up-regulation of IK1 currents in cells from KCa1.1-null mice, while the apparent level of maxi-K currents in cells from KCa3.1-null mice was 2–3 times higher that in the wild-type cells. This suggests that IK1 may play a dominant role in membrane hyperpolarization during stimulated fluid secretion. Indeed, the lack of sensitivity of submandibular fluid secretion to the maxi-K channel blocker TEA is consistent with this observation (Ishikawa et al. 1994). The mechanism by which maxi-K currents in cells from KCa3.1-null mice increased can most easily be explained by an increase in the expression of maxi-K channels; however, we did not detect a change in the level of KCa1.1 transcripts by quantitative RT-PCR (n = 6; not shown). Alternatively, IK1 channel activation has been shown to inhibit maxi-K currents (Thompson & Begenisich, 2006). It was therefore not totally unexpected that the magnitude of maxi-K current increased in IK1-deficient mice.
Remarkably, when both of the K+ channel genes were disrupted, the Vm did not depolarize to the Cl− equilibrium potential, and the submandibular glands continued to secrete fluid, although at a reduced rate. The currently accepted secretion model states that the efflux of Cl− anions from acinar cells to the lumen drives fluid secretion. The electrogenic transport of Cl− across the apical membrane depolarizes the cell and therefore must be counteracted by a hyperpolarizing ion transporter(s). Our results indicate that in the absence of both IK1 and maxi-K channels, the Na+,K+-ATPase can significantly hyperpolarize the acinar cell membrane and therefore can support a low level of fluid secretion (∼27% of normal). Na+,K+-ATPase activity is associated with intracellular K+ accumulation, and hence, in the absence of K channels, there is a requirement for an alternative K+ efflux pathway. This pathway is most likely to be a Vm-independent secretory mechanism, taking into consideration that inhibition of the Na+ pump depolarized cells to near the Cl− equilibrium potential when IK1 and maxi-K currents were inhibited (Fig. 8). A possible candidate is the electroneutral K+–Cl− cotransporter KCC1, expression of which has been reported in rat salivary glands (Roussa et al. 2002).
The most surprising result in submandibular glands was the modest effect of knocking out both K+ channels on the in vivo stimulated fluid secretion (Fig. 6). In contrast, a dramatic decrease in secretion (∼70%) was observed in the parotid glands of KCa3.1−/−/KCa1.1−/− mice (Romanenko et al. 2006). The experimental conditions were the same in these two studies, including intraperitoneal injection of pilocarpine to stimulate secretion (10 mg (kg body weight)−1). Pilocarpine has been widely used in mammalian model systems to activate saliva production in vivo (e.g. Ma et al. 1999; Singh et al. 2001) and is used clinically to treat dry mouth (Wiseman & Faulds, 1995). Systemic administration of pilocarpine can induce salivation through at least two mechanisms: (i) direct – through activation of glandular cholinergic receptors, and (ii) neural – through the stimulation of the central nervous system (Renzi et al. 1993; Cecanho et al. 1999; Renzi et al. 2002; Takakura et al. 2003). Therefore, one possible explanation for this difference between the submandibular and parotid glands in vivo is that they display different sensitivities to cholinergic agonists. Indeed, we found that disruption of the M3 receptor, the predominant muscarinic receptor subtype in both parotid and submandibular glands (Matsui et al. 2000; Bymaster et al. 2003), dramatically reduced pilocarpine-stimulated fluid secretion by the parotid but had no significant effect on submandibular gland fluid production (supplemental material, Fig. S2). These latter results clearly demonstrate that cholinergic agonists have distinctive in vivo pharmacological effects on these two glands independent of glandular cholinergic receptors. Therefore, to eliminate potential systemic effects, we developed an ex vivo perfused gland system. It is interesting to note that the submandibular glands of the different wild-type strains produced considerably different volumes of saliva in vivo in response to cholinergic stimulation (Fig. 6). This difference disappeared in ex vivo stimulated glands (Fig. 7), consistent with the possibility that different strains display different responses to cholinergic agonists in vivo. As predicted for the functional roles of IK1 and maxi-K channels in the fluid secretion model, we found that the ex vivo glands from KCa3.1−/−/KCa1.1−/− mice produced much less fluid than wild-type littermates (Fig. 7), while secretion was not inhibited in the glands of single knockout mice.
In summary, seromucous submandibular acinar cells express IK1- and maxi-K-like currents. Targeted disruption of the KCa3.1 and the KCa1.1 genes confirmed the molecular identities of the channels producing these currents as IK1 and maxi-K channels, respectively. Null mutations of both genes were required to significantly affect the Vm of stimulated submandibular acinar cells. The results from our ex vivo stimulated gland studies supported the role predicted by our electrophysiological measurements of the Ca2+-activated K+ channels. In contrast, the in vivo assay indicated additional complexity for the regulation of the secretory mechanism in an intact animal. The lack of a significant effect on acinar cell secretion or Vm when the expression of only one of these channels was disrupted demonstrates that each of these channels can compensate for the loss of the other channel. Such redundancy indicates the importance of Ca2+-activated K channels to salivary gland physiology.
Acknowledgments
We thank Mark Wagner, Laurie Koek, and Jennifer Scantlin for technical assistance. This work was supported in part by NIH grants DE13539, DE08921 (to J.E.M.) and DE016960 (to T.B.).
Supplemental material
Online supplemental material for this paper can be accessed at:http://jp.physoc.org/cgi/content/full/jphysiol.2006.127498/DC1 and http://www.blackwell-synergy.com/doi/suppl/10.1113/jphysiol.2006.127498
References
- Begenisich T, Nakamoto T, Ovitt CE, Nehrke K, Brugnara C, Alper SL, Melvin JE. Physiological roles of the intermediate conductance, Ca2+-activated potassium channel Kcnn4. J Biol Chem. 2004;279:47681–47687. doi: 10.1074/jbc.M409627200. [DOI] [PubMed] [Google Scholar]
- Bruce JI, Shuttleworth TJ, Giovannucci DR, Yule DI. Phosphorylation of inositol 1,4,5-trisphosphate receptors in parotid acinar cells. A mechanism for the synergistic effects of cAMP on Ca2+ signaling. J Biol Chem. 2002;277:1340–1348. doi: 10.1074/jbc.M106609200. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Carter PA, Yamada M, Gomeza J, Wess J, Hamilton SE, Nathanson NM, McKinzie DL, Felder CC. Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine-induced seizure activity. Eur J Neurosci. 2003;17:1403–1410. doi: 10.1046/j.1460-9568.2003.02588.x. [DOI] [PubMed] [Google Scholar]
- Cecanho R, Anaya M, Renzi A, Menani JV, De Luca LA., Jr Sympathetic mediation of salivation induced by intracerebroventricular pilocarpine in rats. J Auton Nerv Syst. 1999;76:9–14. doi: 10.1016/s0165-1838(99)00002-8. [DOI] [PubMed] [Google Scholar]
- Cook DI, Van Lennep EW, Roberts ML, Young JA. Secretion by the Major Salivary Glands. New York: Raven Press; 1994. [Google Scholar]
- Evans RL, Park K, Turner RJ, Watson GE, Nguyen HV, Dennett MR, Hand AR, Flagella M, Shull GE, Melvin JE. Severe impairment of salivation in Na+/K+/2Cl– cotransporter (NKCC1) -deficient mice. J Biol Chem. 2000;275:26720–26726. doi: 10.1074/jbc.M003753200. [DOI] [PubMed] [Google Scholar]
- Foskett JK, Gunter-Smith PJ, Melvin JE, Turner RJ. Physiological localization of an agonist-sensitive pool of Ca2+ in parotid acinar cells. Proc Natl Acad Sci U S A. 1989;86:167–171. doi: 10.1073/pnas.86.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett JK, Melvin JE. Activation of salivary secretion: coupling of cell volume and [Ca2+]i in single cells. Science. 1989;244:1582–1585. doi: 10.1126/science.2500708. [DOI] [PubMed] [Google Scholar]
- Gray PT. Oscillations of free cytosolic calcium evoked by cholinergic and catecholaminergic agonists in rat parotid acinar cells. J Physiol. 1988;406:35–53. doi: 10.1113/jphysiol.1988.sp017367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray PT. The relation of elevation of cytosolic free calcium to activation of membrane conductance in rat parotid acinar cells. Proc R Soc Lond B Biol Sci. 1989;237:99–107. doi: 10.1098/rspb.1989.0039. [DOI] [PubMed] [Google Scholar]
- Harmer AR, Smith PM, Gallacher DV. Local and global calcium signals and fluid and electrolyte secretion in mouse submandibular acinar cells. Am J Physiol Gastrointest Liver Physiol. 2005;288:G118–G124. doi: 10.1152/ajpgi.00096.2004. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Kunii C, Takahata T, Ishikawa T. ATP-dependent regulation of SK4/IK1-like currents in rat submandibular acinar cells: possible role of cAMP-dependent protein kinase. Am J Physiol Cell Physiol. 2004;286:C635–C646. doi: 10.1152/ajpcell.00283.2003. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Poronnik P, Young JA, Cook DI. The ACh-evoked, Ca2+-activated whole-cell K+ current in mouse mandibular secretory cells. Whole-cell and fluorescence studies. J Membr Biol. 1996;152:253–259. doi: 10.1007/s002329900103. [DOI] [PubMed] [Google Scholar]
- Ishii TM, Silvia C, Hirschberg B, Bond CT, Adelman JP, Maylie J. A human intermediate conductance calcium-activated potassium channel. Proc Natl Acad Sci U S A. 1997;94:11651–11656. doi: 10.1073/pnas.94.21.11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Murakami M. Tetraethylammonium-insensitive, Ca2+-activated whole-cell K+ currents in rat submandibular acinar cells. Pflugers Arch. 1995;429:748–750. doi: 10.1007/BF00373998. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Murakami M, Seo Y. Basolateral K+ efflux is largely independent of maxi-K+ channels in rat submandibular glands during secretion. Pflugers Arch. 1994;428:516–525. doi: 10.1007/BF00374573. [DOI] [PubMed] [Google Scholar]
- Jorgensen TD, Jensen BS, Strobaek D, Christophersen P, Olesen SP, Ahring PK. Functional characterization of a cloned human intermediate-conductance Ca2+-activated K+ channel. Ann N Y Acad Sci. 1999;868:423–426. doi: 10.1111/j.1749-6632.1999.tb11305.x. [DOI] [PubMed] [Google Scholar]
- Latorre R, Oberhauser A, Labarca P, Alvarez O. Varieties of calcium-activated potassium channels. Annu Rev Physiol. 1989;51:385–399. doi: 10.1146/annurev.ph.51.030189.002125. [DOI] [PubMed] [Google Scholar]
- Lau KR, Howorth AJ, Case RM. The effects of bumetanide, amiloride and Ba2+ on fluid and electrolyte secretion in rabbit salivary gland. J Physiol. 1990;425:407–427. doi: 10.1113/jphysiol.1990.sp018111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Nam JH, Kim SJ. Muscarinic activation of Na+-dependent ion transporters and modulation by bicarbonate in rat submandibular gland acinus. Am J Physiol Gastrointest Liver Physiol. 2005;288:G822–G831. doi: 10.1152/ajpgi.00406.2004. [DOI] [PubMed] [Google Scholar]
- Logsdon NJ, Kang J, Togo JA, Christian EP, Aiyar J. A novel gene, hKCa4, encodes the calcium-activated potassium channel in human T lymphocytes. J Biol Chem. 1997;272:32723–32726. doi: 10.1074/jbc.272.52.32723. [DOI] [PubMed] [Google Scholar]
- Ma T, Song Y, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Defective secretion of saliva in transgenic mice lacking aquaporin-5 water channels. J Biol Chem. 1999;274:20071–20074. doi: 10.1074/jbc.274.29.20071. [DOI] [PubMed] [Google Scholar]
- Maruyama Y, Gallacher DV, Petersen OH. Voltage and Ca2+-activated K+ channel in baso-lateral acinar cell membranes of mammalian salivary glands. Nature. 1983;302:827–829. doi: 10.1038/302827a0. [DOI] [PubMed] [Google Scholar]
- Maruyama Y, Nishiyama A, Teshima T. Two types of cation channels in the basolateral cell membrane of human salivary gland acinar cells. Jpn J Physiol. 1986;36:219–223. doi: 10.2170/jjphysiol.36.219. [DOI] [PubMed] [Google Scholar]
- Matsui M, Motomura D, Karasawa H, Fujikawa T, Jiang J, Komiya Y, Takahashi S, Taketo MM. Multiple functional defects in peripheral autonomic organs in mice lacking muscarinic acetylcholine receptor gene for the M3 subtype. Proc Natl Acad Sci U S A. 2000;97:9579–9584. doi: 10.1073/pnas.97.17.9579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melvin JE, Yule D, Shuttleworth T, Begenisich T. Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu Rev Physiol. 2005;67:445–469. doi: 10.1146/annurev.physiol.67.041703.084745. [DOI] [PubMed] [Google Scholar]
- Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem. 2004;279:36746–36752. doi: 10.1074/jbc.M405621200. [DOI] [PubMed] [Google Scholar]
- Morris AP, Gallacher DV, Fuller CM, Scott J. Cholinergic receptor-regulation of potassium channels and potassium transport in human submandibular acinar cells. J Dent Res. 1987;66:541–546. doi: 10.1177/00220345870660022601. [DOI] [PubMed] [Google Scholar]
- Murakami M, Miyamoto S, Imai Y. Oxygen consumption for K+ uptake during post-stimulatory activation of Na+,K+-ATPase in perfused rat mandibular gland. J Physiol. 1990;426:127–143. doi: 10.1113/jphysiol.1990.sp018130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehrke K, Quinn CC, Begenisich T. Molecular identification of Ca2+-activated K+ channels in parotid acinar cells. Am J Physiol Cell Physiol. 2003;284:C535–C546. doi: 10.1152/ajpcell.00044.2002. [DOI] [PubMed] [Google Scholar]
- Pallanck L, Ganetzky B. Cloning and characterization of human and mouse homologs of the Drosophila calcium-activated potassium channel gene, slowpoke. Hum Mol Genet. 1994;3:1239–1243. doi: 10.1093/hmg/3.8.1239. [DOI] [PubMed] [Google Scholar]
- Park K, Case RM, Brown PD. Identification and regulation of K+ and Cl– channels in human parotid acinar cells. Arch Oral Biol. 2001;46:801–810. doi: 10.1016/s0003-9969(01)00047-4. [DOI] [PubMed] [Google Scholar]
- Renzi A, Colombari E, Mattos Filho TR, Silveira JE, Saad WA, Camargo LA, de Luca Junior LA, Derobio JG, Menani JV. Involvement of the central nervous system in the salivary secretion induced by pilocarpine in rats. J Dent Res. 1993;72:1481–1484. doi: 10.1177/00220345930720110401. [DOI] [PubMed] [Google Scholar]
- Renzi A, De Luca LA, Jr, Menani JV. Lesions of the lateral hypothalamus impair pilocarpine-induced salivation in rats. Brain Res Bull. 2002;58:455–459. doi: 10.1016/s0361-9230(02)00817-1. [DOI] [PubMed] [Google Scholar]
- Roberts ML, Iwatsuki N, Petersen OH. Parotid acinar cells: ionic dependence of acetylcholine-evoked membrane potential changes. Pflugers Arch. 1978;376:159–167. doi: 10.1007/BF00581579. [DOI] [PubMed] [Google Scholar]
- Romanenko V, Nakamoto T, Srivastava A, Melvin JE, Begenisich T. Molecular identification and physiological roles of parotid acinar cell maxi-K channels. J Biol Chem. 2006;281:27964–27972. doi: 10.1074/jbc.M603871200. [DOI] [PubMed] [Google Scholar]
- Roussa E, Shmukler BE, Wilhelm S, Casula S, Stuart-Tilley AK, Thevenod F, Alper SL. Immunolocalization of potassium-chloride cotransporter polypeptides in rat exocrine glands. Histochem Cell Biol. 2002;117:335–344. doi: 10.1007/s00418-002-0393-3. [DOI] [PubMed] [Google Scholar]
- Salkoff L, Butler A, Ferreira G, Santi C, Wei A. High-conductance potassium channels of the SLO family. Nat Rev Neurosci. 2006;7:921–931. doi: 10.1038/nrn1992. [DOI] [PubMed] [Google Scholar]
- Singh BB, Zheng C, Liu X, Lockwich T, Liao D, Zhu MX, Birnbaumer L, Ambudkar IS. Trp1-dependent enhancement of salivary gland fluid secretion: role of store-operated calcium entry. FASEB J. 2001;15:1652–1654. doi: 10.1096/fj.00-0749fje. [DOI] [PubMed] [Google Scholar]
- Takahata T, Hayashi M, Ishikawa T. SK4/IK1-like channels mediate TEA-insensitive, Ca2+-activated K+ currents in bovine parotid acinar cells. Am J Physiol Cell Physiol. 2003;284:C127–C144. doi: 10.1152/ajpcell.00250.2002. [DOI] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Laitano SC, De Luca Junior LA, Renzi A, Menani JV. Central muscarinic receptors signal pilocarpine-induced salivation. J Dent Res. 2003;82:993–997. doi: 10.1177/154405910308201211. [DOI] [PubMed] [Google Scholar]
- Thompson J, Begenisich T. Membrane-delimited inhibition of maxi-K channel activity by the intermediate conductance Ca2+-activated K channel. J Gen Physiol. 2006;127:159–169. doi: 10.1085/jgp.200509457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojyo Y, Tanimura A, Nezu A, Matsumoto Y. Activation of β-adrenoceptors does not cause any change in cytosolic Ca2+ distribution in rat parotid acinar cells. Eur J Pharmacol. 1998;360:73–79. doi: 10.1016/s0014-2999(98)00651-7. [DOI] [PubMed] [Google Scholar]
- Wegman EA, Ishikawa T, Young JA, Cook DI. Cation channels in basolateral membranes of sheep parotid secretory cells. Am J Physiol Gastrointest Liver Physiol. 1992;263:G786–G794. doi: 10.1152/ajpgi.1992.263.5.G786. [DOI] [PubMed] [Google Scholar]
- Wiseman LR, Faulds D. Oral pilocarpine: a review of its pharmacological properties and clinical potential in xerostomia. Drugs. 1995;49:143–155. doi: 10.2165/00003495-199549010-00010. [DOI] [PubMed] [Google Scholar]
- Young JA, Van Lennep EW. Morphology and physiology of salivary myoepithelial cells. Int Rev Physiol. 1977;12:105–125. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.